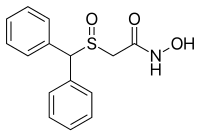
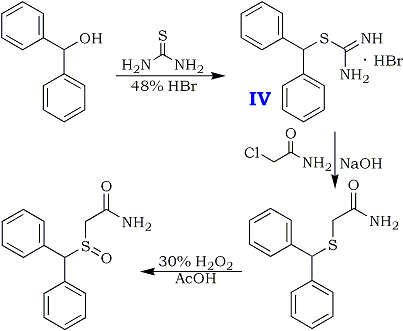
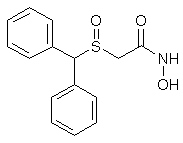
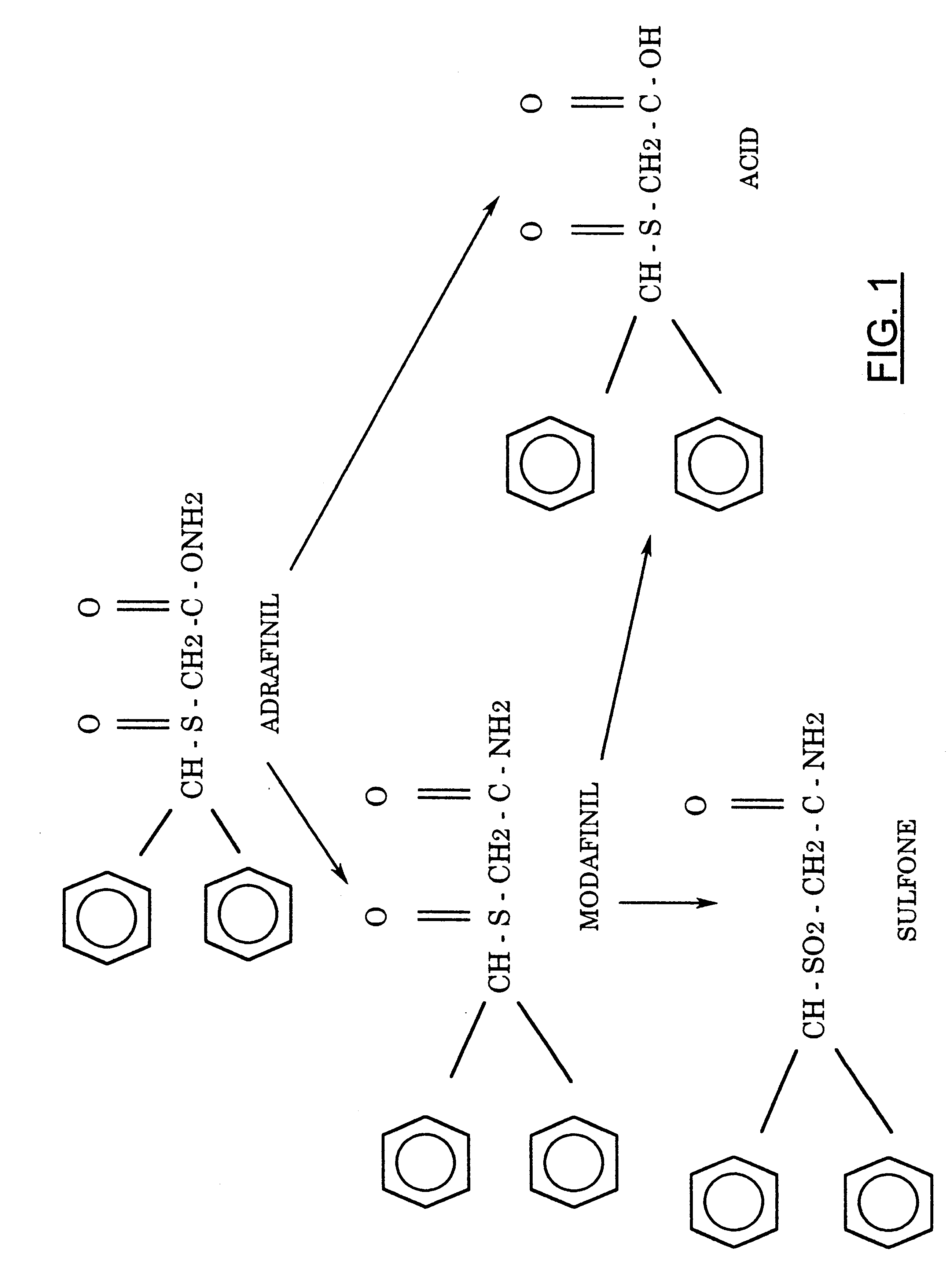
WO 2016173551 China State Institute of Pharmaceutical Industry; Shanghai Institute of Pharmaceutical Industry
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016173551&redirectedID=true
MA, Shuai; (CN).
