Green Chem., 2016, Advance Article DOI: 10.1039/C6GC02346K, Critical Review
Si Amar Dahoumane, Mourad Mechouet, Kushlani Wijesekera, Carlos D. M. Filipe, Clemence Sicard, Dennis A. Bazylinski, Clayton Jeffryes
This review presents an exhaustive and in-depth description of inorganic nanoparticle biosynthesis from photosynthetic organisms, known mechanisms and bio-applications.
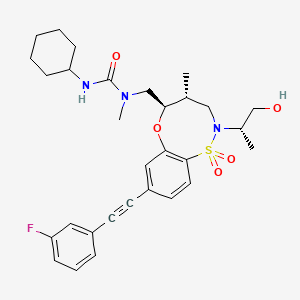
BRD2879 is a potent and cell-active inhibitor of IDH1-R132H with a markedly different structure from previously reported probes with (IC50 = 50 nM for inhibiting IDH1-R132H enzyme). BRD2879 represents a new structural class of mutant IDH1 inhibitors that, with optimization, may prove useful in the study of this enzyme and its role in cancer
Evidence suggests that specific mutations of isocitrate dehydrogenases 1 and 2 (IDH1/2) are critical for the initiation and maintenance of certain tumor types and that inhibiting these mutant enzymes with small molecules may be therapeutically beneficial. In order to discover mutant allele-selective IDH1 inhibitors with chemical features distinct from existing probes, we screened a collection of small molecules derived from diversity-oriented synthesis. The assay identified compounds that inhibit the IDH1-R132H mutant allele commonly found in glioma. Here, we report the discovery of a potent (IC50 = 50 nM) series of IDH1-R132H inhibitors having 8-membered ring sulfonamides as exemplified by the compound BRD2879. The inhibitors suppress (R)-2-hydroxyglutarate production in cells without apparent toxicity. Although the solubility and pharmacokinetic properties of the specific inhibitor BRD2879 prevent its use in vivo, the scaffold presents a validated starting point for the synthesis of future IDH1-R132H inhibitors having improved pharmacological properties.
Discovery of 8-Membered Ring Sulfonamides as Inhibitors of Oncogenic Mutant Isocitrate Dehydrogenase 1
Discovery of 8-Membered Ring Sulfonamides as Inhibitors of Oncogenic Mutant Isocitrate Dehydrogenase 1
Jason M. Law, Sebastian C. Stark, Ke Liu, Norah E. Liang, Mahmud M. Hussain, Matthias Leiendecker, Daisuke Ito, Oscar Verho, Andrew M. Stern, Stephen E. Johnston, Yan-Ling Zhang, Gavin P. Dunn, Alykhan F. Shamji, and Stuart L. Schreiber
Publication Date (Web): August 18, 2016 (Letter)
DOI: 10.1021/acsmedchemlett.6b00264
Green Chem., 2016, Advance Article DOI: 10.1039/C6GC02396G, Communication
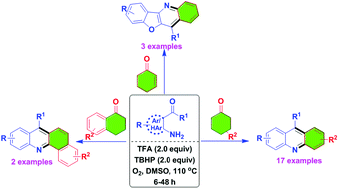
Gopal Chandru Senadi, Ganesh Kumar Dhandabani, Wan-Ping Hu, Jeh-Jeng Wang
We have identified metal-free reaction conditions for the annulation/aerobic oxidative dehydrogenation of cyclohexanones with o-acylanilines to the corresponding acridine derivatives.
Metal-free annulation/aerobic oxidative dehydrogenation of cyclohexanones with o-acylanilines: efficient syntheses of acridines
Metal-free annulation/aerobic oxidative dehydrogenation of cyclohexanones with o-acylanilines: efficient syntheses of acridines
CAS 1402438-74-7
Chemical Formula: C23H22ClN7O3
Molecular Weight: 479.925
Originator Pfizer
ClassAnalgesics; Small molecules
Mechanism of ActionUndefined mechanism
06 Oct 2014 Pfizer plans a phase I trial in Pain (In volunteers) in the Netherlands (NCT02260947)
07 Aug 2014 Discontinued – Phase-I for Pain (In volunteers) in Belgium (PO)
07 Aug 2014 Discontinued – Phase-I for Pain (In volunteers) in Singapore (PO)
PF-06273340 is a Potent, Selective, and Peripherally Restricted Pan-Trk Inhibitor with an excellent LipE profile (IC50 value: Trk-A = 6 nM; Trk-B = 4 nM; Trk-C = 3 nM). PF-06273340 has low metabolic turnover in HLM and hHep is a good substrate for efflux transporters P-gp (ER = 35.7) and BCRP (ER = 4.0) and has moderate passive permeability (RRCK Papp = 5.4 × 10−6 cm s−1). PF-06273340 is well-tolerated was selected as a candidate for clinical development.
Image may be NSFW. Clik here to view.
Tropomyosin-related kinases (Trks) are a family of receptor tyrosine kinases activated by neurotrophins. Trks play important roles in pain sensation as well as tumour cell growth and survival signaling. Thus, inhibitors of Trk receptor kinases might provide targeted treatments for conditions such as pain and cancer. Recent developments in this field have been reviewed by Wang et al in Expert Opin. Ther.
Patents (2009) 19(3): 305-319 and an extract is reproduced below.
“1.1 Trk receptors
As one of the largest family of proteins encoded by the human genome, protein kinases are the central regulators of signal transduction as well as control of various complex cell processes. Receptor tyrosine kinases (RTKs) are a subfamily of protein kinases (up to 100 members) bound to the cell membrane that specifically act on the tyrosine residues of proteins. One small group within this subfamily is the Trk kinases, with three highly homologous isoforms: TrkA, TrkB, and TrkC. All three isofonns are activated by high affinity growth factors named neurotrophins (NT): i) nerve growth factor (NGF), which activates TrkA; ii) brain-derived neurotrophic factor (BDNF) and NT-4/5, which activate TrkB; and iii) NT-3, which activates TrkC. The binding of neurotrophins to the extracellular domain of Trks causes the Trk kinase to autophosphorylate at several intracellular tyrosine sites and triggers downstream signal transduction pathways. Trks and neurotrophins are well known for their effects on neuronal growth and survival.
1.2 Trks and cancer
Originally isolated from neuronal tissues, Trks were thought to mainly affect the maintenance and survival of neuronal cells. However, in the past 20 years, increasing evidence has suggested that Trks play key roles in malignant transformation, chemotaxis, metastasis, and survival signaling in human tumors. The association between Trks and cancer focused on prostate cancer in earlier years and the topic has been reviewed. For example, it was reported that malignant prostate epithelial cells secrete a series of neurotrophins and at least one Trks. In pancreatic cancer, it was proposed that paracrine and/or autocrine neurotrophin-Trk interactions may influence the invasive behavior of the cancer. TrkB was also reported to be overexpressed in metastatic human pancreatic cancer cells. Recently, there have been a number of new findings in other cancer settings. For example, a translocation leads to expression of a fusion protein derived from the W-terminus of the ETV9 transcription factor and the C-terminal kinase domain of TrkC. The resulting ETV6-TrkC fusions are oncogenic in vitro and appear causative in secretory breast carcinoma and some acute myelogenous leukemias (AML). Constitutively active TrkA fusions occurred in a subset of papillary thyroid cancers and colon carcinomas. In neuroblastoma, TrkB expression was reported to be a strong predictor of aggressive tumor growth and poor prognosis, and TrkB overexpression was also associated with increased resistance to chemotherapy in neuroblastoma tumor cells in vitro. One report showed that a novel splice variant of TrkA called TrkAIII signaled in the absence of neurotrophins through the inositol phosphate-AKT pathway in a subset of neuroblastoma. Also, mutational analysis of the tyrosine kinome revealed that Trk mutations occurred in colorectal and lung cancers. In summary, Trks have been linked to a variety of human cancers, and discovering a Trk inhibitor and testing it clinically might provide further insight to the biological and medical hypothesis of treating cancer with targeted therapies.
1.3 Trks and pain
Besides the newly developed association with cancer, Trks are also being recognized as an important mediator of pain sensation. Congenital insensitivity to pain with anhidrosis (CIPA) is a disorder of the peripheral nerves (and normally innervated sweat glands) that prevents the patient from either being able to adequately perceive painful stimuli or to sweat. TrkA defects have been shown to cause CIPA in various ethnic groups.
Currently, non-steroidal anti-inflammatory drugs (NSAIDs) and opiates have low efficacy and/or side effects (e.g., gastrointestinal/renal and psychotropic side effects, respectively) against neuropathic pain and therefore development of novel pain treatments is highly desired. It has been recognized that NGF levels are elevated in response to chronic pain, injury and inflammation and the administration of exogenous NGF increases pain hypersensitivity. In addition, inhibition of NGF function with either anti- NGF antibodies or non-selective small molecule Trk inhibitors has been shown to have effects on pain in animal models. It appears that a selective Trk inhibitor (inhibiting at least NGF’s target, the TrkA receptor) might provide clinical benefit for the treatment of pain. Excellent earlier reviews have covered targeting NGF/BDNF for the treatment of pain so this review will only focus on small molecule Trk kinase inhibitors claimed against cancer and pain. However, it is notable that the NGF antibody tanezumab was very recently reported to show good efficacy in a Phase II trial against osteoarthritic knee pain.”
International Patent Application publication number WO2009/012283 refers to various fluorophenyl compounds as Trk inhibitors; International Patent Application publication numbers WO2009/152087, WO2008/080015 and WO2008/08001 and WO2009/152083 refer to various fused pyrroles as kinase modulators; International Patent Application publication numbers WO2009/143024 and WO2009/143018 refer to various pyrrolo[2,3-d]pyrimidines substituted as Trk inhibitors; International Patent Application publication numbers WO2004/056830 and WO2005/1 16035 describe various 4-amino-pyrrolo[2,3- d]pyrimidines as Trk inhibitors. International Patent Application publication number WO201 1/133637 describes various pyrrolo[2,3-d]pyrimidines and pyrrolo[2,3-b]pyridines as inhibitors of various kinases.
US provisional application US61/471758 was filed 5th April 2012 and the whole contents of that application in it’s entirety are herewith included by reference thereto.
Thus Trk inhibitors have a wide variety of potential medical uses. There is a need to provide new Trk inhibitors that are good drug candidates. In particular, compounds should preferably bind potently to the Trk receptors in a selective manner compared to other receptors, whilst showing little affinity for other receptors, including other kinase and / or GPC receptors, and show functional activity as Trk receptor antagonists. They should be non-toxic and demonstrate few side-effects. Furthermore, the ideal drug candidate will exist in a physical form that is stable, non-hygroscopic and easily formulated. They should preferably be e.g. well absorbed from the gastrointestinal tract, and / or be injectable directly into the bloodstream, muscle, or subcutaneously, and / or be metabolically stable and possess favourable pharmacokinetic properties.
Among the aims of this invention are to provide orally-active, efficacious, compounds and salts which can be used as active drug substances, particularly Trk antagonists, i.e. that block the intracellular kinase activity of the Trk, e.g. TrkA (NGF) receptor. Other desirable features include good HLM/hepatocyte stability, oral bioavailability, metabolic stability, absorption, selectivity over other types of kinase, dofetilide selectivity. Preferable compounds and salts will show a lack of CYP inhibition/induction, and be CNS- sparing.