ZHOU, Weicheng; (CN)
WO2016173551, IPRAGLIFLOZIN PREPARATION METHOD
CHINA STATE INSTITUTE OF PHARMACEUTICAL INDUSTRY [CN/CN]; 4th Floor, Building 1, No.1111 Halley Road,pudong New Area Shanghai 201203 (CN).
SHANGHAI INSTITUTE OF PHARMACEUTICAL INDUSTRY [CN/CN]; No.1320,West Beijing Road,Jing’an District Shanghai 200040 (CN)

MACHINE TRANSLATED FROM CHINESE……
Ignatius column Net (English name: Ipragliflozin) by Astellas Pharma Ltd. (Astellas) new sodium life Pharmaceutical Co., Ltd. (Kotobuki) R & D – glucose cotransporter (Sodium glucose co-transporters, referred to as SGLT-2 ) inhibitor, on January 17, 2014 in the Japanese market for the treatment of patients with type ⅱ diabetes; tradename Suglat, currently provide 25mg and 50mg tablets.
Chemical Name column Ignatius net is (1S) -1,5- dehydration -1-C- [3- (1- benzothien-2-yl-methyl) -4-fluorophenyl] -D-glucose alcohols of the formula the C 21 the H 21 the FO 5 the S, the CAS No. 761423-87-4, as the structure of formula 2, as a column for medicinal Eagle with L- proline net clinical eutectics, such as a structural formula FIG.
Ignatius column in the process of preparation of the net, the chiral synthesis of β glycoside bond synthetic route key points. Currently, Ignatius column net of synthetic methods reported in the literature there are several of these methods for the synthesis of chiral β-glucoside bonds mainly relates to hydroxy-protected D- glucose lactone ester carbonyl nucleophilic addition reaction.
Route One: Patent WO2004080990 synthetic route reported net Ignatius column is as follows:
This route, [1-benzopyran-2-yl (5-bromo-2-fluorophenyl) methoxy] (tert-butyl) dimethylsilane (Compound 10) with n-butyl lithium at -78 deg.] C (or minus 78 deg.] C) the reaction of an organolithium reagent and then with 2,3,4,6-tetra -O- benzyl -D- glucose lactone (compound 9) nucleophilic addition at low temperature -78 ℃ to obtain compound 8, followed by removal of the silicon compound 8 hydroxy group is protected with tetrabutylammonium fluoride (of TBAF) to give compound 7, triethylsilane and then reducing the compound 7 obtained with chiral β glycosidic bond Ignatius column net intermediate 6, the last off at -78 ℃ intermediate ring 6 sugar hydroxyl protecting groups to obtain the desired product – Ignatius column net (compound 2). Compound 10 was prepared by the target product – Ignatius column net synthesis route yield 9.94%, net Ignatius column purity not reported. The disadvantage of this method is that a long synthetic route, after every step of the reaction were purified by column chromatography, and the yield is low. Deprotecting the hydroxy group on two key steps chiral β glycosidic bond synthesis and sugar ring need to be at a low temperature at -78 deg.] C, clearly, it is difficult to meet the needs of industrial production.