(5-Chloropyridin-2-yl)acetic acid (26.1 g, 152 mmol) (see Preparation 90) was added to (5-aminopyridin- 3-yl){7-(2-{[ferf-butyl(dimethyl)silyl]oxy}-1 , 1-dimethylethyl)-2-[(2,4-dimethoxybenzyl)ami
d]pyrimidin-5-yl}methanone (75.0 g, 130 mmol ) (see Preparation 51 ), 1-propylphosphonic acid cyclic anhydride (187 mL, 317 mmol, 50% solution in EtOAc) and triethylamine (61.9 mL, 444 mmol ) in THF (423 mL). The mixture was stirred at 25°C for 2 hours then saturated aqueous sodium bicarbonate (400 mL) was added and the organic layer was separated. The aqueous phase was extracted with EtOAc (400 mL) and all organic phases were combined and dried over sodium sulfate then evaporated in vacuo. The residue brown solid was dissolved in trifluoroacetic acid (300 mL) and the solution was stirred at 50°C for 3 hours then evaporated in vacuo. Methanol (1800 mL) was added to the residue and the mixture was filtered. The filtrate was evaporated in vacuo and azeotroped with ethanol (3 x 200 mL).
Potassium carbonate (87.7 g, mmol) was added to the crude trifluoroacetamide in methanol (300 mL) and the mixture was stirred at room temperature for 16 hours. The mixture was poured into water (2000 mL) and filtered. The solid was washed with water (200 mL) then triturated with ethanol (2 x 200 mL at room temperature then 380 mL at 50°C) to afford the title compound as a yellow solid in 48% yield, 29.9 g. H NMR (400 MHz, DMSO-c/6) δ: 1.64 (s, 6H), 3.90 (d, 2H), 3.95 (s, 2H), 5.05 (t, 1 H), 6.54 (br s, 2H), 7.49 (d, 1 H), 7.69 (s, 1 H), 7.92 (dd, 1 H), 8.40 (m, 1 H), 8.56 (m, 1 H), 8.64 (d, 1 H), 8.94 (d, 1 H), 8.96 (s, 1 H), 10.71 (s, 1 H); LCMS (System 3): Rt = 9.92 min; m/z 480 [M+H]+.
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
The neurotrophin family of growth factors, comprised of nerve growth factor (NGF), brain derived neurotrophic factor (BDNF), neurotrophin 3 (NT3), and neurotrophin 4 (NT4), is implicated in the physiology of chronic pain. Given the clinical efficacy of anti-NGF monoclonal antibody (mAb) therapies, there is significant interest in the development of small molecule modulators of neurotrophin activity. Neurotrophins signal through the tropomyosin related kinase (Trk) family of tyrosine kinase receptors, hence Trk kinase inhibition represents a potentially “druggable” point of intervention. To deliver the safety profile required for chronic, nonlife threatening pain indications, highly kinase-selective Trk inhibitors with minimal brain availability are sought. Herein we describe how the use of SBDD, 2D QSAR models, and matched molecular pair data in compound design enabled the delivery of the highly potent, kinase-selective, and peripherally restricted clinical candidate PF-06273340.
ADDITIONAL INFORMATION
The aqueous solubility of PF-06273340 is 131 μM, much improved over previous analogues, it is highly kinase-selective (Gini score of 0.92) and has no measurable activity at the hERG channel. PF-06273340 was profiled in a series of in vitro safety assays, showing little cytotoxicity in THLE or HepG2 cell lines (IC50 > 42 μM and >300 μM, respectively) and was evaluated for broader pharmacological activity in a panel of receptors, ion channels, and enzymes. In this broad panel, all IC50/Ki values were >10 μM except for COX-1 (IC50 = 2.7 μM) and dopamine transporter assays (Ki = 5.2 μM) and PDEs 4D, 5A, 7B, 8B, and 11 (54−89% inhibition at 10 μM). PF-06273340 was screened in the Invitrogen wide kinase panel of 309 kinases, and all were inhibited by <40% when tested at 1 μM except the following: MUSK (IC50 53 nM), FLT-3 (IC50 395 nM), IRAK1 (IC50 2.5 μM), MKK (90% @ 1 μM), and DDR1 (60% @ 1 μM).
REFERENCES
1: Skerratt SE, Andrews MD, Bagal SK, Bilsland J, Brown D, Bungay PJ, Cole S,
Gibson KR, Jones R, Morao I, Nedderman A, Omoto K, Robinson C, Ryckmans T,
Skinner K, Stupple PA, Waldron G. The Discovery of a Potent, Selective and
Peripherally Restricted Pan-Trk Inhibitor (PF-06273340) for the Treatment of
Pain. J Med Chem. 2016 Oct 21. [Epub ahead of print] PubMed PMID: 27766865.
The aberrant activation of B-cells has been implicated in several types of cancers and hematological disorders. BTK and PI3Kδ are kinases responsible for B-cell signal transduction, and inhibitors of these enzymes have demonstrated clinical benefit in certain types of lymphoma. Simultaneous inhibition of these pathways could result in more robust responses or overcome resistance as observed in single agent use. We report a series of novel compounds that have low nanomolar potency against both BTK and PI3Kδ as well as acceptable PK properties that could be useful in the development of treatments against B-cell related diseases.
Image may be NSFW. Clik here to view.
Monali Banerjee
Director, R&D
Ms. Banerjee has more than 10 years of research experience, during which she has held positions of increasing responsibility. Her past organizations include TCG Lifesciences (Chembiotek) and Sphaera Pharma. Ms. Banerjee is a versatile scientist with a deep understanding of the fundamental issues that underlie various aspects of drug discovery. At Curadev, she has been responsible for target selection, patent analysis, pharmacophore design, assay development, ADME/PK and in vivo and in vitro pharmacology. Ms. Banerjee holds a Masters in Biochemistry and a Bachelors in Chemistry both from Kolkata University.
Image may be NSFW. Clik here to view.Nidhi Adlaka & Neha Munjal are developing a bioprocess for butanediol. Over the next few decades, chemical routes of manufacture will gradually be replaced by more environment friendly biological methods.
ENHANCED ANALYTICAL METHOD CONTROL STRATEGY CONCEPT
The benefits of quality by design (QbD) concepts related to both product (ICH Q8)1 and drug substance (ICH Q11)2 are well-established, particularly in regards to the potential to use knowledge to affect process changes without major regulatory hurdles, i.e., revalidation/regulatory filing, etc. Less wellestablished, but potentially of significant value, is the application of the same concepts to analytical methods.
Analytical methods play an obvious key role in establishing the quality of final product as they establish conformance with product acceptance criteria (i.e., specifications) and indicate the integrity of the product through indication of product stability. Analytical methods are validated, like manufacturing processes, but what if the operational ranges could be established during method validation when demonstrating fitness for purpose?
Would it be possible to drive method improvement, especially post validation in the same way that the concept of continuous improvement is a key driver for…
Zopiclone (Imovance), 4-methyl-1-piperzinecarboxylic acid 6-(5-chloro-2- pyridinyl)-6,7-dihydro-7-oxo-5H-pyrrolo[3,4-b]pyrazin-5-yl ester (Figure 1) is one of the non benzodiazepine sedative-hypnotics of the cyclopyrrolone class, sold by Rhone-Poulene Company in France since 1987. Although structurally unrelated to benzodiazepines, its pharmacological profile is similar, exhibiting sedative-hypnotic, anxiolytic, myorelaxant, and anticonvulsant activity.[1] Other than the first generation barbiturates and the second-generation benzodiazepines, zopiclone, which is widely used in Europe as well as other regions worldwide,[2,3] as a representative of the third generation sedative-hypnotic drugs, has been shown to be free from residual effects on performance and psychological function the day after intake and from the risks of accumulation because of its short elimination half-life (3.5 to 6.5 hours).[3,4] It is indicated for the short term treatment of insomnia, transient, situational or chronic insomnia, and insomnia secondary to psychiatric disturbances.[3]
REFERENCES 1. Mann, K.; Bauer, H.; Hiemke, C.; Ro¨schke, J.; Wetzel, H.; Benkert, O. Acute, subchronic and discontinuation effects of zopiclone on sleep EEG and nocturnal melatonin secretion. Eur. Neuropsychopharm. 1996, 6 (3), 163– 168. Structure Elucidation of Sedative-Hypnotic Zopiclone 359 Downloaded by [Dalhousie University] at 22:10 19 December 2012
2. Le´ger, D.; Janus, C.; Pellois, A.; Quera-Salva, M.A.; Dreyfus, J.P. Sleep, morning alertness and quality of life in subjects treated with zopiclone and in good sleepers. study comparing 167 patients and 381 good sleepers. Eur. Psychiat. 1995, 10 (973) Suppl. 3, 99s – 102s.
3. Piperaki, S.; Parissi-Poulou, M. Enantiomeric separation of zopiclone, its metabolites and products of degradation on a b-cyclodextrin bonded phase. J. Chromatogr. A 1996, 729 (1 – 2), 19 – 28
Spectral Data Analyses and Structure Elucidation of Sedative‐Hypnotic Zopiclone
Percent Composition: C 69.63%, H 11.04%, N 9.02%, O 10.31%
Literature References: Description: Bockmühl, Schaumann, Dtsch. Med. Wochenschr.54, 270 (1928). Pharmacokinetics and metabolism: H. Uehleke, M. Brinkschulte-Freitas, Arch. Pharmacol.302, 11 (1978). TLC determn in urine: E. Klug, P. Toffel, Arzneim.-Forsch.29, 1651 (1979).
Properties: White powder, mp 75-76°. Sol in 120 parts water; freely sol in alcohol, ether.
Acetylcholine chloride is obtained as white or off-white hygroscopic crystals, or as a crystalline powder. The salt is odorless, or nearly odorless, and is a very deliquescent powder. Acetylcholine bromide is obtained as deliquescent crystals, or as a white crystalline powder. The substance is hydrolyzed by hot water and alkali
Image may be NSFW. Clik here to view.
Acetylcholine is an organic chemical that functions in the brain and body of many types of animals, including humans, as a neurotransmitter—a chemical released by nerve cells to send signals to other cells. Its name is derived from its chemical structure: it is an ester of acetic acid and choline. Parts in the body that use or are affected by acetylcholine are referred to as cholinergic. Substances that interfere with acetylcholine activity are called anticholinergics.
Acetylcholine is the neurotransmitter used at the neuromuscular junction—in other words, it is the chemical that motor neurons of the nervous system release in order to activate muscles. This property means that drugs that affect cholinergic systems can have very dangerous effects ranging from paralysis to convulsions. Acetylcholine is also used as a neurotransmitter in the autonomic nervous system, both as an internal transmitter for the sympathetic nervous system and as the final product released by the parasympathetic nervous system.
Inside the brain, acetylcholine functions as a neuromodulator—a chemical that alters the way other brain structures process information rather than a chemical used to transmit information from point to point. The brain contains a number of cholinergic areas, each with distinct functions. They play an important role in arousal, attention, and motivation.
Partly because of its muscle-activating function, but also because of its functions in the autonomic nervous system and brain, a large number of important drugs exert their effects by altering cholinergic transmission. Numerous venoms and toxins produced by plants, animals, and bacteria, as well as chemical nerve agents such as Sarin, cause harm by inactivating or hyperactivating muscles via their influences on the neuromuscular junction. Drugs that act on muscarinic acetylcholine receptors, such as atropine, can be poisonous in large quantities, but in smaller doses they are commonly used to treat certain heart conditions and eye problems. Scopolamine, which acts mainly on muscarinic receptors in the brain, can cause delirium and amnesia. The addictive qualities of nicotine derive from its effects on nicotinic acetylcholine receptors in the brain.
Acetylcholine is a choline molecule that has been acetylated at the oxygen atom. Because of the presence of a highly polar, charged ammonium group, acetylcholine does not penetrate lipid membranes. Because of this, when the drug is introduced externally, it remains in the extracellular space and does not pass through the blood–brain barrier. A synonym of this drug is miochol.
When applying for a Certificate of Suitability (CEP) for an API, detailed information has to be provided regarding the synthesis stages, the starting material and the intermediates. In the event that the starting materials or the intermediates are already covered by a CEP, the EDQM has recently published a “Public Document” entitled “Use of a CEP to describe a material used in an application for another CEP”. The document contains regulations on how to reference the “CEP X” of a starting material or an intermediate in the application for the “CEP Y” of an API. The requirements for both scenarios are described as follows:
We would like to share our ~13 yrs of practical experience in the field of product development using statistical tools. But first, what compelled us to pursue six-sigma. Most of us started our career as a process chemist after completing PhD and it was during those initial days we realized the importance of “first time right” during commercialization. This enabled not only first mover advantage but also ensured timely and un-interrupted supply of our products into the market. Another aspect of the process development is its robustness, which ensures sustainable margins in whatever products we manufacture. Above achievement was possible only because of the six-sigma tools that we learned and applied at R&D stage. Latter we shifted our focus to the legacy products running in the plants, which we again studied using six-sigma tools to beat the eroding margins and this was possible because of few chemical engineers with six-sigma black belt joined the team.
As a chemist we were never trained on statistical tools hence, it was really a Herculean task for us to understand it. Another problem we faced was the statistical software, we would like to confess that we were never comfortable using these software as we were aware of “garbage in and garbage out” concept very well. We were never confident of the calculations thrown by software because we were not acquainted with the statistics. To site some examples
We were using regression analysis on five variables and found that we were getting an R Sq. of ~0.99 by including all five variables. We were happy about the results but we failed to realize that Adj. R Sq. has decreased while we added 4th and 5th variable, ending with a regression equation with un-necessary terms in it. As a result, we un-necessarily proposed control strategies for those insignificant variables which involved investment.
Another mistake we often made is to ignore the outliers during the Design of Experiments (DOE)! But we always wonder why we were ignoring these outliers? Just because we wanted to have a good regression equation? Are we not doubting our own experimental data? Later on we learned that if we keep ignoring the outliers just to have a good model, we would ultimately be modeling the system noise rather than modeling the effect. It is better to investigate the cause of outlier rather than ignoring it.
Above examples made us realize that having theoretical knowledge of six-sigma is not enough, it is the practical experience that really matters. Real challenge is the correct analysis of the experiments data so that the product could be scaled up without any problem. Learning to do the correct statistical analysis using any software was the mantra of the game. We should be confident that whatever output we are getting from the software is correct and this is possible only if we have good understanding of the statistical concepts. We are not saying that we should master the statistics but we must have clear understanding of the concepts before we use any software. It took us too long to understand these fundamentals aspects of applied statistics, main reason being the absence of statistical guru with adequate industrial experience. But major hurdle was to find a good tutor or at least a good book which can explain the concepts without involving too much of the statistics. We started looking for applied statistics courses and we found some solace in the “research methodology” module of MBA courses. Having gone through it, it gave us the confidence that six sigma tools can be learned without having in-depth knowledge of statistics.
During last 7-8 years we developed our own way of learning applied statistics with the help of diagrams and figures. During this journey we also found that each statistical topic have some connections with other topics and we can’t study any topic in isolation.
How normal distribution and hypothesis testing is working behind the scene in ANOVA, DoE, regression analysis and control charts.
Having gone through these hardship, I decided to share the experience with all those who like to understand the six sigma tools but are reluctant in doing so because of the statistics involved. Our website would help all six-sigma aspirants to understand the statistical concepts with the help of figures and diagrams. We would also be helping you in understanding the relationship between two unrelated topics like hypothesis testing and control charts.
Another feature that will help you is the solved example from the industry. Hence this would be an ideal website if you wish to appear for green/black belt exam from a reputed institute. We are saying this because we ourselves are ASQ certified six-sigma black belt and we want to share one important thing about the exam that we experienced, you can’t clear the exam unless you have understood the statistical concepts behind every six sigma tools. When we are saying understanding the statistical concepts, it doesn’t means learning pure statistics but only the concepts behind any tool, their advantages and limitations. This becomes important as ASQ never asks direct questions but questions are applied in nature. For example
A bulb production process found to follow normal distribution. A sample of 100 bulbs were drawn from a batch of 1000000 at random and found to have a mean life time of 1525 hrs. Historical mean life time was found to be 1548 hrs. with a standard deviation of 200 hrs. What is the percentage of bulbs having a life span of exactly 1548 hrs. from the current batch?
A manufacturing process was under optimization in a plant and a sample to 10 bags were selected at random from each batch. There were 5 batches in total and the mean weight (in Kgs) of the samples (10 bags) withdrawn are 100.5, 101.1, 99.8, 100.2 and 99.95. The range (in Kg) for these consecutive 5 batches were found to be 0.7, 0.9, 0.8, 0.9 and 1 Kg. Calculate the control limits for the chart.
Problem looks simple, in first case just calculate the z-value to tell the percentage and in the second case appears to be a direct question where we can easily calculate the control limits. But there is a catch, in first case probability for z = any number is zero! it is always about finding the probability between two numbers for a continuous probability distribution. In second case, if you missed the opening statement “under optimization” you are wasting your time in calculating the control limits, as control charts are always calculated for the stable process. In either of the question if you start the calculation, we can ensure that you won’t be finishing the exam in time!
Another major issue during applying six-sigma is the “use of right tool at the right place”. Hence our focus would not only to understand the concepts behind any statistical tools but also about selecting an appropriate tool for a given situation.
This website will start posting the six sigma topics (mainly statistical portion) from first week of January, 2017. Hence get registered on this course as soon as possible. The way we are planning to run the course is by posting one topic every week so that we can understand it well before taking subsequent topic. We are doing it in a slow pace because once we are on some advanced topic say “normal distribution” then at that time we should not be struggling with topics like variance, mean, z-transformation etc. Each topic will be followed by real life examples so that one can understand not only the concepts but also the use of appropriate tools. At the end of each topic we will also be demonstrating the use of excel sheet in resolving statistical problems. We are emphasizing on excel sheet as it is available to all. This would be our main USP during the course.
Sugammadex (brand name Bridion) is marketed by Merck Sharp and Dohme, and was approved by the United States FDA on December 15, 2015.
Sugammadex sodium was first approved by European Medicine Agency (EMA) on July 25, 2008, then approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jan 20, 2010, and approved by the US. food and drug administration (FDA) on Dec 15, 2015. It was developed and marketed as Bridion® by Merck Sharp & Dohme.
Sugammadex is reversal of neuromuscular blockade without relying on inhibition of acetylcholinesterase. It is the first selective relaxant binding agent (SRBA). It is indicated in neuromuscular blockade induced by rocuronium or vecuronium in adults.
Bridion® is available as injection solution for intravenous use, containing 100 mg /mL of free Sugammadex. The recommended dose is 4 mg/kg for adults if recovery has reached at least 1-2 post-tetanic counts (PTC) following rocuronium or vecuronium induced blockade.
Sugammadex Sodium is the sodium salt form of the biologically inert, selective relaxant binding agent (SRBA) sugammadex, a modified, anionic gamma cyclodextrinderivative containing a hydrophilic exterior and a hydrophobic core, with neuromuscular blocking drug (NMBD) reversal activity. Upon administration, the negatively charged carboxyl-thio-ether groups of sugammadex selectively and reversibly bind to the positively charged quaternary nitrogen of a steroidal NMBD, which was administered at an earlier time for anesthetic purposes. The encapsulation of the NMBD by sugammadex blocks its ability to bind to nicotinic receptors in the neuromuscular junction and thereby reverses the NMBD-induced neuromuscular blockade. Sugammadex binds rocuronium, vecuronium, and to a lesser extent pancuronium.
Sugammadex is a selective relaxant binding agent indicated for reversal of neuromuscular blockade induced by rocuronium bromide and vecuronium bromide during surgery in adults. Rocuronium bromide and vecuronium bromide are neuromuscular blocking medications that cause temporary paralysis and are especially useful for general anesthesia, ventilation, or tracheal intubation that patients may require for surgery. Sugammadex provides a new treatment option to reverse the effects of those medications and possibly help patients recover sooner post-surgery. Sugammadex (brand name Bridion) is marketed by Merck Sharp and Dohme, and was approved by the United States FDA on December 15, 2015.
Sugammadex is a modified γ-cyclodextrin, with a lipophilic core and a hydrophilic periphery. This gamma cyclodextrin has been modified from its natural state by placing eight carboxyl thio ether groups at the sixth carbon positions. These extensions extend the cavity size allowing greater encapsulation of the rocuronium molecule. These negatively charged extensions electrostatically bind to the quaternary nitrogen of the target as well as contribute to the aqueous nature of the cyclodextrin. Sugammadex’s binding encapsulation of rocuronium is one of the strongest among cyclodextrins and their guest molecules. The rocuronium molecule (a modified steroid) bound within sugammadex’s lipophilic core, is rendered unavailable to bind to the acetylcholine receptor at the neuromuscular junction.
Left: Schematic of a sugammadex molecule encapsulating a rocuronium molecule.
Right: Space-filling model of a sugammadex sodium molecule in the same orientation.
When muscle relaxant with rapid onset and short duration of action is required, there has been little choice apart from suxamethonium but this drug has important contraindications; for example, it can trigger malignant hyperthermia in susceptible individuals, it has a prolonged duration of action in patients with pseudocholinesterase deficiency and it causes an increase in plasma potassium concentration which is dangerous in some circumstances. Rocuronium has a comparably quick onset in high dose (0.6 mg kg−1 to 1 mg kg−1) and can be rapidly reversed with sugammadex (16 mg kg−1), so this drug combination offers an alternative to suxamethonium.
‘Recurarisation’, a phenomenon of recurrence of neuromuscular block, may occur where the reversal agents wear off before a neuromuscular blocking drug is completely cleared. This is very unusual with all but the longest acting neuromuscular blocking drugs (such as gallamine, pancuronium or tubocurarine). It has been demonstrated to occur only rarely with sugammadex, and only when insufficient doses were administered.[1] The underlying mechanism is thought to be related to redistribution of relaxant after reversal. It may occur for a limited range of sugammadex doses which are sufficient for complex formation with relaxant in the central compartment, but insufficient for additional relaxant returning to central from peripheral compartments.[2]
Sugammadex has been shown to have affinity for two other aminosteroid neuromuscular blocking agents, vecuronium and pancuronium. Although sugammadex has a lower affinity for vecuronium than for rocuronium, reversal of vecuronium is still effective because fewer vecuronium molecules are present in vivo for equivalent blockade: vecuronium is approximately seven times more potent than rocuronium. Sugammadex encapsulates with a 1:1 ratio and therefore will adequately reverse vecuronium as there are fewer molecules to bind compared to rocuronium.[3] Shallow pancuronium blockade has been successfully reversed by sugammadex in phase III clinical trials.[4]
Efficacy
A study was carried out in Europe looking at its suitability in rapid sequence induction. It found that sugammadex provides a rapid and dose-dependent reversal of neuromuscular blockade induced by high-dose rocuronium.[5]
A Cochrane review on sugammadex concluded that “sugammadex was shown to be more effective than placebo (no medication) or neostigmine in reversing muscle relaxation caused by neuromuscular blockade during surgery and is relatively safe. Serious complications occurred in less than 1% of the patients who received sugammadex. The results of this review article (especially the safety results) need to be confirmed by future trials on larger patient populations”.[6]
Tolerability
Sugammadex was generally well tolerated in clinical trials in surgical patients or healthy volunteers. In pooled analyses, the tolerability profile of sugammadex was generally similar to that of placebo or neostigmine plus glycopyrrolate.[7]
History
Sugammadex was discovered by the pharmaceutical company Organon at the Newhouse Research Site in Scotland.[8] Organon was acquired by Schering-Plough in 2007; Schering-Plough merged with Merck in 2009. Sugammadex is now owned and sold by Merck.
Sugammadex (Trade name: Bridion) is first disclosed in US6670340 assigned to Akzo Nobel. Sugammadex sodium was approved in EMEA as an agent for reversal of neuromuscular blockade by the agent rocuronium in general anaesthesia in 2008 and is the first selective relaxant binding agent (SRBA).