Route II: Patent WO2008075736 Ignatius column reported net synthetic route is as follows:
The route of 2- (5-bromo-2-fluorobenzyl) benzothiophene (compound 15) with n-butyl lithium at -43.5 ~ -33.3 ℃ reaction of an organolithium reagent and then with 2,3,4 , 6-tetrafluoro -O- trimethylsilyl -D- glucose lactone (compound 14) nucleophilic addition reactions at -72.6 ~ -65 ℃ to give compound 13, compound 13 and then acetylation, reduction Ignatius column net intermediates prepared with chiral β glycoside bond of 11, finally deacetylated to obtain the desired product of intermediate 11 – Ignatius column net (compound 2). Compound 15 was prepared by the Scheme 2 the desired product in a yield of 72.46%, a purity of compound 2 was 99%. The disadvantage of this method is that the route Ignatius column net synthesis requires at a low temperature of -72.6 ℃ to react and involve nucleophilic addition reaction, a hydroxyl group on the terminal carbon methylation, acetylation of hydroxyl groups on the sugar ring, the end methoxy groups on carbon reduction, the reaction and post-treatment process is very complicated, more difficult to industrial production, and on the terminal carbon-methoxy-reducing agent used in the reduction – t-butyldimethylsilyl more expensive, increasing the whole synthetic route costs.
Route III: Patent WO2015012110 Ignatius column reported net synthetic route is as follows:![]()
On the basis of patent WO2015012110 patent WO2008075736 reported synthetic route for the synthesis net Ignatius column primarily relates to the further improvements: namely: 2- (5-bromo-2-fluorobenzyl) benzothiophene (Compound 15) three butylmagnesium lithium at -12 ~ -26 ℃ organomagnesium reagent prepared by the reaction – compound 16, and then with 2,3,4,6-tetra -O- trimethylsilyl -D- glucose lactone (compound 14) carried out at -12 ~ -16 ℃ nucleophilic addition reaction Ignatius column net key intermediates – compounds 13, this step is nucleophilic addition reaction temperature was raised to -26 ℃, avoid the use of other organic lithium reagent required -78 ℃ low temperature reactions. The disadvantage of this method is that Ignatius column net synthesis still need to involve nucleophilic addition reaction, a hydroxyl group on the terminal carbon methylation, acetylation of hydroxyl groups on the sugar ring, a methoxy group on the terminal carbon reduction reaction and post-treatment very complicated problem is not resolved; in addition, tributyltin lithium magnesium used in the route in the country not commercially available, and can be prepared before the experiment, the manufacturing process is more complex, more difficult to industrial production.
Skilled in the art knows the energy super low temperature chemical reaction operations is considerable. Generally, the reaction temperature at -40 ℃ over the operation of the more conventional reactor in the plant can be relatively easy to do; but lower than the reaction below -40 ℃ the need to use special equipment or a special reactor is required with liquid nitrogen as the cooling source, the higher the cost. For ultra-low temperature improvements often become enlarged or when the process of large-scale, process optimization of key points.
In the background art described in this article about the Ignatius column net three synthetic route, the “connection” between the main synthon mainly related to the organometallic reagents – such as organic lithium or magnesium organic lithium reagents protected hydroxy D- glucose ester carbonyl lactone on nucleophilic substitution reaction with hydroxyl groups to form the corresponding glucose derivative on the terminal carbon; then after hydroxy or derivatives thereof – methoxy reduced to hydrogen, to give the title with β-type hand glycoside bond Ignatius column net key intermediate structure; and finally the removal of hydroxy protecting groups on the pyranose ring to give Ignatius column net. In these types of synthetic route, operation and post-processing reaction steps are more complicated, the cost is high. For example, in Scheme 1 and 2, both the use of ultra-low temperature organolithium reagent – minus 78 ℃; several synthetic route in addition, most of the intermediate purification using column chromatography, such process is not suitable for plant production is amplified. Therefore, an urgent need to find new Ignatius column net synthesis method, and enables industrial production.