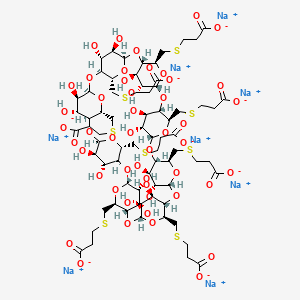
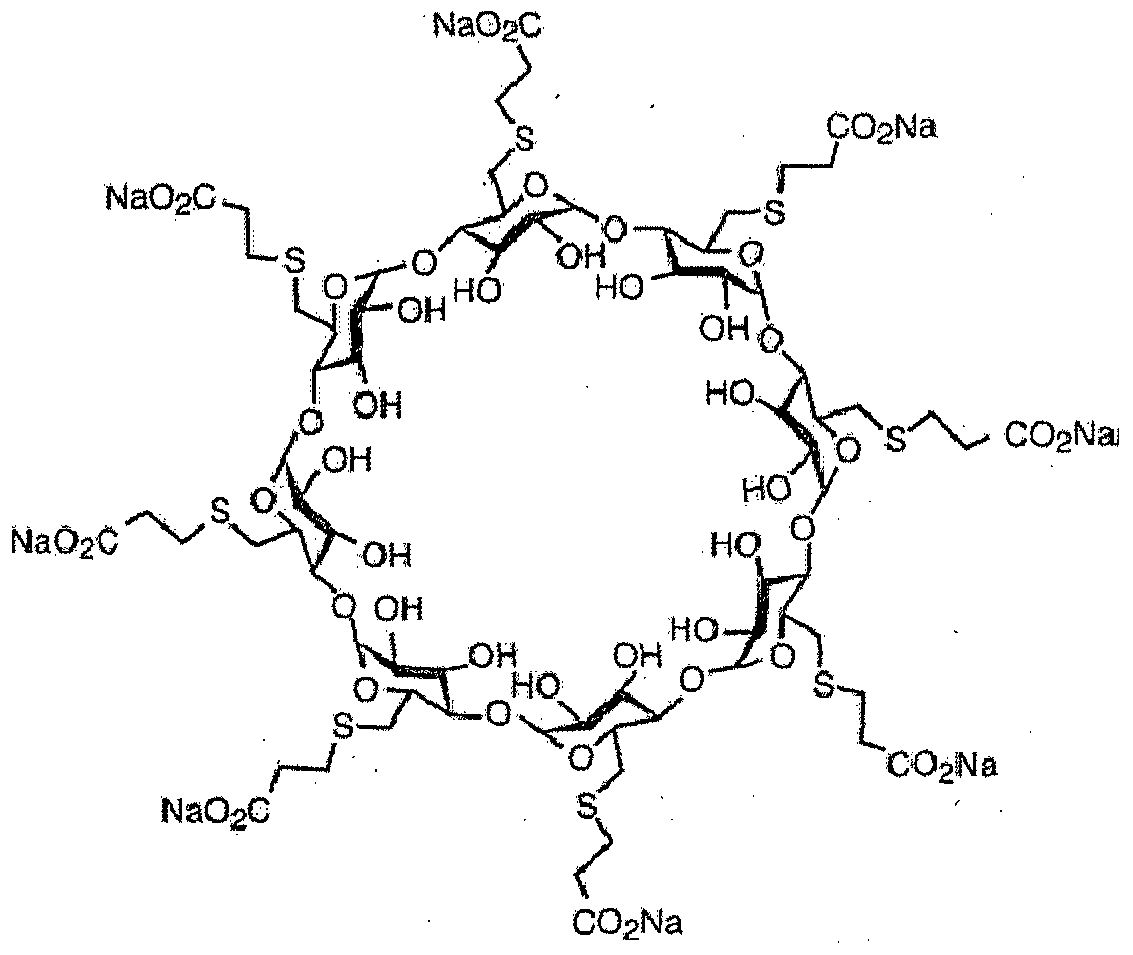
Sugammadex sodium contains 8 recurring glucose units each with 5 asymmetric carbon atoms, in total 40 asymmetric carbon atoms for the whole molecule. Sugammadex is a modified γ-cyclodextrin, with a lipophilic core and a hydrophilic periphery. The gamma cyclodextrin has been modified from its natural state by placing eight carboxyl thio ether groups at the sixth carbon positions.
The US Patent 6670340 assigned to Akzo Nobel discloses a process for preparing Sugammadex sodium as depicted in Scheme-I:
Sugammadex Sodium The first step in the process in the scheme-I involves the preparation of Vilsmeier Hack reagent by the reaction , of DMF, triphenylphosphine and Iodine. The triphenylphosphine oxide is formed as a byproduct of the first step. Removal of triphenylphosphine oxide from the product is very difficult from the reaction mass as it requires repeated washing with DMF under argon atmosphere, which leads to inconsistency in yield of final product Sugammadex. The second step involves the reaction of 6-perdeoxy-6-per-Iodo-Gamma cyclodextrin with 3-mercapto propionic acid in presence of alkali metal hydrides in an organic solvent to give 6-per-deoxy-6-per-(2-carboxyethyl)thio-y- cyclodextrin sodium salt.
The PCT publication WO2012/025937 discloses preparation of Sugammadex involving the reaction of gamma cyclodextrin with phosphorous halide in presence of organic solvent, thereby overcomes the formation of triphenyl phosphine oxide. The publication also discloses the use of 6-per deoxy-6-per- chloro-y-cyclodextrin in the preparation of the Sugammadex.
The purification techniques in the prior arts employ column chromatographic / membrane dialysis techniques which are costly and not convenient in large scale operations.
Image may be NSFW. Clik here to view.
Image may be NSFW. Clik here to view.
Sugammadex is a modified γ-cyclodextrin, with a lipophilic core and a hydrophilic periphery.
Sugammadex (designation Org 25969, trade name Bridion) is an agent for reversal of neuromuscular blockade by the agent rocuronium in general anaesthesia. It is the first selective relaxant binding agent (SRBA). This gamma cyclodextrin has been modified from its natural state by placing eight carboxyl thio ether groups at the sixth earbon positions. These extensions extend the cavity size allowing greater encapsulation of the rocuronium molecule. These negatively charged extensions electrostatically bind to the positively charged ammonium group as well as contribute to the aqueous nature of the cyclodextrin. Sugammadex’s binding encapsulation of rocuronium is one of the strongest among cyclodextrins and their guest molecules. The rocuronium molecule (a modified steroid) bound within Sugammadex’s lipophilic core, is rendered unavailable to bind to the acetylcholine receptor at the neuromuscular junction. Sugammadex sodium contains 8 recurring glucose units each with 5 asymmetric carbon atoms, in total 40 asymmetric carbon atoms for the whole molecule.
The Sugammadex was disclosed in US6670340 by Akzo Nobel. The process for preparing Sugammadex is there outlined as follows: (Scheme-I)
above process step-1 involves the preparation of Vilsmeier Hack reagent by the reaction of DMF, triphenylphosphine and Iodine. Drawback associated with this step is formation of triphenylphosphine oxide as a byproduct. Removal of triphenylphosphine oxide is very difficult from the reaction; it requires repeated washing with DMF under argon atmosphere and leads to inconsistency in yield of final product. Due to this, process is lengthy and not feasible on commercial scale.
[0018] The alkali metal hydrides are selected from the group consisting of sodium hydride, lithium hydride, potassium hydride preferably sodium hydride. [0019] The advantage of the present process is there that there is no formation of by product such as triphenylphosphine oxide, as present in prior art process. So, purification is not required which leads to better purity and yields for the intermediate as well as for final product.
[0020] Another advantage of the present invention is the significant difference between molecular weight of 6-per deoxy-6-per-chloro-y-cyclodextrin (Mol. wt. 1444) and the final product (Mol. wt. 2178). The use of 6-per deoxy-6-per- chforo-y-cyclodextrin instead of 6-per deoxy-6-per-bromo-y-cyclodextrin (Mol. wt. 1800) in the final stage‘ of the process would extend the scope of selection of appropriate dialysis membranes with precise molecular weight cut off and there by facilitate efficient purification of Sugammadex. The invention is further illustrated with following non-limiting examples:
Example: 1 Preparation of 6-perdeoxy-6-per-bromo Gamma Cyclodextrin
[0021] A portion of phosphorous pentachloride (256.5 g) was added in DMF (300 ml) at 0-5°C. Mixture was stirred at 20-25°C for lhr. A solution of gamma- cyclodextrin (50 g) in DMF (400ml) was added to above solution at 5-10°C under nitrogen. Mixture was stirred at 65 -70°C 14 hrs. The reaction mixture was cooled to 20 – 25°C and DMF was removed under vacuum. The viscous residue was diluted with water. 5M NaOH solution was added dropwise to the above solution at 5-10°C until PH=8, the resulting slurry was stirred for one hour at 20-25°C. The slurry was filtered under vacuum and washed with water and dried. The crude product was diluted with water and resulting slurry was stirred at 20-25uC for one hour. The slurry was filtered under vacuum and the solid dried at 55- 60°C under vacuum for 12hrs. (Yield – 94 – 98%, purity-98.5% by HPLC) Example: 2 Preparation of Sugammadax
[0022] To a mixture of sodium hydride (24.4 g) in DMF (150 ml) at 0-5°C, a solution of 3-mercapto propionic acid (23.7 ml, 10 eq) in DMF (50 ml) was added slowly under argon maintaining the temperature below 10 C. The resulting mixture was stirred at 20 -25°C for 30 mins. Then 6-deoxy-6-chloro gamma cyclodextrin (40 g) in DMF (400 ml) was added slowly at 5-10°C under argon and the resulting mixture was heated to 70-75°C for 12 hrs. Reaction mixture was cooled to 20 -25°C and DMF removed partially under vacuum and the reaction mixture is diluted with ethanol (600 ml). The resulting precipitate was stirred at 20 – 25°C for 1 hr and filtered under vacuum and the solid dried to afford the crude Sugammadex (wet) (100 g). The crude product was purified over silica gel and sephadex G-25 column using water as eluent. (Yield 60%)
CLIP
Sugammadex Sodium
Synonyms:ORG-25969
ATC:V03AB35
Use:Lorem
Chemical name:Lorem ipsum dolor sit amet, consetetur sadipscing elitr, sed diam
EXAMPLES Example: 1 Preparation of 6-perdeoxy-6-per-chloro Gamma Cyclodextrin
256.8 g (0.62 Moles) of Phosphorous pentachloride was added to 400 ml of Dimethyformamide (DMF) at 25-30 °C and mixture was maintained for 1 hour at the same temperature. 100 g (0.04 Moles) of Gamma-cyclodextrin was gradually added to the reaction mixture at 25-30 °C under nitrogen. The temperature of the reaction mixture was raised to 65 -70 °C and maintained at the same temperature for 14 to 16 hrs. The reaction mixture was then slowly added to chilled water at 0- 15 °C. The pH of the reaction mass was adjusted to 7-8 with 30% solution of sodium hydroxide in water. The contents were stirred at 25-30 °C at 2 hours. The resultant solid was filtered and washed with water (200 ml). The wet solid was repeatedly washed with purified water at 25-30 °C and dried at 65-70 °C till the moisture level was reduced to less than 4.0%. The yield of the obtained product was 90% Example: 2 Preparation of Sugammadex sodium
To a mixture of 1 10.2 g, (15 equ.) 3-mercapto propionic acid and 800 ml Dimethyl formamide (DMF) , a 30% solution of sodium methoxide (373.9 g, 30 equ) in methanol was added at 20-25°C and stirred for 1 hour at the same temperature. The compound from example- 1 (100 g) was added to the reaction mixture at 25-30°Cand heated to 75-80°C and maintained at the 75-80°C for 12 to 14 hours. After completion of the reaction, the reaction mass was cooled to 20- 25°C, then methanol (1000 ml) was added to the reaction mass and stirred for 2 hours at the same temperature. The resultant solid was filtered, washed with methanol (200 ml) and dried for 60-65°C for 8 hrs.
The crude product was dissolved in water (294 ml) and methanol (294 ml), treated with activated carbon (39.2 g, 20 % w/w) and was filtered through celite, washed the carbon cake with purified water (98 ml). The filtrate was heated to 50-55°C and slowly methanol (2646 ml) was added at the same temperature. The contents were cooled to 20 to 25°C and stirred for 2 hours at the same temperature. The resulted solid was washed with methanol (200 ml) and dried under vacuum at 60-65°C for 14 hours. The obtained product had yield of 70.34% and HPLC purity of 99.43 %.
Example-3
The Sugammadex prepared from example-2 was, dissolved in water ( 150 ml) and methanol (1 50 ml), treated with activated carbon (20 g) and filtered the carbon cake through celite bed and the carbon cake was washed with purified water (50 mL). The filtrate was heated to 50-55°C and added methanol (1350 ml) at the same temperature. The contents were cooled to 20 to 25°C and stirred for 2 hours at the same temperature. The resultant solid was washed with methanol (200 ml) and dried in vacuum at 70 – 75°C for 24 hrs. The obtained yield was 63%.
ugammadex [6A,6B,6C,6D,6E,6F,6G,6H-octakis-S- (2-carboxyethyl)-6A,6B,6C,6D,6E,6F,6G,6H- octathio-γ-cyclodextrin octasodium salt] is a modified γ-cyclodextrin [Figure 1]. Chemical formula is C 72 H 104 Na 8 O 48 S 8. “Su” stands for sugar and “gammadex” stands for structural molecule gamma-cyclodextrin. [1] Cyclodextrins are cyclic dextrose units joined through 1-4 glycosyl bonds that are produced from starch or starch derivates using cyclodextrin glycosyltransferase. The three natural unmodified cyclodextrins are α-, β-or γ-cyclodextrin. Compared with α-and β-cyclodextrins, γ-cyclodextrin exhibits more favorable properties in terms of the size of its internal cavity, water solubility and bioavailability. To have a better fit of the larger rigid structure of the aminosteroid muscle relaxant molecule (e.g., rocuronium or vecuronium) within the cavity of γ-cyclodextrin, the latter was modified by adding eight side chains to extend the cavity. This modification allowed the four hydrophobic steroidal rings of rocuronium to be better accommodated within the hydrophobic cavity. In addition, adding negatively charged carboxyl groups at the end of the eight side chains served two purposes. First, the repellent forces of the negative charges keep propionic acid side chains from being disordered, thereby allowing the cavity to remain open and maintain structural integrity. Second, adding these negatively charged carboxyl groups enhances electrostatic binding to the positively charged quaternary nitrogen of rocuronium [Figure 1]. Three-dimensional structure resembles a hollow, truncated cone or a doughnut. [9]
Jump up^Eleveld DJ; Kuizenga, K; Proost, JH; Wierda, JM (2008). “A Temporary Decrease in Twitch Response During Reversal of Rocuronium-Induced Muscle Relaxation with a Small Dose of Sugammadex”. Anesth Analg. 104 (3): 582–4. doi:10.1213/01.ane.0000250617.79166.7f. PMID17312212.
Jump up^Welliver M (2006). “New drug sugammadex; A selective relaxant binding agent”. AANA J74(5): 357–363. PMID 17048555
Jump up^Decoopman M (2007). “Reversal of pancuronium-induced block by the selective relaxant binding agent sugammadex”. Eur J Anaesthesiol. 24(Suppl 39):110-111.
Jump up^Pühringer FK, Rex C, Sielenkämper AW, et al. (August 2008). “Reversal of profound, high-dose rocuronium-induced neuromuscular blockade by sugammadex at two different time points: an international, multicenter, randomized, dose-finding, safety assessor-blinded, phase II trial”. Anesthesiology. 109 (2): 188–97. doi:10.1097/ALN.0b013e31817f5bc7. PMID18648227.
Jump up^Abrishami A, Ho J, Wong J, Yin L, Chung F. (October 2009). Abrishami, Amir, ed. “Sugammadex, a selective reversal medication for preventing postoperative residual neuromuscular blockade”. Cochrane Database of Systematic Reviews (4): CD007362. doi:10.1002/14651858.CD007362.pub2. PMID19821409.
1: Takazawa T, Mitsuhata H, Mertes PM. Sugammadex and rocuronium-induced anaphylaxis. J Anesth. 2016 Apr;30(2):290-7. doi: 10.1007/s00540-015-2105-x. Epub 2015 Dec 8. Review. PubMed PMID: 26646837; PubMed Central PMCID: PMC4819478.
2: Abad-Gurumeta A, Ripollés-Melchor J, Casans-Francés R, Espinosa A, Martínez-Hurtado E, Fernández-Pérez C, Ramírez JM, López-Timoneda F, Calvo-Vecino JM; Evidence Anaesthesia Review Group. A systematic review of sugammadex vs neostigmine for reversal of neuromuscular blockade. Anaesthesia. 2015 Dec;70(12):1441-52. doi: 10.1111/anae.13277. Review. PubMed PMID: 26558858.
3: Ledowski T. Sugammadex: what do we know and what do we still need to know? A review of the recent (2013 to 2014) literature. Anaesth Intensive Care. 2015 Jan;43(1):14-22. Review. PubMed PMID: 25579285.
4: Partownavid P, Romito BT, Ching W, Berry AA, Barkulis CT, Nguyen KP, Jahr JS. Sugammadex: A Comprehensive Review of the Published Human Science, Including Renal Studies. Am J Ther. 2015 Jul-Aug;22(4):298-317. doi: 10.1097/MJT.0000000000000103. Review. PubMed PMID: 25299638.
5: Jahr JS, Miller JE, Hiruma J, Emaus K, You M, Meistelman C. Sugammadex: A Scientific Review Including Safety and Efficacy, Update on Regulatory Issues, and Clinical Use in Europe. Am J Ther. 2015 Jul-Aug;22(4):288-97. doi: 10.1097/MJT.0000000000000092. Review. PubMed PMID: 25299637.
6: de Boer HD, Shields MO, Booij LH. Reversal of neuromuscular blockade with sugammadex in patients with myasthenia gravis: a case series of 21 patients and review of the literature. Eur J Anaesthesiol. 2014 Dec;31(12):715-21. doi: 10.1097/EJA.0000000000000153. Review. PubMed PMID: 25192270.
7: Tsur A, Kalansky A. Hypersensitivity associated with sugammadex administration: a systematic review. Anaesthesia. 2014 Nov;69(11):1251-7. doi: 10.1111/anae.12736. Epub 2014 May 22. Review. PubMed PMID: 24848211.
8: Luxen J, Trentzsch H, Urban B. [Rocuronium and sugammadex in emergency medicine: requirements of a muscle relaxant for rapid sequence induction]. Anaesthesist. 2014 Apr;63(4):331-7. doi: 10.1007/s00101-014-2303-1. Review. German. PubMed PMID: 24595442.
9: Fuchs-Buder T, Meistelman C, Raft J. Sugammadex: clinical development and practical use. Korean J Anesthesiol. 2013 Dec;65(6):495-500. doi: 10.4097/kjae.2013.65.6.495. Epub 2013 Dec 26. Review. PubMed PMID: 24427454; PubMed Central PMCID: PMC3888841.
10: Dubois PE, Mulier JP. A review of the interest of sugammadex for deep neuromuscular blockade management in Belgium. Acta Anaesthesiol Belg. 2013;64(2):49-60. Review. PubMed PMID: 24191526.
11: Van Gestel L, Cammu G. Is the effect of sugammadex always rapid in onset? Acta Anaesthesiol Belg. 2013;64(2):41-7. Review. PubMed PMID: 24191525.
12: Schaller SJ, Fink H. Sugammadex as a reversal agent for neuromuscular block: an evidence-based review. Core Evid. 2013;8:57-67. doi: 10.2147/CE.S35675. Epub 2013 Sep 25. Review. PubMed PMID: 24098155; PubMed Central PMCID: PMC3789633.
13: Nag K, Singh DR, Shetti AN, Kumar H, Sivashanmugam T, Parthasarathy S. Sugammadex: A revolutionary drug in neuromuscular pharmacology. Anesth Essays Res. 2013 Sep-Dec;7(3):302-6. doi: 10.4103/0259-1162.123211. Review. PubMed PMID: 25885973; PubMed Central PMCID: PMC4173552.
14: Karalapillai D, Kaufman M, Weinberg L. Sugammadex. Crit Care Resusc. 2013 Mar;15(1):57-62. Review. PubMed PMID: 23432503.
15: Øberg E, Claudius C. [Possible clinical potential in reverting muscular block with sugammadex in anaesthesia and surgery]. Ugeskr Laeger. 2013 Feb 11;175(7):428-32. Review. Danish. PubMed PMID: 23402253.
16: Della Rocca G, Di Marco P, Beretta L, De Gaudio AR, Ori C, Mastronardi P. Do we need to use sugammadex at the end of a general anesthesia to reverse the action of neuromuscular bloking agents? Position Paper on Sugammadex use. Minerva Anestesiol. 2013 Jun;79(6):661-6. Epub 2012 Nov 29. Review. PubMed PMID: 23192221.
17: Stair C, Fernandez-Bustamante A. Sugammadex, the first selective relaxant binding agent for neuromuscular block reversal. Drugs Today (Barc). 2012 Jun;48(6):405-13. doi: 10.1358/dot.2012.48.6.1813474. Review. PubMed PMID: 22745926.
18: Baldo BA, McDonnell NJ, Pham NH. The cyclodextrin sugammadex and anaphylaxis to rocuronium: is rocuronium still potentially allergenic in the inclusion complex form? Mini Rev Med Chem. 2012 Jul;12(8):701-12. Review. PubMed PMID: 22512555.
20: Baldo BA, McDonnell NJ, Pham NH. Drug-specific cyclodextrins with emphasis on sugammadex, the neuromuscular blocker rocuronium and perioperative anaphylaxis: implications for drug allergy. Clin Exp Allergy. 2011 Dec;41(12):1663-78. doi: 10.1111/j.1365-2222.2011.03805.x. Epub 2011 Jul 7. Review. PubMed PMID: 21732999.
BRIDION (sugammadex) injection, for intravenous use, contains sugammadex sodium, a modified gamma cyclodextrin chemically designated as 6A,6B,6C,6D,6E,6F,6G,6H-Octakis-S-(2-carboxyethyl)6A,6B,6C,6D,6E,6F,6G,6H-octathio-γ-cyclodextrin sodium salt (1:8) with a molecular weight of 2178.01. The structural formula is:
Image may be NSFW. Clik here to view.
BRIDION is supplied as a sterile, non-pyrogenic aqueous solution that is clear, colorless to slightly yellow-brown for intravenous injection only. Each mL contains 100 mg sugammadex, which is equivalent to 108.8 mg sugammadex sodium. The aqueous solution is adjusted to a pH of between 7 and 8 with hydrochloric acid and/or sodium hydroxide. The osmolality of the product is between 300 and 500 mOsmol/kg.
BRIDION may contain up to 7 mg/mL of the mono OH-derivative of sugammadex [see CLINICAL PHARMACOLOGY]. This derivative is chemically designated as 6A,6B,6C,6D,6E,6F,6G-Heptakis-S-(2carboxyethyl)-6A,6B,6C,6D,6E,6F,6G-heptathio-γ-cyclodextrin sodium salt (1:7) with a molecular weight of 2067.90. The structural formula is:
FDA approves Intrarosa for postmenopausal women experiencing pain during sex
The U.S. Food and Drug Administration approved Intrarosa (prasterone) to treat women experiencing moderate to severe pain during sexual intercourse (dyspareunia), a symptom of vulvar and vaginal atrophy (VVA), due to menopause. Intrarosa is the first FDA approved product containing the active ingredient prasterone, which is also known as dehydroepiandrosterone (DHEA).