(1), from 4-fluoro-3- (2-benzothienyl) phenyl methyl halide (Compound 5) as a starting material, the compound 5 in a suitable solvent, is reacted with an alkyl lithium, followed by reaction with zinc an organic zinc reagent – bis [4-fluoro-3- (2-benzothienyl) methyl phenyl] zinc, and then with 2,3,4,6-tetra -O- pivaloyl bromo -α-D- Generation glucopyranose (compound 4) nucleophilic substitution reaction of intermediate net Ignatius column – compound 3;
(2), compound 3 by an organic base off pivaloyl protecting group to obtain Eagle column net (Compound 2);
Wherein in the 4-fluoro-3- (2-benzothienyl) phenyl methyl halide (Compound 5) Structure X is selected from bromo or iodo;
Synthetic route is as follows:
Example 1, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Preparation fluorophenyl] glucitol (compound 3) –
Zinc bromide (0.676 g) and lithium bromide (0.261 g) was added n-butyl ether (8mL), stirred and heated to 50 deg.] C 2h, cooling backup. Under nitrogen, was added 2- (5-iodo-2-fluorobenzyl) benzothiophene (2.21g) in toluene (5mL), n-butyl ether (5mL), cooled to -25 deg.] C, was slowly added dropwise 1.6mol / L n-hexyl lithium hexane solution (4.13 ml), to control the internal temperature does not exceed -10 deg.] C, after the addition was complete the reaction was incubated at -20 ℃ 0.5h, a solution of n-butyl ether was added to the backup lithium bromide and zinc bromide, at 10 ℃ reaction was stirred 3h. Was added 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (3.48 g of) in toluene (10 mL) solution and heated to 80 deg.] C the reaction was stirred 6h, TLC analysis after completion of the reaction, was added 1mol / L dilute hydrochloric acid (7mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, concentrated, and n-heptane (5mL) and methanol (15mL) recrystallized 3.452g 3 of solid compound, yield: 77.65%. Purity: 99.45%. Melting point: 128.9 ~ 130.5 ℃. 1 the H-NMR (CDCl 3 ): [delta] 7.72 (IH, D), 7.64 (IH, D), 7.21-7.30 (4H, m), 7.04 (IH, T), 6.96 (IH, S), 5.40 ( 1H, t), 5.27 (2H , m), 4.36 (1H, d), 4.08-4.21 (4H, m), 3.82 (1H, dd), 1.19 (9H, s), 1.16 (9H, s), 1.11 (9H, s), 0.85 ( 9H, s).
Example 2, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Preparation fluorophenyl] glucitol (compound 3) –
Zinc bromide (0.676 g) and lithium bromide (0.261 g) was added n-butyl ether (8mL), stirred and heated to 50 deg.] C 2h, cooling backup. Under nitrogen, was added 2- toluene (5mL) (5- iodo-2-fluorobenzyl) benzothiophene (2.21g) in n-butyl ether (5mL), cooled to – 50 ℃, was slowly added dropwise 2.5mol / L n-butyllithium hexane solution (2.64 mL), controlling the internal temperature does not exceed -30 deg.] C, 6h after the addition was complete the reaction was kept at -50 deg.] C, was added a solution of n-butyl ether in said auxiliary zinc bromide and lithium bromide, the reaction was stirred 8h at -20 ℃. Was added 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (6.954g) in toluene (12mL) solution, heated to 25 deg.] C the reaction was stirred 24h, after completion of the reaction by TLC, was added 1mol / L dilute hydrochloric acid (8mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, concentrated, and n-heptane (5mL) and methanol (15mL) recrystallized 3.237g 3 of solid compound, yield: 72.81%. Purity: 99.36%.
Example 3, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Preparation fluorophenyl] glucitol (compound 3) –
Zinc iodide (1.915g) and lithium iodide (0.803 g) in n-butyl ether was added (10mL), stirred and heated to 50 deg.] C 1.5h, cool reserve. Under nitrogen, was added 2- (5-iodo-2-fluorobenzyl) benzothiophene (2.21g) in toluene (9mL), n-butyl ether (3mL), cooled to -30 deg.] C, was slowly added dropwise 1.6mol / L n-hexyl lithium hexane solution (4.13mL), controlling the internal temperature does not exceed -20 ℃, n-butyl ether solution after the addition was complete the reaction was kept at -30 ℃ at 5h, zinc iodide was added to the backup and lithium iodide the mixture was stirred at 25 ℃ reaction 1h. After addition of 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (4.346g) in toluene (10 mL) solution, the reaction was heated to reflux for 145 ℃ 0.5h, TLC detection completion of the reaction , was added 1mol / L dilute hydrochloric acid (8mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, concentrated, and n-heptane (5mL) and methanol (15mL) recrystallized 3.552 3 g of a solid compound in a yield of 79.9%. Purity: 99.41%.