The U.S. Food and Drug Administration approved Intrarosa (prasterone) to treat women experiencing moderate to severe pain during sexual intercourse (dyspareunia), a symptom of vulvar and vaginal atrophy (VVA), due to menopause. Intrarosa is the first FDA approved product containing the active ingredient prasterone, which is also known as dehydroepiandrosterone (DHEA).
During menopause, levels of estrogen decline in vaginal tissues, which may cause a condition known as VVA, leading to symptoms such as pain during sexual intercourse.
“Pain during sexual intercourse is one of the most frequent symptoms of VVA reported by postmenopausal women,” said Audrey Gassman, M.D., deputy director of the Division of Bone, Reproductive, and Urologic Products (DBRUP) in the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research (CDER). “Intrarosa provides an additional treatment option for women seeking relief of dyspareunia caused by VVA.”
Efficacy of Intrarosa, a once-daily vaginal insert, was established in two 12-week placebo-controlled clinical trials of 406 healthy postmenopausal women, 40 to 80 years of age, who identified moderate to severe pain during sexual intercourse as their most bothersome symptom of VVA. Women were randomly assigned to receive Intrarosa or a placebo vaginal insert. Intrarosa, when compared to placebo, was shown to reduce the severity of pain experienced during sexual intercourse.
The safety of Intrarosa was established in four 12-week placebo-controlled trials and one 52-week open-label trial. The most common adverse reactions were vaginal discharge and abnormal Pap smear.
Although DHEA is included in some dietary supplements, the efficacy and safety of those products have not been established for diagnosing, curing, mitigating, treating or preventing any disease.
Intrarosa is marketed by Quebec-based Endoceutics Inc.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Chapter 5 of the EC GMP Guide for the area of production was updated last year. This chapter contains concrete information about the conditions when testing and sampling of APIs and excipients can be reduced. Read more here about the sections 5.35 and 5.36 of the EU GMP Guide.
Chapter 5 of the EC GMP Guide for the area of production was already updated last year. However, not everybody really knows that it contains concrete information about the conditions when testing and sampling of APIs and excipients can be reduced. Particularly sections 5.35 and 5.36 include requirements and thus show possibilities for a reduction.
Basically, the manufacturers of finished products are responsible for every testing of starting materials as described in the marketing authorisation dossier. Yet, part of or complete test results from the approved starting material manufacturer can be used, but at least their identity has to be tested…
When applying for a Certificate of Suitability (CEP) for an API, detailed information has to be provided regarding the synthesis stages, the starting material and the intermediates. In the event that the starting materials or the intermediates are already covered by a CEP, the EDQM has recently published a “Public Document” entitled “Use of a CEP to describe a material used in an application for another CEP”. The document contains regulations on how to reference the “CEP X” of a starting material or an intermediate in the application for the “CEP Y” of an API. The requirements for both scenarios are described as follows:
Now online – Stimuli article on the proposed USP General Chapter “The Analytical Procedure Lifecycle <1220>”
A Stimuli Article to the Revision Process regarding the proposed New USP General Chapter “The Analytical Procedure Lifecycle <1220>” has been published. Read more about the new concept for the lifecycle managment of analytical methods.
Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.
The General Chapters—Chemical Analysis Expert Committee is currently developing a new general chapter <1220> The Analytical Procedure Lifecycle. The purpose of this new chapter will be to more fully address the entire procedure lifecycle and define concepts that may be useful.
A Stimuli article on the proposed General Chapter <1220> has been approved for publication in Pharmacopeial Forum 43(1) [Jan.-Feb. 2017]. USP is providing this Stimuli article in advance of its publication to provide additional time for comments.
In addition to offering a preview of the proposed general chapter, the General Chapters—Chemical Analysis Expert Committee and the Validation and…
SUCCESS QUOTIENT: Lupin chairman DB Gupta (sitting) with managing director Kamal K Sharma (centre), directors Vinita Gupta (right) and Nilesh Gupta.
The present invention provides a novel process for preparation N-[(2,3,4,5,6- Pentafluorophenoxy)phenoxyphosphinyl]-L-alanine 1-methylethyl ester (formula 2) and resolving the formula 2 in the presence base to form N-[(S)-(2,3,4,5,6- Pentafluorophenoxy)phenoxyphosphinyl]-L-alanine 1-methylethyl ester (formula 2′).
Sofosbuvir is chemically named as (S)-isopropyl 2-((S)-(((2R,3R,4R,5R)-5-(2,4- dioxo3,4-dihydropyrimidin-l(2H)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran- 2yl)methoxy)-(phenoxy)phosphorylamino)propanoate and is represented by the following chemical structure:
Image may be NSFW. Clik here to view.
Formula 1
PCT publications WO2011123645 and WO2010135569 describes process for preparation of compound of formula 2′ by reacting isopropyl (chloro(phenoxy)phosphoryl)-L-alaninate and pentaflurophenol in the presence of base.
Image may be NSFW. Clik here to view.
Formula 2′
Example-1:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium hydride
10.2g of sodium hydride was dissolved in 100 ml anhydrous THF. This solution was slowly added to a solution of pentafluorophenol (50g) in THF (100ml), Reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained solid was dried under vacuum at 45-50°C (yield=55g, confirmed by IR)
Example-2:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium methoxide
2,3,4,5, 6-pentafluorophenol (lOg) was dissolved in methanol (100ml), solution was cooled to 5-10°C. To this was added a solution of sodium methoxide in methanol. The reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained residue was striped with toluene. Obtained solid was dried under vacuum at 45-50°C (yield=l lg)
Example 3:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium hydroxide
2,3,4,5, 6-pentafluorophenol (lOOg) dissolved in methanol (—ml), solution was cooled to 5-10°C. To this was added a solution of sodium hydroxide (— g) in methanol. The reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained residue was striped with dichloromethane. Obtained solid was dried under vacuum at 45-50°C (yield=— g)
Example 4:
Preparation of (2S)-isopropyl-2-((chloro(phenoxy)posphoryl)amino)propanoate:
phenyl phosphodichloridate (30.6g) was dissolved in dichloromethane , to this was added a solution of 1-alanine isopropyl ester free base (19.16g) in dichloromethane at-60°C under nitrogen. Solution of triethylamine (20.7ml) was added to above reaction mass. Reaction mass was stirredat -60°C for 30 min and then temperature was raised to 25 °C. Reaction mass was stirred at 20-25 °C for 60 min & filtered and washed with dichloromethane. Clear filtrate was distilled under reduced pressure obtained residue was stirred with diisopropyl ether & filtered. Clear filtrate was distilled under reduced pressure to get (2S)-isopropyl-2-((chloro(phenoxy)posphoryl)amino)propanoate compound.
Example 5:
Preparation of isopropyl ((perfluorophenoxy)(phenoxy)phosphoryl)-L-alaninate (formula 2):
Image may be NSFW. Clik here to view.
(Formula 2)
Obtained (2S)-isopropyl-2-((chloro(phenoxy)phosphoryl)amino)propanoate (1.2 mol eq.) was dissolved in dichloromethane and cooled to 0-5°C under nitrogen atmosphere. To this was added solution of sodium 2,3,4,5,6-pentaflurophemolate (1 mol eq.) in tetrahydrofuran . Temperature of reaction mass was raised to 25°C and reaction mass was stirred for 3 hrs. After completion of reaction, reaction mass was distilled under reduced pressure & obtained residue was dissolved I ethyl acetate. Ethyl acetate layer was washed with water, dried over sodium sulfate & distilled off under reduced pressure. Diisopropyl ether was added to obtained residue and stirred for 60 min at 25 °C, obtained mass was filtered & washed with diisopropyl ether. Solid product was dried under vacuum at 40-45 °C .(yield=20g, enantiomer purity=93.45%)
Example 6:
Preparation of (S)-isopropyl 2-(((S)- (perfluorophenoxy)phenoxy)phosphoyl)amino)propanoate (Formula 2′):
Image may be NSFW. Clik here to view.
Formula 2′
(2S)-isopropyl-2-((chloro(phenoxy)phosphoryl)amino)propanoate (1.2 mol eq.) was dissolved in tetrahydrofuran (3.5 volumes). The reaction mass was cooled to -10°C. Solution of sodium salt of pentafluorophenol (1 mol eq.) in tetrahydrofuran (3.5 volumes) was added dropwise to the reaction mass at -10°C. After completion of the reaction solvent was distilled off. Ethyl acetate and water were added to the reaction mass. Reaction mass was stirred, ethyl acetate layer was separated and washed with sodium bicarbonate solution and brine. Ethyl acetate layer was concentrated under reduced pressure. Reaction mass was stripped with n-hepatane to get crude product. Crude product was dissolved in Methyl tert-butyl ether and n-heptane (1 : 1 ratio). The pH of reaction mass was adjusted to pH 8 by using triethylamine. Reaction mass was stirred overnight. Solid mass was filtered and washed with a mixture of methyl tertiary-butyl ether: n-heptane (1 : 1). The obtained product was dissolved in ethyl-acetate and washed with water and 20% brine solution. Ethyl acetate layer was separated; solvent was distilled off under reduced pressure. Reaction mass was stripped with diisopropyl ether. Di-isopropyl ether was added to the reaction mass. Reaction mass was stirred at 45-50°C. Reaction mass was cooled to 5-10°C and stirred. The titled compound was isolated by filtration and washed with di-isopropyl ether. The titled compound was dried under reduced pressure at 40°C. Yield 66.81%.
Image may be NSFW. Clik here to view.
Vinita Gupta, CEO, Lupin Pharmaceuticals Inc
Image may be NSFW. Clik here to view.
Desh Bhandu Gupta- Founder and chairman of Lupin Limited
////////////LUPIN LIMITED, WO 2016181313, NEW PATENT, SOFOSBUVIR
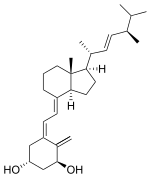
Literature References: Synthetic vitamin D prohormone. Prepn: H.-Y. P. Lam et al.,Science186, 1038 (1974); eidem,Steroids30, 671 (1977); H. E. Paaren et al.,J. Org. Chem.45, 3253 (1980). Comparative activity and toxicity: G. Sjöden et al.,Proc. Soc. Exp. Biol. Med.178, 432 (1985). Metabolism to bioactive form: J. C. Knutson et al.,Endocrinology136, 4749 (1995). Pharmacology: J. W. Coburn et al.,Nephrol. Dial. Transplant.11, Suppl. 3, 153 (1996). Clinical trial for suppression of secondary hyperparathyroidism in hemodialysis: J. M. Frazao et al.,ibid.13, Suppl. 3, 68 (1998).
Properties: Crystals, mp 138-140°. uv max (ethanol): 265 nm (e 18300). LD50 orally in rats: 3.5-6.5 mg/kg (Sjöden).
Melting point: mp 138-140°
Absorption maximum: uv max (ethanol): 265 nm (e 18300)
Toxicity data: LD50 orally in rats: 3.5-6.5 mg/kg (Sjöden)
Therap-Cat: Antihyperparathyroid.
Keywords: Antihyperparathyroid.
Image may be NSFW. Clik here to view.
CLIP
Image may be NSFW. Clik here to view.
Doxercalciferol (1α-hydroxyvitamin D2) is a commercially approved vitamin D derivative used to treat chronic kidney disease (CKD) patients whose kidneys cannot metabolically introduce a hydroxyl group at C1. A new process for the production of doxercalciferol from ergocalciferol was developed using a continuous photoisomerization of a known vitamin D intermediate as the key step, thus circumventing the limitations of batch photoisomerization processes. Doxercalciferol is produced in an overall yield of about 10% from ergocalciferol.
Jump up^Sprague S M; Ho L T (2002). “Oral doxercalciferol therapy for secondary hyperparathyroidism in a peritoneal dialysis patient”.Clinical nephrology. 58 (2): 155–160. PMID12227689.
The present invention provides novel crystalline Form of Nintedanib and process for its preparation. The present invention also provides to a novel process for the preparation of Nintedanib. The present invention further provides to novel intermediates used in the preparation of Nintedanib and process for their preparation.
Image may be NSFW. Clik here to view.
Nintedanib inhibits multiple receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (nRTKs).The chemical name of Nintedanib is lH-Indole-6-carboxylic acid, 2,3-dihydro-3-[[[4-[methyl-(4-methyl-l-iperazinyl)acetyl]amino]phenyl]amino]phenylmethylene] -2-oxo-,methylester, (3Z)-, ethanesulfonate (1 : 1) and is structurally represented by compound of Formula I.
Image may be NSFW. Clik here to view.
Formula I
Nintedanib is marketed in the United States under the trade name OFEV and is indicated for the treatment of Idiopathic Pulmonary Fibrosis (IPF).
Nintedanib was first described and claimed in U.S. Pat.No. 6,762, 180 and EP 1224 170. These patents disclose a process for the preparation of Nintedanib as depicted in scheme I given below:
Image may be NSFW. Clik here to view.
U.S.Pat.No. 8,067,617 discloses a process for the preparation of Nintedanib intermediate Enolindole derivative), which is shown in the scheme-II given below:
Polymorphism, the occurrence of different crystal forms, is a property of some molecules and molecular complexes. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, X-ray diffraction pattern, infrared absorption fingerprint and solid state NMR spectrum. One polymorph may give rise to thermal behaviour different from that of another polymorph. Thermal behaviour can be measured in the laboratory by such techniques as capillary melting point, thermo gravimetric analysis (“TGA”) and differential scanning calorimetry (“DSC”), which have been used to distinguish polymorphic forms.
The differences in the physical properties of different polymorphs results from the orientation and intermolecular interactions of adjacent molecules or complexes in the bulk solid. Accordingly, polymorphs are distinct solids sharing the same molecular Formula yet having distinct advantageous physical properties compared to other polymorphs of the same composition or complex. Hence there remains a need for polymorphic forms which have properties suitable for pharmaceutical processing on a commercial scale.
Considering the importance of Nintedanib, there exists a need to develop an alternate and improved process for the preparation of Nintedanib with better yield. Further, the process involved should be simple, convenient and cost-effective for large scale production. The inventors of the present invention during their continuous efforts also developed a novel high melting stable polymorphic form of Nintedanib ethanesulfonate.
EXAMPLES
Example 1: Process for the preparation of Nintedanib Monoethane Sulfonate:
Step-1: Preparation of methyl-3-(hydroxy(phenyl)methylene)-2-oxoindoline-6-carboxylate: To the suspension of methyl 2-oxoindoline-6-carboxylate (50 gm, 0.261 mol) in IPA (350 ml) was added slowly SMO-powder (33.8 gm, 0.626 mol) and stirred for about 15 min. Benzyl chloride (44 g, 0.313 mol) was added after completion of the reaction at a reaction temperature of -5 to -10°C for about 5hrs. The reaction mixture was quenched into ice-water (700 ml) and acidified with Cone. HC1 (2.0-2.5 ml). Filtered the reaction mixture, washed with water (2X100 ml) and dried the precipitate to obtain crude product which can be recrystallized from acetonitrile (28 ml) to obtain methyl-3-(hydroxy(phenyl)methylene)-2- oxoindoline-6-carboxylate pure crystalline solid (32 gm) (61%) (HPLC purity >97%). The filtrate was evaporated in vacuum to give unreacted methyl 2-oxoindoline-6-carboxylate. MR: 216-223°C; IR (KBr, cm“1): 3178, 1711, 1651; 1H-NMR (400 MHz, DMSO): δ 3.80 (s, 3H), 7.17 (s, 1H), 7.28-7.31 (m, 2H), 7.46-7.50 (m, 3H), 7.72 (d, 2H, J = 6.0 Hz), 9.52 (s, 1H), 11.53 (s, 1H); 13C-NMR (100 MHz, DMSO): δ 22.12, 52.41, 101.13, 111.13, 119.23, 123.06, 126.65, 127.06, 128.65, 129.21, 132.26, 134.47, 136.99, 166.58, 172.52 and 175.80; MS: m/z 294 [M]“1
Step-2: Preparation of methyl-3-(acetoxy(phenyl)methylene)-l-acetyl-2-oxoindoline-6-carboxylate (Acetyl derivative):
To the suspension of methyl-3-(hydroxy(phenyl)methylene)-2-oxoindoline-6-carboxylate (45 gm, 0.1512 mol) in acetic anhydride (300 ml) was added pyridine (4.5g) slowly (drop-wise) and stirred the reaction at temperature of 0-5°C for about30 min. After completion of the reaction raised the temperature of the reaction mass to 75-80°C and stirred for about lhr. Cooled the reaction mass and stirred for about 30 min at 25-28°C, filtered, washed with hexane (100ml) and dried the precipitate to obtain methyl-3-(acetoxy(phenyl)methylene)-l-acetyl-2-oxoindoline-6-carboxylate.
Step-3: Preparation of methyl- l-acetyl-3-(((4-(2-chloro-N-methylacetamido)phenyl)amino) (phenyl)methylene)-2-oxoindoline-6-carboxylate) (Chloroacetyl derivative) :
Suspension of methyl-3-(acetoxy(phenyl)methylene)- l-acetyl-2-oxoindoline-6-carboxylate (Acetyl derivative) (49gm, 0.129mol) and N-(4-aminophenyl)-2-chloro-N-methylacetamide(25.66gm, 0.129 mol) in a mixture of methanol (350 ml) and DMF (88 ml) was heated to 60-65°C stirred for about 12hr at the same temperature. After completion of the reaction cooled the reaction mass to room temperature and stirred for about 30min. Filtered the reaction mixture, washed with methanol (2X50ml) and dried the precipitate to obtainmethyl-l-acetyl-3-(((4-(2-chloro-N-ethylacetamido)phenyl)amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate).
Step-4: Preparation of (Z)-methyl-3-(((4-(N-methyl-2-(4-methylpiperazin-lyl)acetamide) phenyl) amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate (Nintedanib free base):
Suspension of methyl- l-acetyl-3-(((4-(2-chloro-N-methylacetamido)phenyl)amino) (phenyl)methylene)-2-oxoindoline-6-carboxylate)(40 gm, 0.077ml) and N-methylpiperidine (23.24 gm, 0.232 mol) in a mixture of DMF (160 ml) was heated to a reaction temperature of 45-50°C for about l-2hrs. The reaction mixture was quenched into ice-water (1.6 Lt) and stirred for about lhr at 15-20°C. Filtered the reaction mixture mass washed with water and dried the precipitate to obtain crystalline crude solid (36 gm). Purified with acetonitrile to obtain Nintedanib free base (34 gm) as a yellow crystals (93.74%) (HPLC purity: >98%). MR: 240-246°C; IR (KBr, cm“1): 3559, 3455, 2940, 2810, 1711, 1657; 1H-NMR (400 MHz, DMSO): δ 2.09 (s, 3H), 2.17 (s, 8H), 2.68 (s, 2H), 3.05 (s, 3H), 3.76 (s, 3H), 5.80 (d, 1H, J = 7.56 Hz), 6.86 (d, 2H, J = 6.72 Hz), 7.11 (d, 1H, J = 6.48 Hz), 7.17 (d, 2H, J = 7.68 Hz), 7.42-7.57 (m, 6H), 10.98 (s, 1H), 12.23 (s, 1H) ; 13C-NMR (100MHz, DMSO): δ 37.17, 46.18, 52.24, 52.79, 55.05, 59.68, 98.10, 109.94, 117.75, 121.96, 124.29, 124.52, 128.06, 128.90, 129.40, 129.92, 130.91, 132.50, 136.72, 140.66, 158.81, 166.84, 169.04 and 170.66; MS: m/z 540 [M]+1.
Step-5: Preparation of (Z)-methyl-3-(((4-(N-methyl-2-(4-methylpiperazin-lyl)acetamide) phenyl) amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate ethane sulfonate salt:
Suspension of (Z)-methyl-3-(((4-(N-methyl-2-(4-methylpiperazin-l-yl)acetamide)phenyl) amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate (36 gm, 0.066 mol) in methanol (237 ml) and water (2.88 ml)was heated to 60-65°C and aq. ethane sulfonic acid was added to the reaction mixture. The resulting solution was cooled to 50°C, seeds diluted with isopropanol (237 ml) was added. The reaction mixture was cooled at 0°C for lhr. Filtered the precipitate, washed with mixture of methanol and isopropanol (50 ml), dried to obtain crude Nintedanib monoethane sulfonate (36.6 gm) and crystallized from methanol (5 Vol) to
obtained pure Nintedanib monoethane sulfonate salt as yellow crystals (33 gm) (80%) (HPLC purity >99%).
Example 2: Process for the preparation of Polymorph Form S of Nintedanib monoethanesulf onate :
Crude Nintedanib monoethane sulfonate was dissolved in methanol and heated to 60-64°C for about 15 min. After completion of the reaction cooled to room temperature for about lhr. Filtered the precipitate, washed with mixture of methanol (20ml) and isopropanol (30 ml) and dried to obtain pure crystalline solid (28 gm) (yield: 93.3%) with HPLC purity 99.72% and individual impurities 0.09%, 0.02% and 0.04%.
SUVEN, Chief executive and chairman Venkat Jasti
Image may be NSFW. Clik here to view.
Image may be NSFW. Clik here to view.
Image may be NSFW. Clik here to view.
//////////WO2016178064, POLYMORPH, NINTEDANIB ETHANESULPHONATE, PROCESSES, INTERMEDIATES, suven, new patent
Heart failure is a very high mortality syndrome, for patients with heart failure, so far no drug can significantly improve mortality and morbidity, and thus a new type of therapy is necessary. AHU-377 (CAS No. 149709-62-6) is an enkephalinase inhibitor, which is a prodrug ester groups can be lost through hydrolysis, converted to pharmaceutically active LBQ657, inhibit endorphin enzyme (NEP) the role of the main biological effects of NEP is to natriuretic peptides, bradykinin and other vasoactive peptide degradation failure. AHU-377 and angiotensin valsartan composition according to the molar ratio of 1 LCZ696. LCZ696 is an angiotensin receptor enkephalinase inhibitors, which can lower blood pressure, treat heart failure may become a new drug. Clinical data show, LCZ696 is more effective for the treatment of hypertension than valsartan alone.
Patents US 5,217,996 and US 5,354,892 reported the first synthesis of AHU-377, the synthetic route is as follows:
Image may be NSFW. Clik here to view.
Reaction with unnatural D-tyrosine derivative as a substrate, more expensive, while the second step in the synthesis is necessary to use Pd-catalyzed Suzuki coupling reaction, whereby preparative route costs than the AHU-377 high.
Patent US 8,115,016 above routes also reported the departure from the pyroglutamate, through multi-step process for preparing a reaction AHU-377, which is more difficult methylation reaction, and the yield is not high. Patent US 8,580,974 also reported a carbonyl group of the a- introducing N, N- dimethyl enamine is converted to methyl, however, there are some problems in the route for constructing methyl chiral centers, are not suitable for scale-up synthesis route as follows:
Image may be NSFW. Clik here to view.