Example 4, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Preparation fluorophenyl] glucitol (compound 3) –
Zinc bromide (0.676 g) and lithium bromide (0.261 g) was added n-butyl ether (7mL), stirred and heated to 50 deg.] C 2h, cooling backup. Under nitrogen atmosphere, 2- (5-bromo-2-yl) benzothiophene (1.927g) was added toluene (6mL), n-butyl ether (4mL), cooled to -30 deg.] C, was slowly added dropwise 2.5mol / L n-butyllithium hexane solution (2.88 mL), controlling the internal temperature does not exceed -20 deg.] C, 3h after the addition was complete the reaction was kept at -30 deg.] C, was added a solution of n-butyl ether in said auxiliary zinc bromide and lithium bromide, the reaction was kept at -5 ℃ 4h, was added 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (4.346g) in toluene (7mL) solution, stirred and heated to 120 ℃ The reaction 4h, after completion of the reaction by TLC, was added 1mol / L dilute hydrochloric acid (8mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, and concentrated under reduced pressure, n-heptane (5mL ) and methanol (15mL) recrystallized 2.783g solid compound 3, yield: 62.6%. Purity: 99.29%.
EXAMPLE 5, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Preparation fluorophenyl] glucitol (compound 3) –
Zinc bromide (0.676 g) and lithium bromide (0.261 g) was added cyclopentyl ether (8mL), stirred and heated to 50 deg.] C 2h, cooling backup. Under nitrogen, was added 2- (5-iodo-2-fluorobenzyl) benzothiophene (2.21g) in toluene (6mL), cyclopentyl methyl ether (6mL), cooled to -30 deg.] C, was slowly added dropwise 1.6 mol / L hexane solution of n-hexyl lithium (4.5mL), controlling the internal temperature does not exceed -20 ℃, after the addition was complete the reaction was kept at -30 ℃ at 3h, added to the backup lithium bromide and zinc bromide cyclopentylmethyl the ether solution, the reaction incubated at -5 ℃ 4h, was added 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (4.346g) in toluene (8mL) solution, heated to 120 ℃ reaction was stirred 4h, after completion of the reaction by TLC, was added 1mol / L dilute hydrochloric acid (8mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, and concentrated under reduced pressure, with n-heptane dioxane (5mL) and methanol (15mL) recrystallized 2.088g solid compound 3, yield: 47%. Purity: 99.3%.
6, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Example Preparation fluorophenyl] glucitol (compound 3) –
Zinc bromide (0.676 g) and lithium bromide (0.261 g) was added methyl t-butyl ether (8mL), was heated to 50 ℃ stirred 3h, cooling backup. Under nitrogen, was added 2- toluene (6mL), methyl t-butyl ether (4mL) (5- iodo-2-fluorobenzyl) benzothiophene (2.21g), cooled to -40 deg.] C, was slowly added dropwise 1.6 mol / L n-hexyl lithium hexane solution (3.94mL), controlling the internal temperature does not exceed -30 ℃, after the addition was complete the reaction was kept at -40 ℃ at 4h, was added to the lithium bromide and zinc bromide spare methyl tert-butyl ether solution, the reaction incubated at 5 ℃ 7H, was added 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (3.48 g of) in toluene (8mL) solution, heated to 90 ℃ reaction was stirred 6h, after completion of the reaction by TLC, was added 1mol / L dilute hydrochloric acid (8mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, and concentrated under reduced pressure, with n-heptane dioxane (5mL) and methanol (15mL) recrystallized 2.792g solid compound 3, yield: 62.8%. Purity: 99.44%.
Example 7, (1S) -1,5- anhydro-1- [3- (1-methyl-thiophen-2-yl) -4-fluorophenyl] -D-glucitol (Compound 2) preparation
Compound 3 (7.41g) was added methanol (35mL), was added sodium methoxide (2.161g), heated at reflux for 5H reaction, after completion of the reaction by TLC, concentrated and the residue was added methanol (10 mL), water (10 mL), acetic acid ( 3g), was added seed crystal (0.1g), stirred at 5 ℃ crystallization, filtration, the filter cake washed with cold (methanol: (5mL) was washed with 1) solvent to give an off-white solid 3.89g compound 2: water = 1 , yield: 96.2%. Purity: 99.29%. 1 the H-NMR (the CD 3 the OD): [delta] 7.70 (IH, D), 7.63 (IH, D), 7.43 (IH, dd), 7.34-7.38 (IH, m), 7.21-7.26 (2H, m) , 7.08 (1H, t), 7.01 (1H, s), 4.18-4.28 (2H, m), 4.12 (1H, d), 3.88 (1H, dd), 3.70 (1H, dd), 3.30-3.50 (4H , m).
Example 8, (1S) -1,5- anhydro-1- [3- (1-methyl-thiophen-2-yl) -4-fluorophenyl] -D-glucitol (Compound 2) preparatio
Methanol was added (15mL) of the compound 3 (7.41g) was added sodium hydroxide (2g) in water (10 mL) solution was heated to 50 deg.] C the reaction was stirred 10h, TLC detection after completion of the reaction, water (10mL), 2mol / L hydrochloric acid (2mL), stirred at room temperature for crystallization, white solid was suction filtered, the filter cake washed with water (5mL) was washed and dried to give 3.806g of compound 2, yield: 94.1%. Purity: 99.31%.
Preparation 9, Ignatius column eutectic net L- proline (Compound 1) Example
Net Ignatius column (compound 2) (4.04g) was added ethanol (25mL), was added L- proline (1.15 g of), the reaction was heated at reflux for 1h, cooled to room temperature, filtered, the filter cake washed with cold ethanol, and dried to give white solid 4.67g of compound 1. Yield: 90%. Purity: 99.51%. Melting point: 194.0 ~ 202.1 ℃. The MS-ESI (m / Z): 427.16 [the M + of Na] + . 1 the H-NMR (the CD 3 the OD): [delta] 7.75 (IH, D), 7.67 (IH, D), 7.45 (IH, dd), 7.37 (IH, m), 7.24-7.31 (2H, m), 7.10 (1H, t), 7.07 ( 1H, s), 4.23-4.32 (2H, m), 4.13 (1H, d), 3.98 (1H, t), 3.89 (1H, d), 3.71 (1H, dd),3.31-3.50 (5H, m), 3.21-3.27 (1H, m), 2.27-2.34 (1H, m), 2.09-2.17 (1H, m), 1.95-2.02 (2H, m).
Claims
Ignatius one kind of column and net synthesis process, comprising the steps of: (1), from 4-fluoro-3- (2-benzothienyl) methyl-5-phenyl-halide as a raw material, in an appropriate solvent 5 is reacted with an alkyl lithium, followed by reaction with the zinc salt prepared organozinc reagents – bis [4-fluoro-3- (2-benzothienyl) methyl phenyl] zinc, and then with 2,3,4,6-tetra -O- pivaloyl -α-D- glucopyranose 4-bromo nucleophilic substitution reaction of intermediate net Ignatius column 3; (2), compound 3 by an organic base off pivaloyl protecting group prepared net Ignatius column 2; wherein, in the 4-fluoro-3- (2-benzothienyl) methyl-5-phenyl halide of structure X is selected from bromo or iodo; synthesis route is as follows:![]()
////////WO 2016173551, China State Institute of Pharmaceutical Industry; Shanghai Institute of Pharmaceutical Industry, IPRAGLIFLOZIN, NEW PATENT,
Filed under: PATENT, PATENTS Tagged: China State Institute of Pharmaceutical Industry; Shanghai Institute of Pharmaceutical Industry, Ipragliflozin, NEW PATENT, WO 2016173551
























.JPG)