About the latest AHU377 synthesis intermediates, Patent WO2014032627A1 reported using a Grignard reagent to react with epichlorohydrin, a quicker been important intermediates, synthetic route Compound AHU377 synthesized as follows:
Image may be NSFW. Clik here to view.
However, the second step of the synthetic route use succinimide nitrogen atoms introduced by Mitsunobu reaction with hydrochloric acid hydrolysis to remove, then converted to Boc protected at the end of the synthesis process AHU377 Boc will have to take off protection, then any connection with succinic anhydride reaction product introduced into the structure of succinic acid portion, so that this method of atom economy and the economy of the steps are low.
Example 1
Synthesis of Compound 2
Image may be NSFW. Clik here to view.
In inert atmosphere, a solution of three 500mL flask was added compound 1 (10g, 1eq), dissolved after 90mL THF, was added CuI (4.814g, 0.1eq), the system moves to the low temperature in the cooling bath to -20 ℃ when, biphenyl magnesium bromide dropwise addition, the internal temperature was controlled not higher than -10 ℃. Bi closed refrigeration drop, return to room temperature overnight. Completion of the reaction, the reaction solution was poured into saturated the NH 4 of Cl (10vol, 100 mL) was stirred at room temperature for 0.5h. Suction filtered, the filter cake was rinsed with a small amount of EA, and the filtrate was transferred to a separatory funnel carved, and the aqueous phase was extracted with EA (10vol × 2,100mL × 2) and the combined organic phases with saturated NaHC [theta] 3 , the NH 4 of Cl, each Brine 150mL (15vol) washed once, dried over anhydrous over MgSO 4 dried, suction filtered, and concentrated to give a white solid. Product obtained was purified by column 15.2g, yield 78%.
In an inert gas, at room temperature was added to the flask 500mL three Ph3P (18.54g, 2eq), 240mL DCM dissolution, butyryl diimide (of 6.44 g), compound 2 (15g), an ice-water bath cooling to 0 ℃ or so, was added dropwise DIAD (14mL) was complete, the reaction go to room temperature.Starting material the reaction was complete, the system was added to water (100 mL) quenched the reaction was stirred for 10min; liquid separation, the aqueous phase was extracted with DCM (100mL × 2), the combined organic phases with saturated Brine 100mL × 2), dried over anhydrous over MgSO 4 dried , filtration, spin dry to give a white solid; product was purified by column 15.4g, yield 82%.
Protection of inert gas, at room temperature was added to the flask 1L three compound 3 (18.81g), 470mL EtOH was dissolved, was added Pd / C, replaced the H 2 three times, move heated on an oil bath at 60 ℃ reaction. Raw reaction was complete, the system was removed from the oil bath, the reaction solution was suction filtered through Celite and concentrated to give the crude product. It was purified by column pure 11.8g, a yield of 81.2%.
Protection of inert gas, at room temperature to a 25mL flask was added three Dess-Martin oxidant (767.7mg), 10mL DCM was dissolved, the system was cooled down to -10 deg.] C, was added 4 (500mg). Starting material the reaction was complete, to the system was added saturated NaHCO3 and Na2S2O3 each 5mL, quench the reaction stirred for 10min; aqueous phase was extracted with DCM (10mL × 3) and the combined organic phases with saturated NaHCO3, Brine 30mL each wash, dried over anhydrous MgSO4, filtration, spin dried to give the crude product used directly in the next reaction cast.
Example 5
Synthesis of Compound 8
Image may be NSFW. Clik here to view.
Inert gas, at room temperature for three to 500mL flask 7 (497.5mg), 10mL DCM to dissolve an ice water bath to cool, added phosphorus ylide reagent (880.6mg), the system was removed from the ice water bath at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. Liquid separation, the aqueous phase was extracted with DCM (10mL × 2), organic phases were combined, washed with saturated Brine 20mL × 2, dried over anhydrous MgSO4, filtration, spin crude done. Product obtained was purified by column 563mg, 90% yield.
Protection of inert gas, at room temperature to a 50mL flask was added three 8 (365mg, 1eq), 9mL of ethanol and stirred to dissolve, the system was replaced with hydrogen three times, was added Pd / C (25% w / w) at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. The reaction mixture was suction filtered through Celite and concentrated to give the crude product. Product was purified by column, yield 80.2%, purity 97.2%.
Example 7
Synthesis of Compound 10
Image may be NSFW. Clik here to view.
Equipped with Compound 9 (100mg) acetic acid A reaction flask (9mL), hydrochloric acid (1mL). The reaction was heated oil bath at 80 deg.] C. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. After saturated NaHCO3 and extracted with EA and concentrated to give crude product. Product obtained was purified by column 90mg, yield 84%.
Example 8-1: The reaction flask was added compound 4 (1eq) was added water (2VOL), concentrated hydrochloric acid (2VOL), 110 ℃ reaction was heated in an oil bath overnight, complete conversion of starting material, the HPLC peak area 97%. 10% NaOH solution was added to adjust the pH to about 10, filtration products. Yield 85%.
Example 8-2: The reaction flask was added compound 4 (1eq) was added ethanol (5 vol), water (5 vol), potassium hydroxide (8 eq), was heated in an oil bath overnight at 110 ℃ reaction, complete conversion of the starting material, the HPLC peak area 99%. Water was added (5Vol), filtered to obtain the product. Yield 95%. Product was dissolved in toluene, was added ethanolic hydrochloric acid, the precipitated hydrochloride Compound 5.
To the reactor was added compound 5, was added absolute ethanol (3vol). Temperature of the outer set 30 ℃ heating, stirring was continued after the temperature reached 25 ℃ 20min. Was added 30% NaOH aqueous solution (1.1eq). External temperature 65 ℃ heating provided, after the internal temperature reached 60 deg.] C was slowly added (of Boc) 2 O (1.1 eq). Stirring 0.5h, reaction monitoring. After completion of the reaction, water was added slowly dropwise (8vol), turn off the heating and natural cooling. The system temperature was lowered to 25 deg.] C and continue stirring for 2h. Filter cake at 50 ℃ blast oven drying to obtain the product.
Synthesis of Intermediate Compound 6 to Compound 10, i.e., the AHU-377, a synthetic route in the background of the present invention, the cited patent application WO2014032627A1 loaded in detail, not in this repeat.
Example 10
Synthesis of Compound 2
Image may be NSFW. Clik here to view.
Benzyl glycidyl ether preparation (50g) in THF (200mL) was added. Under inert gas protection, the biphenyl magnesium bromide (365mmol) was added to THF (1020mL) was added the reaction flask is placed in a low temperature bath -40 ℃ cooling. Cuprous iodide (O.leq) when the internal temperature dropped to -9 ℃. Continued to decrease the temperature of -23 ℃ dropwise addition of benzyl glycidyl ether in THF was added dropwise to control the internal temperature process of not higher than -15 deg.] C, 47 min when used, the addition was completed the cooling off the reaction was stirred overnight. The cooling system to -20 ℃ quenched with 1N HCl aqueous solution, <10 ℃ Go stirred 30min at room temperature. Liquid separation, the aqueous phase was extracted with THF, the combined THF phases. Respectively saturated ammonium chloride (250mL), saturated brine (250mL) washed. Rotary evaporation to remove THF, and water (200 mL) Continue rotary evaporation 1h, cool to precipitate a solid. Suction crude. Crude n-heptane was added 2Vol beating, suction filtration to obtain the product in a yield of 90 ~ 95%, HPLC peak area 94%. In another column purification was pure, columned yield 88.6%, HPLC 99.1%.
Example 11
Synthesis of Compound 3
Image may be NSFW. Clik here to view.
Preparation Example 9, said compound taking the embodiment 2 (5g) added to the reaction flask, the reaction flask was added toluene (80mL), phthalimide (2.55 g of) and triphenylphosphine (5.35g of), the nitrogen was replaced protection. An ice-salt bath cooling to -5 deg.] C, was added dropwise DIAD (4.12g), dropwise addition was exothermic, the temperature was raised to 5 ℃. The reaction was continued 1h sampling HPLC test material substantially complete reaction. Join 12g silica spin column done to collect the product (including DIEA derivative).
Example 12
Synthesis of Compound 11
Image may be NSFW. Clik here to view.
Compound 3 (3g) was added to the reaction flask embodiment taken in Preparation Example 10, was added ethanol (30 mL), with stirring. Was added hydrazine hydrate (2g) was heated in an oil bath reflux 1h, when supplemented with 20mL ethanol was stirred difficulties, the reaction was continued to 2.5h, HPLC showed the starting material the reaction was complete. Add EA / H2O 100mL each liquid separation, the EA phase was washed with water (100mL) and the combined organic phases were washed with water (100mL) and saturated brine (100mL) washed. Anhydrous magnesium sulfate and filtered spin column was done product 1.88g, yield 88%, HPLC 94%.
To the toluene solution of the compound 2 was added phthalimide (1.1 eq), triphenylphosphine (1.3 eq) with stirring. External bath set -10 ℃, to cool the system, the internal temperature dropped to 0 ~ 5 ℃, start dropping DIAD (1.3eq), control the internal temperature -5 ~ 5 ℃. Completion of the dropwise addition, the cooling bath was turned off outside the reaction was stirred at room temperature. The reaction was stirred for 1 to 4 hours. The reaction solution to give compound 3, administered directly in the next reaction. To the above reaction mixture was added hydrazine hydrate (6 eq), heated to 70 ~ 80 ℃, to complete the reaction, filtered hot, the filtrate. Aqueous sodium hydroxide solution (20vol 10%) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. Water was added (20vol) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. The toluene phase was added hydrochloric acid (20vol, 3N), stirred for 0.5h, to form a solid precipitate. Filtration and drying to obtain a product, i.e. compound 11, the hydrochloride salt, yield 60% in two steps.
Weigh Compound 11 (1.38g) was added to the reaction flask. To the reaction flask plus DCM (14ml) and Et3N (462mg, 0.73ml). Weighed (of Boc) 2O (1.23 g of) was added to DCM (5ml) was dissolved. Room temperature (8 ℃), a solution (of Boc) 2 DCM solution O was added dropwise to the reaction, (2ml) rinsed with DCM. The reaction mixture was stirred at room temperature, detected by HPLC, the reaction ends 4h. Reaction mixture was washed (15ml) 3 times with Brine (15ml) The reaction solution was washed 1 times. Inorganic sulfate, concentrated and purified by column PE:EA = 15:1 give product 560mg, yield 30.8%, HPLC 99.92%.
Weigh Compound 12 (250mg) and methanol (9ml) was added to the reaction flask. Added Pd / C (138mg, 1 / 4w / w, water content 55%). The H 2replaced 3 times, 50 ℃ stirred and heated. HPLC detection reaction, the reaction end 30h. Filtered off Pd / C, 40 ℃ concentrated under reduced pressure to remove methanol. PE:EA = 3:1 florisil column to give the product 196mg, 100% yield, 99.34% purity.
Method for preparing the AHU-377, characterized by comprising the steps of: (a) Compound (1) S- benzyl glycidyl ether and biphenyl Grignard reagent produced by the reaction of the compound (2) in an organic solvent; ( b) compound (2) with a succinimide or phthalimide Mitsunobu reaction occurs in an organic solvent to form a compound (3); (C) compound (3) in an organic solvent in the role of a catalyst under removal debenzylation protected form compound (4); (D) compound (4) with an oxidizing agent oxidation reaction occurs in an organic solvent to form a compound (7); (E) compound (7) with a phosphorus ylide reagent in an organic solvent to give the compound (8); (F.) compound (8) in an organic solvent in the selective catalytic hydrogenation of the compound (9); and (g) of the compound (9) in an organic solvent in the hydrolysis reaction of the amide compound occurs in the presence of an acid ( 10), i.e., AHU-377;Image may be NSFW. Clik here to view.
 Green Chemistry International
Green Chemistry International
 Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW.