![str1]()
![Figure]()
![SCHEMBL2786493.png]()
BMS-741672
N-((1R,2S,5R)-5-(Isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide BMS-741672
N-((lR,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6- (trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-l-yl)cyclohexyl)acetamide
N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide;
C25 H33 F3 N6 O2, 506.56
Acetamide, N-[(1R,2S,5R)-5-[methyl(1-methylethyl)amino]-2-[(3S)-2-oxo-3-[[6-(trifluoromethyl)-4-quinazolinyl]amino]-1-pyrrolidinyl]cyclohexyl]-
CAS 1004757-96-3
PHASE 2, , Treatment of Type 2 Diabetes, Agents for Neuropathic Pain
Chemokine CCR2 (MCP-1 Receptor) Antagonists
![Image result for Bristol-Myers Squibb]()
| Molecular Formula: |
C25H33F3N6O2 |
| Molecular Weight: |
506.574 g/mol |
![Image result for bristol myers squibb headquarters]()
- Originator Bristol-Myers Squibb
- Class Analgesics; Antihyperglycaemics
- Mechanism of Action CCR2 receptor antagonists
- Discontinued Diabetic neuropathies; Type 2 diabetes mellitus
Most Recent Events
- 10 Apr 2007 Preclinical trials in Inflammation in USA (unspecified route)
BMS-741672, 1 , is a highly selective CCR2 antagonist (IC50 = 1.4 nM) featuring a complex array of four stereocenters. The key synthetic challenge was efficient assembly of the densely functionalized 1,2,4-triaminocyclohexane (TACH) core in a minimum number of linear steps.
![Figure]()
N-((1R,2S,5R)-5-(Isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide BMS-741672
Mp 161.3 °C.
1H NMR (400 MHz, CDCl3) δ 9.50–9.20 (1H), 9.04 (s, 1H), 8.68 (s, 1H), 8.41 (d, J = 7.1 Hz, 1H), 7.87 (s, 1H), 5.04 (dt, J = 1.3, 7.3 Hz, 1H), 4.9 (m, 1H), 4.07 (dt, J = 3.7, 12.9 Hz, 1H), 3.53 (dt, J = 1.4, 9.9 Hz, 1H), 3.44–3.30 (m, 2H), 2.39 (dq, J = 13.6, 8.4 Hz, 1H), 2.26 (m, 1H), 2.21 (s, 3H), 2.17 (q, J = 2.9 Hz, 1H), 2.03–1.91 (m, 5H), 1.71–1.54 (m, 5H), 1.04 (s, br., 6H).
13C NMR (100 MHz, d6-DMSO) δ 171.46, 169.49, 159.62, 156.92, 151.22, 129.28, 128.27 (q, 4JCF = 3 Hz), 125.78 (q, 2JCF = 32 Hz), 124.11 (q, 1JCF = 272 Hz), 121.57 (q, 3JCF = 4 Hz), 114.33, 54.83, 53.54, 52.36, 47.34, 46.94, 43.13, 30.76, 30.24, 26.94, 26.38, 23.28, 20.87, 17.65 (br.), 16.73 (br.).
13C NMR (100 MHz, CDCl3) δ 172.17. 170.73, 159.89, 156.91, 151.16, 128.68, 128.06 (q,4JCF = 3.0 Hz), 127.25 (q, 2JCF = 32 Hz), 123.98 (q, 1JCF = 272 Hz), 121.78 (q, 3JCF = 4 Hz), 115.11, 54.89, 53.21, 52.40, 47.40, 46.98, 43.72, 30.84, 30.70, 29.96, 27.80, 23.55, 19.96, 17.70 (2C).
LCMS (ESI, pos.): 508 (16.8), 507 (66.2), 254 (5.0). HR-ESI(pos)-MS: calcd for C25H34F3N6O2 507.2690 [M + H]+, found 507.2694.
IR (KBr): ν = 3428 (m, br.), 2966![(w)]() , 1686 (s), 1635 (m), 1584 (s), 1540 (m), 1334 (m), 1307 (s), 1164 (m), 1121 (m), 870
, 1686 (s), 1635 (m), 1584 (s), 1540 (m), 1334 (m), 1307 (s), 1164 (m), 1121 (m), 870![(w)]() , 845
, 845![(w)]() .
.
[α]20D−187.9 (c 1.0, CHCl3).
Anal. Calcd for C25H33F3N6O2: C, 59.28; H, 6.57; F, 11.25; N, 16.59. Found: C, 59.21; H, 6.43; F, 11.07; N, 16.53.
![Image result for Bristol-Myers Squibb]()
PATENT
WO 2008014381
http://www.google.ch/patents/WO2008014381A2?cl=en&hl=de
EXAMPLE 1
N-((lR,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6- (trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-l-yl)cyclohexyl)acetamide
![Figure imgf000072_0001]()
[00212] Example 1, Step 1: (IR, 2S, 5R)-tert-Butyl 2-benzyloxycarbonylamino- 7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate (89.6 g, 0.24 mol, see: P. H. Carter, et al. PCT application WO 2005/021500) was dissolved in ethyl acetate (1.5 L) and the resulting solution was washed with sat. NaHCCh (2 x 0.45 L) and sat. NaCl (I x 0.45 L). The solution was dried (Na2SO4) and then filtered directly into a 3 -necked 3 L round-bottom flask. The solution was purged with direct nitrogen injection before being charged with 10% Pd/C (13.65 g) under nitrogen atmosphere. The flask was evacuated and back-filled with hydrogen; this was repeated twice more. Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated, back-filled with nitrogen, and charged with fresh catalyst (6 g of 10% Pd/C). Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated and back-filled with nitrogen. The mixture was filtered through Celite; the filter pad was then washed with ethyl acetate. The filtrate (-1.6 L EtOAc volume) was diluted with acetonitrile (0.3 L) and charged sequentially with Z-N-Cbz- methionine (68 g, 0.24 mol), TBTU (77 g, 0.24 mol), and Ν,Ν-diisopropylethylamine (42 mL, 0.24 mol). The reaction was stirred at room temperature for 4 h, during which time it changed from a suspension to a clear solution. The reaction was quenched with the addition of sat. NH4Cl (0.75 L) and water (0.15 L); the mixture was diluted further with EtOAc (0.75 L). The phases were mixed and separated and the organic phase was washed with sat. Na2Cθ3 (2 x 0.9 L) and sat. NaCl (1 x 0.75 L). The solution was dried (Na2SO4), filtered, and concentrated in vacuo to give (IR,2S,5R)- tert-butyl 2-((5)-2-(benzyloxycarbonylamino)-4-
(methylthio)butanamido)-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate as an oil, which was taken into the next step without further purification. LC/MS for primary peak: [M-Boc+H]+ = 406.3; [M+Naf = 528.3. 1H-NMR (400 MHz, d4-Me0H): δ 7.36 (m, 5H), 5.11 (s, 2H), 4.32 (m, IH), 4.2 (m, IH), 4.0 (m, IH), 2.5 – 2.7 (m, 3H), 2.25 (m, IH), 2.11 (s, 3H), 2.05 (m, 4H), 1.9 (m, IH), 1.7 (m, 2H), 1.54 (s, 9H). Also present are EtOAc [1.26 (t), 2.03 (s), 4.12 (q)] and N,N,N,N-tetramethylurea [2.83
(S)].
[00213] Example 1, Step 2: A sample of (1^,25,5^)- tert-butyl 2-((5)-2- (benzyloxycarbonylamino)-4-(methylthio)butanamido)-7-oxo-6-aza- bicyclo[3.2. l]octane-6-carboxylate (0.24 mol assumed; see previous procedure) was dissolved in iodomethane (1,250 g) and stirred for 48 h at room temperature. The reaction was concentrated in vacuo. The residue was dissolved in dichloromethane and concentrated in vacuo. This was repeated twice more. The resultant sludge was dissolved in dichloromethane (0.4 L) and poured into a rapidly stirring solution of MTBE (4.0 L). The resultant yellow solids were collected via suction filtration and dried under high vacuum to afford the sulfonium salt (179 g). This material was taken into the next step without further purification. LC/MS for primary peak: [M- Me2S+H]+ = 458.4; [M]+ = 520.4. 1H-NMR (400 MHz, d4-Me0H): δ 7.35 (m, 5H), 5.09 (s, 2H), 4.33 (m, IH), 4.28 (m, IH), 3.98 (m, IH), 3.3 – 3.45 (m, 2H), 2.97 (s, 3H), 2.94 (s, 3H), 2.78 (m, IH), 2.0 – 2.3 (m, 4H), 1.7 (m, 2H), 1.52 (s, 9H). Also present are MTBE [1.18 (s), 3.2 (s)] and traces of N,N,N,N-tetramethylurea [2.81 (s)]. [00214] Example 1, Step 3: All of the sulfonium salt from the previous step (0.24 mol assumed) was dissolved in DMSO (2.0 L). The resultant solution was stirred under nitrogen at room temperature and charged with cesium carbonate (216 g) portionwise. The suspension was stirred at room temperature for 3 h and then filtered to remove the solids. The solution was divided into -0.22 L portions and worked up as follows: the reaction mixture (-0.22 L) was diluted with ethyl acetate (1.5 L) and washed successively with water (3 x 0.5 L) and brine (1 x 0.3 L). The organic phase was dried (Na2SO4), filtered, and concentrated in vacuo. The desired (\R,2S,5R)- tert-bvXyl 2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-7-oxo-6- azabicyclo[3.2.1]octane-6-carboxylate (90.8 g, 83%) was obtained as a microcrystalline foam, free from tetramethyl urea impurity. LC/MS for primary peak: [M-Boc+H]+ = 358.4; [M+Na]+ = 480.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.12 (s, 2H), 4.35 (m, 2H), 4.2 (m, IH), 3.6 (m, IH), 3.3 (m, IH), 2.64 (m, IH), 2.28 – 2.42 (m, 2H), 2.15 (m, IH), 1.7 – 2.0 (m, 5H), 1.55 (s, 9H). If desired, this material can be isolated as a solid by dissolving in MTBE (1 volume), adding to heptane (3.3 volumes), and collecting the resultant precipitate.
[00215] Example 1, Step 4: A stirring solution of (\R,2S,5R)- tert-butyl 2-((S>3- (benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-7-oxo-6-azabicyclo[3.2.1]octane-6- carboxylate (108 g, 0.236 mol) in THF (1 L) was charged with lithium hydroxide monohydrate (21.74 g, 0.519 mol). Water (0.3 L) was added slowly, such that the temperature did not exceed 20 0C. The reaction was stirred at room temperature overnight and the volatiles were removed in vacuo. The pH was adjusted to -4 through the addition of IN HCl (450 mL) and NaH2PO4. The resultant white precipitates were collected by filtration and washed with water (2 x 1 L). The solid was dissolved in dichloromethane (1.5 L) and water (~ 1 L). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was dissolved in EtOAc (0.7 L) and the resultant solution was heated at reflux for 1 h. Solids separated after cooling to RT, and were collected via filtration. These solids were purified by recrystallization in isopropanol to afford the desired (\R,2S,5R)-2-((S)-3- (benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxylic acid as a white solid (104.5 g, 93% yield). LC/MS for primary peak: [M-tBu+H]+ = 420.2; [M-Boc+H]+ = 376.2; [M+H]+ = 476.2. 1H-NMR (400 MHz, d4-Me0H): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.35 (m, 2H), 3.71 (m, IH), 3.45 – 3.6 (m, 2H), 2.99 (m, IH), 2.41 (m, IH), 2.15 (m, IH), 2.0 (m, 2H), 1.6 – 1.9 (m, 4H), 1.46 (s, 9H).
[00216] Example 1, Step 5: A 3 L round bottom flask was charged with (lR,25′,5R)-2-((5)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxylic acid (75.5 g, 0.158 mol), EDOHCl (33.5 g, 0.175 mol), 1 -hydroxybenzotriazole (23.6 g, 0.175 mol), and dichloromethane (1 L). The reaction was stirred at room temperature for 2 h, during which time it changed from a white suspension to a clear solution. Ammonia (gas) was bubbled into the solution until the pH was strongly basic (paper) and the reaction was stirred for 10 min; this ammonia addition was repeated and the reaction was stirred for an additional 10 min. Water was added. The organic phase was washed with sat. NaHCθ3, NaH2PO4, and brine before being concentrated in vacuo. The residue was slurried with acetonitrile (0.5 L) and then concentrated in to give (lR,2S,5R)-2-((5)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxamide as a white solid (75.9 g, -100%), which was used in the next step without further purification. LC/MS for primary peak: [M-Boc+H]+ = 375.3; [M+H]+ = 475.4; [M-tBu+H]+ = 419.3. 1H-NMR (400 MHz, Cl4-MeOH): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.25 (m, 2H), 3.70 (m, IH), 3.6 (m, IH), 3.45 (m, IH), 2.91 (m, IH), 2.38 (m, IH), 2.12 (m, IH), 1.9 – 2.05 (m, 2H), 1.65 – 1.9 (m, 4H), 1.46 (s, 9H).
[00217] Example 1, Step 6: The reaction was run in three equal portions and combined for aqueous workup. A 5 L, 3-necked round bottom flask was charged with (lR,2S,5R)-2-((5)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-l-yl)-5-(tert- butoxycarbonylamino)cyclohexanecarboxamide (25.3 g, 53 mmol), acetonitrile (1.9 L), and 2.6 L of water/ice. The mixture was stirred and cooled to 0 0C. Iodobenzene diacetate (25.77 g, 80 mmol) was added and the reaction was stirred for 2 h; another 0.5 eq of iodobenzene diacetate was added. The reaction was stirred for 9 h (reaction temp < 10 0C). The mixture was charged with 8 eq N,N-diisopropylethylamine and 2 eq acetic anhydride. Over the next thirty minutes, 4 eq N,N-diisopropylethylamine and 2 eq acetic anhydride were added every ten minutes, until the reaction had proceeded to completion (HPLC). The acetonitrile was removed in vacuo; some solid separated from the residue, and this was collected by filtration. The remaining residue was extracted with dichloromethane (3 L, then 1 L). The organic phase was washed sequentially with water, sat. NaHCθ3, and brine. The collected solids were added to the organic phase, along with activated carbon (15 g). The mixture was stirred for 30 minutes at 40 0C before being filtered and concentrated in vacuo. The residue was dissolved in EtOAc (1 L), and the resultant solution was stirred at 75 0C for 1 h before being allowed to cool to room temperature. A solid separated and was collected by filtration. This solid was purified further by recrystallization: it was first dissolved in 0.5 L CH2CI2, then concentrated in vacuo, then re-crystallized from 1 L EtOAc; this was repeated three times. The solids obtained from the mother liquors of the above were recrystallized three times using the same method. The combined solids were recrystallized twice more from acetonitrile (0.7 L) to provide 66 g (84%) of tert-bυXyl (lR,3R,45)-3-acetamido-4-((5)-3-(benzyloxycarbonylamino)-2- oxopyrrolidin-l-yl)cyclohexylcarbamate (purity >99.5% by HPLC). LC/MS for primary peak: [M+H]+ = 489.4; [M-tBu+H]+ = 433.3. 1H-NMR (400 MHz, d4– MeOH): δ 7.3 – 7.4 (m, 5H), 5.11 (s, 2H), 4.35 (m, IH), 4.15 (m, IH), 4.04 (m, IH), 3.8 (m, IH), 3.6 (m, 2H), 2.44 (m, IH), 2.12 (m, IH), 1.87 – 2.05 (m, 4H), 1.87 (s, 3H), 1.55 – 1.7 (m, 2H), 1.46 (s, 9H). The stereochemical fidelity of the Hofmann rearrangement was confirmed through X-ray crystal structure analysis of this compound, as shown in Figure 1. [00218] Example 1, Step 7: A stirring solution of tert-butyl (\R,3R,4S)-3- acetamido-4-((5′)-3 -(benzyloxycarbonylamino)-2-oxopyrrolidin- 1 – yl)cyclohexylcarbamate (66 g, 0.135 mol) in dichloromethane (216 mL) was charged with trifluoroacetic acid (216 mL). The reaction was stirred for 2 h at room temperature and concentrated in vacuo. The residue was dissolved in methanol and the resultant solution was concentrated in vacuo; this was repeated once. Benzyl («S)-l-((l«S,2R,4R)-2-acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate was obtained as an oil and used directly in Step 8 below. LC/MS found [M + H]+ = 389.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3 – 7.4 (m, 5H), 5.12 (s, 2H), 4.41 (br. s, IH), 4.15 (m, IH), 4.00 (t, J= 9.3 Hz, IH), 3.81 (t, J= 9.1 Hz, IH), 3.65 (q, J= 8.4 Hz, IH), 3.3 – 3.4 (m, IH), 2.45 (m, IH), 1.95 – 2.24 (m, 5H), 2.00 (s, 3H), 1.6 – 1.8 (m, 2H). [00219] Example 1, Step 8: A stirring solution of benzyl (S)- 1-(( \S,2R,4R)-2- acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (-0.135 mol) in methanol (675 mL) was charged sequentially with acetone (37.8 g, 4 eq), sodium acetate (33.2 g, 3 eq), and sodium cyanoborohydride (16.9 g, 2 eq). The mixture was stirred at room temperature for 6 h and filtered. The filtrate was dissolved in dichloromethane (1 L); this solution was washed with IN NaOH (1 L). The solids collected in the filtration were dissolved in IN NaOH (IL) at 0 0C and then extracted with dichloromethane (1 L). The organic extracts were combined and extracted with aqueous HCl (200 mL IN HCl + 800 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then IN NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl (S)-I- ((lS,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2-oxopyrrolidin-3- ylcarbamate as an oil. LC/MS found [M + H]+ = 431.45. 1H-NMR (400 MHz, d4– MeOH): δ 7.3 – 7.4 (m, 5H), 5.12 (s, 2H), 4.31 (m, IH), 4.24 (t, J= 9.4 Hz, IH), 4.11 (m, IH), 3.61 (t, J= 9.1 Hz, IH), 3.52 (q, J= 8.6 Hz, IH), 3.04 (br. s, IH), 2.96 (sep, J= 6.3 Hz, IH), 2.40 (m, IH), 2.15 (m, IH), 1.92 (s, 3H), 1.7 – 1.9 (m, 5H), 1.65 (m, IH), 1.12 (app. dd, J= 6.3, 1.1 Hz, 6H).
[00220] Example 1, Step 9 (See Alternative Step 9, below): A stirring solution of benzyl (S)-I -((lS’,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2- oxopyrrolidin-3-ylcarbamate (-115 mmol) in dichloromethane (600 mL) was cooled to 0 0C and charged sequentially with formaldehyde (18.6 g, 37 wt% solution), triethylamine (23 mL), and sodium triacetoxyborohydride (28.7 g). The mixture was stirred at room temperature for 30 minutes and diluted with dichloromethane (up to 1.2 L). This solution was washed thrice with 500 mL sat. NaHCθ3 + NaOH (sat. NaHCO3, pH to 11 w/ IN NaOH). The organic layer was extracted with aq. HCl (200 mL IN HCl + 600 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then IN NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (1.2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl {S)-\-{{\S,2R,AR)-2- acetamido-4-(isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate as an oil, which was used directly in Step 10 below. LC/MS found [M + H]+ = 445.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3 – 7.4 (m, 5H), 5.12 (s, 2H), 4.33 (br s, IH), 4.25 (t, J= 9.2 Hz, IH), 4.11 (br s, IH), 3.5 – 3.6 (m, 2H), 2.77 (v br s, 2 H), 2.41 (m, IH), 2.26 (s, 3H), 2.0 – 2.1 (m, 2H), 1.92 (s, 3H), 1.7 – 1.9 (m, 5H), 1.10 (app. dd, J = 17, 6.4 Hz, 6H). [00221] Example 1, Step 10: To a solution of benzyl (S)- 1-(( 15″,2R,4R)-2- acetamido-4-(isopropyl(methyl)amino)-cyclohexyl)-2-oxopyrrolidin-3 -ylcarbamate (-0.115 mol) in methanol (600 mL) was added 10% Pd/C (6 g of 50% wet catalyst). The flask was evacuated and back-filled with hydrogen. The mixture was stirred under 1 atm H2 for 2 h and the catalyst was removed by filtration through Celite. The filtrate was concentrated in vacuo to provide N-((li?,25,5i?)-2-((S)-3-amino-2- oxopyrrolidin-l-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide as an oil, which was taken on to the next step without further purification. LC/MS found [M + H]+ = 311.47. 1H-NMR (400 MHz, (I4-MeOH): δ 4.39 (br s, IH), 4.00 (m, IH), 3.3 –
3.5 (m, 4H), 2.73 (m, IH), 2.38 (m, IH), 2.25 (s, 3H), 2.0 – 2.2 (m, 3H), 1.94 (s, 3H),
1.6 – 1.75 (m, 4H), 1.07 (app. dd, J= 21, 6.4 Hz, 6H). [00222] Example 1, Step 11: To a solution of N-((lR,25′,5R)-2-((S)-3-amino-2- oxopyrrolidin-l-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide (~35 g, 0.115 mol) in isopropanol (600 mL) was added 4-chloro-6-(trifluoromethyl)quinazoline (32 g, 0.138 mol, 1.2 eq, see: P.H. Carter et al, PCT application WO 2005/021500). The mixture was stirred at room temperature overnight before being charged with triethylamine (46 g, 0.46 mol, 4 eq). The mixture was stirred at 60 0C for 10 h. The solvent was removed under reduced pressure to give an oil. Azeotropic distillation with isopropanol was performed twice. The residue was dissolved in dichloromethane (600 mL) and extracted with water (250 mL, containing 4 eq acetic acid). Dichloromethane (600 mL) was added to the combined aqueous washes, and the mixture was cooled to 0 0C. Aqueous NaOH (50% by weight) was added with stirring until the pH reached 11. The water layer was extracted with dichloromethane twice (2 x 600 mL). The combined organic extracts were dried (Na2SO4), filtered, and concentrated in vacuo to give the amorphous free base of the title compound (99% purity by HPLC). LC/MS found [M+H]+ = 507.3. 1H-NMR (400 MHz, U4– MeOH): δ 8.82 (s, IH), 8.59 (s, IH), 8.05 (dd, J= 8.8, 1.8 Hz, IH), 7.9 (d, J= 8.7 Hz, IH), 5.28 (t, J= 8.6 Hz, IH), 4.58 (br s, IH), 4.06 (m, IH), 3.52 – 3.68 (m, 2H), 3.43 (m, IH), 2.76 (br s, IH), 2.55 (m, IH), 2.28 (s, 3H), 2.1 – 2.3 (m, 3H), 2.0 (s, 3H), 2.0 (m, IH), 1.65 – 1.8 (m, 3H), 1.09 (app. dd, J= 24, 6.4 Hz, 6 H).
Example 1, Alternative Step 9
![Figure imgf000079_0001]()
[00223] Example 1, Alternative step 9a1: To a hydrogenator were charged ethyl (7R,SS)-S-((S)- l-phenyl-ethylamino)-l,4-dioxa-spiro[4.5]decane-7-carboxylate A- toluenesulfonate salt I A (1417 g, 2.8 moles, c.f : WO2004098516, prepared analogous to US Pat.6,835,841), ethanol (200 proof, 11.4 L), and 10% Pd/C catalyst (50% wet, 284 g). The mixture was inerted with nitrogen, then pressurized with hydrogen gas (45 psig) and agitated vigorously at approx. 40 0C until starting material was consumed (HPLC). The suspension was cooled, purged with nitrogen gas and the catalyst was removed by filtration while inerted. The spent catalyst was washed with ethanol (4.3 L). The filtrate and washings were combined and concentrated under vacuum to a volume of 2-3 L while maintaining the batch between 40°-60 0C. Isopropyl acetate (5 L) was charged and the mixture was concentrated to a volume of ~2 L until most ethanol was removed (<0.5%) and residual moisture content was <l,000 ppm. Batch volume was adjusted to -7.5 L by the addition of isopropyl acetate. The mixture was heated to 80 0C until clear, then cooled 65°-70 0C. Seed crystals of 1 (5 g) were added and the batch was cooled to 500C over 2 hours, then further cooled to 20 0C over 4 hours and held for ~10 hours. The resulting slurry was filtered and the cake was washed with isopropyl acetate (2 L). The product was dried under vaccum at -35 0C until volatiles were recduced below -1% (LOD). Ethyl (7R,85′)-8-amino-l,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 was obtained as a white, crystalline solid (936 g, 83% yield; HPLC purity: 99.8%). 1H-NMR: (300MHz, CDCl3) 8.14-7.89 (brs, 3H), 7.75 (d, J 9.0Hz, 2H), 7.15 (d, J 8.0Hz, 2H), 4.22-4.04 (m, 2H), 4.01-3.77 (m, 4H), 3.55-3.43 (m, IH,), 3.20-3.13 (m, IH), 2.40-2.27 (m, 4H), 2.21-1.94 (m, 2H), 1.81-1.51 (m, 3H), 1.23 (t, J 7.0Hz, 3H); HPLC: Waters Xterra MS C18 4.6 mm x 150 mm Ld., 3.5μm particle size, 0.05% NH40H (5% ACN, 95% H2O, solvent A), to 0.05% NH4OH (95% ACN, 5% H2O, solvent B), 5% B to 20% B in 10 minutes, changed to 95% B in 25 minutes, and then changed to 5% B in 1 minute; 11.1 minutes (aminoester 1).
![Figure imgf000080_0001]()
Example 1, Alternative Step 9a”: Aminoester 1 (63g, 0.16M, leq.; the product of reductive deprotection of a known compound – (See e.g. R. J. Cherney, WO 2004/098516 and G. V. Delucca & S. S. Ko, WO 2004/110993) was placed in a round bottom flask and MeCN (50OmL) was added. EDAC (33.1g, 0.17M, l. leq), HOBt-H2O (21.2g, 0.16M, l.Oeq) and N-Cbz-Z-methionine (46.7g, 0.17M, 1.05eq) were then added followed by TEA (48.OmL, 0.35M, 2.2eq). An exotherm to 38 0C was observed. The reaction mass was left to stir at RT. After 30mins, HPLC indicated complete conversion. The reaction mass was diluted with EtOAc (2.5L) and washed with KHCO3 (4x500mL, 20wt% aq. solution) and brine (50OmL). The organic phase was separated, dried over MgSO4 and concentrated. The residue was dissolved in TBME and reconcentrated to give ethyl (7R,85)-8- {(2S)-2-benzyloxycarbonylamino- 4-methylsulfanyl-butyr-yl-amino}-l,4-dioxa-spiro[4.5]decane-7-carboxylate 2 as a sticky semi-solid (76.2g, 98% yield, 93AP purity). 1H-NMR: (300MHz, CDCl3) δ 7.36-7.30 (m, 5H), 7.03 (d, J9.0Hz, IH), 5.66 (d, J 8.0Hz, IH), 5.10 (s, 2H), 4.35- 4.25 (m, 2H), 4.19-4.04 (m, 2H,), 3.98-3.86 (m, 4H), 2.87-2.80 (m, IH), 2.55-2.45 (m, 2H), 2.18 (dd, J 14.0Hz, 7.0Hz, IH), 2.08 (s, 3H), 2.05-1.67 (m, 6H), 1.26 (t, J 7.0Hz, 3H). HPLC: YMC-Pack Pro C18 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. lO.Olmin (Compound 2, 93.1 AP). HRMS: m/z 495.2166 [CaIc: C24H35N2O7S 495.2165].
![Figure imgf000081_0001]()
2 3 [00224] Example 1, Alternative Step 9b: Methionine amide 2 (75.Og, 0.15M) was dissolved in MeI (225mL, 3mL/g) – some off gassing was noted but no exotherm. The reaction mass was left to stir in the dark for 16.5h. After this time a thick light yellow precipitate had formed. The flask was then evacuated to 200mmHg and some of the MeI removed. The remaining material was slurried in TBME (50OmL), after a 30min stir-out the slurry was filtered, the cake washed with TBME (50OmL). NMR analysis of this material indicated a small amount of MeI remaining. The cake was re-slurried in TBME (50OmL), filtered, washed with TBME (50OmL) and dried under vacuum to give [(35)-3-benzyloxycarbonylamino-3-{(7R,85′)-7- ethoxycarbonyl-l,4-di-oxa-spiro[4.5]dec-8-ylcarbamoyl}-propyl]-dimethylsulfonium iodide 3 as a free flowing off-white solid (93.5g, 97%, 99 area% purity). 1H-NMR: (300MHz, CDCl3) δ 7.75 (d, J 9.0Hz, IH), 7.38-7.27 (m, 5H), 6.40 (d, J 7.0Hz, IH), 5.10 (s, 2H), 4.76-4.65 (m, IH), 4.48-4.39 (m, IH), 4.14-3.85 (m, 6H), 3.84-7.73 (m, IH), 3.68-3.55 (m, IH), 3.21 (s, 3H), 3.12 (s, 3H), 2.90-2.83 (s, IH), 2.52-1.55 (m, 8H), 1.24 (t, J7.0Hz, 3H). HPLC: YMC-Pack Pro C18 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 2.45min (I-), 8.14min (Compound 3, 43.6AP, I“ 54.6AP). HRMS: m/z 509.2341 [CaIc: C25H37N2O7S 509.2321].
![Figure imgf000082_0001]()
[00225] Example 1, Alternative Step 9c: Cs2CO3 (61.5g, 0.19M, 1.5eq) was placed in an round bottom flask and anhydrous DMSO (2.4L) was added. Sulfonium salt 3 (80.Og, 0.13M, 1.Oeq) was then added portionwise. Once the addition was complete the reaction mass was left to stir in the dark for 2Oh. The reaction mass was then split in half and each half worked up separately: the reaction mass was diluted with EtOAc (2.0L) and washed with brine (2L), the organic phase was washed with brine (50OmL). The combined aq. layers were then washed EtOAc (50OmL). The combined organic phases were then washed with brine (3x750mL). The second half of the reaction mass was treated in an identical manner and the combined organics dried over MgSO4 and concentrated to give ethyl (7R,8S)-8-{(3S>3- Benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl}-l,4-dioxa-spiro[4.5]decane-7- carboxylate 4 as a light colored oil (56.5g, 0.13M, -100 area-% purity) pure by NMR analysis. 1H-NMR: (300MHz, CDCl3) δ 7.38-7.30 (m, 5H), 5.37 (br d, J4.0Hz, IH), 5.11 (s, 2H), 4.27-4.18 (m, IH), 4.17-3.82 (m, 8H), 3.32 (td, J 10.0Hz, 60.0Hz, IH), 3.23 (q, J5.0Hz, IH), 2.63-2.57 (m, IH), 2.42-2.25 (m, 2H), 1.94-1.68 (m, 5H), 1.25 (t, J 7.0Hz, 3H). HPLC: YMC-Pack Pro Cl 8 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 8.99min (Compound 5, produced on column, 4.2AP), 9.48 (Compound 4, 74.3AP). HRMS: m/z 447.2127 [CaIc: C23H31N2O7 447.2131].
![Figure imgf000083_0001]()
4 5
[00226] Example 1, Alternative Step 9d: Pyrrolidinone 4 (50.Og, 0.1 IM) was dissolved in acetone (50OmL) and IN HCl (50OmL) was added. The reaction mass was then heated to 65°C. After 20mins HPLC indicated complete reaction. The reaction mass was allowed to cool to RT and the acetone was removed on a rotary evaporator. During this distillation the product precipitated from solution as a white solid. This was isolated by filtration and the cake washed with water. The cake was then dried azeotropically with toluene (3x3OOmL) to give ethyl (\R,2S)-2-((3S)-3- Benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-oxo-cyclohexanecarboxylate 5 as a white solid (39.8g, 88%, 97 area-% purity). 1H-NMR: (300MHz, CDCl3) δ 7.37- 7.32 (m, 5H), 6.65 (br d, J4.0Hz, IH), 5.12 (s, 2H), 4.54-4.47 (m, IH), 4.34-4.26 (m, IH), 4.18 (dq, J 11.0Hz, 7.0Hz, IH), 4.09 (dq, J 11.0Hz, , 7.0Hz, IH), 3.36-3.20 (m, 3H), 2.70-2.35 (m, 6H), 2.05-1.96 (m, IH), 1.81 (quin., J l l.OHz, IH), 1.24 (t, J 7.0Hz, 3H). HPLC: YMC-Pack Pro C18 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 8.95min (Compound 5). HRMS: m/z 403.1864 [CaIc: C2iH27N2O6403.1869].
![Figure imgf000083_0002]()
[00227] Example 1, Alternative Step 9e: Cyclohexanone 5 (22.5g, 0.06M, leq), DMSO (3OmL) and Ti(O-ZPr)4 (33.7mL, 0.1 IM, 2.04eq) were placed in a round bottom flask. N-isopropyl-N-methylamine (11.6mL, 0.1 IM, 2.0eq) was then added in one portion. The mixture was left to stir for 30mins at room temperature before being cooled to <3°C in ice/water. MeOH (3OmL) was then added followed by the portionwise addition OfNaBH4 (4.33g, 0.1 IM, 2.04eq) – temperature kept <8°C. 30mins after the addition was completed the reaction mass was diluted with methylene chloride (30OmL) and then NaOH (IN, 4OmL). The resulting slurry was filtered through Celite, and the cake washed with methylene chloride (10OmL). The resulting liquor was concentrated under reduced pressure and the residue dissolved in EtOAc (50OmL). This solution was extracted with IN HCl (2x400mL), the combined aqueous layers were then basified with Na2CO3. Extraction with EtOAc (4x250mL) provided a clear and colorless organic phase which was dried over Na2SO4 and concentrated to give a white powder (24.6g, 96%, 7: 1 d.r.). This material was then slurried overnight in hexane (67OmL). The solid was isolated by filtration and dried under reduced pressure to give ethyl (lR,25′,5R)-2-((3S)-3-benzyloxycarbonylamino- 2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylate 6 as a while solid (20.9g, 81%, 24: 1 d.r.). 1H-NMR: (300MHz, CDCl3) δ 7.37-7.28 (m, 5H), 5.55 (d, J4.5, IH), 5.10 (s, 2H), 4.42 (q, J4.5, IH), 4.23-4.12 (m, IH), 4.08 (dq, J 10.5, 7.0, IH), 4.02 (dq, J 10.5, 7.0, IH), 3.84 (t, J9.0, IH), 3.46-3.36 (m, IH), 3.04 (septet, J6.5, IH), 2.86-2.80 (m, IH), 2.63-2.48 (m, 2H), 2.17 (s, 3H, Me), 2.10-1.63 (m, 7H), 1.22 (t, J 7.0, 3H), 1.00 (d, J 6.5, 3H), 0.97 (d, J 6.5, 3H). HPLC: YMC- Pack Pro C18 5μm 4.6 x 150 mm, 0.01M NH4OAc (MeOH:water 20:80) to 0.01M NH4OAc (MeOH:water:MeCN 20:5:75) 10 to 100% 15min gradient. 8.23 (Compound 6), 8.88 (5-e/«-Compound 6). HRMS: 460.2798 [CaIc: C25H38N3O5 460.2811].
![Figure imgf000084_0001]()
[00228] Example 1, Alternative Step 9f: The aminoester 6 (9.76 g, 2.12 mmol) was dissolved in 2N HCl (80 mL), then heated to -55 0C under inert atmosphere. The reaction was stirred for 20 h, then cooled to room temperature. The reaction solution was washed twice with toluene (25 mL portions), neutralized to pH 6 – 7 by the addition of KOH pellets, then extracted eight times with methylene chloride (100 mL portions). The combined extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure to 50 mL total volume. The concentrated solution was then slowly added to methyl tert-butyl ether (300 mL) over 15 min in an addition funnel with vigorous stirring. The resulting white slurry was stirred at ambient temperature for Ih, then cooled to 0 0C and stirred for Ih. The product was filtered, and washed twice with methyl tert-butyl ether (25 mL portions). Water from the wet cake was removed by azeotropic distillation with acetonitrile (300 mL). The product was dried under reduced pressure to provide (li?,25r,5R)-2-((35′)-3-Benzyloxycarbonylamino-2- oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylic acid 7, (7.69 g, 84% yield) as a white foam. 1H-NMR: (400 MHz, 500C, CDCl3) δ 7.44-7.32 (m, 5H), 6.10 (broad s, IH), 5.19 (app s, 2H), 4.42 (dd, J= 15.6, 7.8 Hz, IH), 4.29-4.23 (m, IH), 3.68-3.60 (m, 2H), 3.33-3.27 (m, 2H), 3.20 (broad s, IH), 2.99 (broad s, IH), 2.51 (s, 3H), 2.49-2.45 (m, 3H), 2.33-2.31 (m, IH), 2.00 (ddd, J= 9.0, 8.6, 3.9 IH), 1.95-1.78 (m, 2H), 1.36-1.21 (m, 6H). LCMS: m/z 432.20 [CaIc: C23H34N3O5 432.25].
NHCbz
![Figure imgf000085_0001]()
![Figure imgf000085_0002]()
![Figure imgf000085_0003]()
[00229] Example 1, Alternative Step 9g: Amino acid 7 (6.3g, 14.7mmol, l.Oeq) was dissolved in THF (8OmL) under N2 and NaH (584mg, 14.7mmol, l.Oeq, 60wt% dispersion in mineral oil) was added portionwise. When the addition was complete, and the evolution of gas had ceased, the reaction mass was concentrated under reduced pressure and the resulting solid azeotroped with toluene (50 mL) to give a white solid (KF 0.59wt%). This solid was slurried in toluene (100 mL) under N2and heated to 900C. DPPA (3.32 mL, 15.3 mmol, 1.05 eq) was added dropwise over ~2min. After ~5min all the solids had dissolved, after lOmins precipitation of a white solid was observed. After 30mins HPLC analysis indicated complete reaction. The reaction mass was allowed to cool to RT before being filtered, the cake was washed with toluene. The liquors where then slowly added into ACOH/AC2O (80/20, 168mL) solution at 900C. After 45mins HPLC still indicated some isocyanate. At 1.15h , the reaction mass was cooled to RT and diluted with toluene (10OmL) and water (10OmL). The organic layer was removed and the toluene washed with IN HCl
(10OmL). The combined aq. phases were then basified with K2Cθ3(s) and brought to pH 12 with NaOH (10N), keeping the temperature below 200C. The aq layer was then extracted with methylene chloride (4xl50mL), the combined organic layers dried over K2CO3 and concentrated to give benzyl (S)-l-((lS,2R,4R)-2-acetamido-4- (isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate 8 as a white foam (4.5g, 70%, 94AP purity). The 1H-NMR was identical to material obtained from the route described above (Example 1, Step 9). HPLC: YMC-Pack Pro Cl 8 5μm 4.6 x 150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% lOmin gradient. 7.20min (Compound 8), 7.85min (urea dimer). HRMS: 445.2809 [CaIc: C24H37N4O4 445.2815].
Alternative Preparation of Example 1
2 3
[00230] Example 1, Alternative Preparation, Step 1: Ethyl (7R,85)-8-amino- l,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 (450. Ig), was combined with l-ethyl-3-(3-dimethyl-amino-propyl)carbo-diimide hydrochloride (236.3g), 1-hydroxy benzotriazole hydrate (171.9g), N-carbobenzyloxy-Z -methionine (333.4g) and acetonitrile (3.1 L). To the stirred mixture was added triethylamine (249.5g) below 30 0C. Upon reaction completion (HPLC), the mixture was diluted with ethyl acetate (8.2 L) and washed with aqueous 25% potassium bicarbonate solution (2×4.5 L) followed by water (4.5 L). The organic phase was separated and concentrated under reduced pressure to obtain a solution of ethyl (7R,85)-8-((5)-2- benzyloxycarbonylamino-4-methylsulfanyl-butyrylamino)-l,4-dioxa- spiro[4.5]decane-7-carboxylate 2 (1.4 L). Methyl iodide (2.39 kg) was added, the vessel was shielded from light and the mixture was held under slow agitation for approx. 24 h. To the thick yellow precipitate was added methyl tert-butyl ether (2.7 L) and the mixture was held for approx. 1 h. The product was isolated by filtration and the cake was washed with methyl tert-butyl ether (2×1.4 L), then dried under vacuum, yielding [(5)-3-benzyloxy-carbonylamino-3-((7R,8«S’)-7-ethoxycarbonyl-l,4- dioxa-spiro[4.5]dec-8-ylcarbamoyl)-propyl]-dimethylsulfonium iodide 3 (671.4 g, -94% yield) as an off-white solid (HPLC purity 99.9%).
![Figure imgf000087_0001]()
[00231] Example 1, Alternative Preparation, Step 2: Sulfonium salt 3 (619.4 g), and cesium carbonate (416.8 g) and anhydrous dimethyl sulfoxide (6.2 L) were combined in a reactor equipped with a scrubber to neutralize volatile sulfides.
Vigorous agitation was maintained until complete conversion was obtained (HPLC). Ethyl acetate (12.4 L) was added, followed by 20 % brine (3 L). The organic phase was separated, washed twice with brine (2×3 L) and evaporated to obtain a solution of ethyl (7R,8«S)-8-((«S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-l,4-dioxa- spiro[4.5]decane-7-carboxylate 4 in ethyl acetate (~0.8 L). Acetone (2.55 L) was added, followed by aqueous 0.5 M hydrochloric acid solution (2.3 L). With good mixing, the solution was heated to 50 to 60 0C until conversion of 4 to ethyl (IR,2S)- 2-((5)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-oxo- cyclohexanecarboxylate 5 was complete (HPLC). The mixture was concentrated under reduced pressure while below 40 0C, cooled to -30 0C, and water (4.1 L) was added. The resulting slurry was cooled to 5 to 10 0C and agitated for ~1 hour. The product was filtered and the cake was washed with water (2×2.5 L). Upon deliquoring, the cake was dried to a constant weight below 40 0C in a vacuum oven. Cyclohexanone 5 (272g, 70% yield) was obtained (HPLC purity 98.7%).
![Figure imgf000088_0001]()
[00232] Example 1, Alternative Preparation, Step 3: Cyclohexanone 5 (206 g) was dissolved in dichloromethane (1.1 L) and charged to a hydrogenator. Titanium tetraisopropoxide (218.2 g) and N-isopropyl N-methylamine (63.64 g) were added and the mixture was stirred at ambient temperature (23 to 25 0C) for at least 5 h. Platinum catalyst (5% Pt/S/C, 15 g, approx. 7.5 % relative to 5) was added and hydrogenation was performed at -30 psig for at least 6 h, yielding a mixture of ethyl (lR,25′,5R)-2-((5)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl- methyl-amino)-cyclohexanecarboxylate 6 and its 5-epz-isomer (-7%). The catalyst was removed by filtration and the filtrate was concentrated under reduced pressure to approx. -600 mL. Wet ethyl acetate (-3% water, 2.0 L) was added with vigorous agitation over a period of at least 1.5 h. Stirring was continued for at least an additional 6 h. The slurry was filtered. Filter cake was washed with ethyl acetate (1.0 L) and discarded. The combined filtrate and washings were concentrated to -400 mL. Toluene (2.0 L) was added and the solution was washed with 2M aqueous hydrochloric acid (2 x 400 mL). The aqueous layer was warmed to 50° to 60 0C for approx. 20 h or hydrolysis of 6 was deemed complete (HPLC). Aqueous sodium hydroxide solution was added to adjust to pH -10, and mixture was extracted with toluene (3×600 mL). The organic phase was discarded and pH was readjusted to ~6 by addition of aqueous hydrochloric acid. The aqueous phase was concentrated to -600 mL under reduced pressure and extracted with methylene chloride (at least 3×2.0 L). The combined methylene chloride layers were evaporated under reduced pressure and continuously replaced with THF to obtain a solution of (\R,2S,5R)-2- ((5*)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)- cyclohexane carboxylic acid 7 (-148 g) in THF (-4 L). Seed crystals of 8 were added, followed by 25 % solution of sodium methoxide in methanol (81.24 g) below 25 0C. The slurry was held for at least additional 16h with agitation. The product was isolated by filtration and the cake was washed with THF (4×200 mL) and dried to a constant weight in vacuo below 30 0C. Dry (lR,25′,5R)-2-((5)-3-benzyloxycarbonyl- amino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl-amino)-cyclohexane-carboxylate sodium salt 8 was obtained (139g, -60% yield from 5).
![Figure imgf000089_0001]()
[00233] Example 1, Alternative Preparation, Step 4: Aminoester sodium salt 8 (10Og), diphenyl phosphate (3.86g), tert-BuOH (1275 mL) and toluene (225 mL) were combined and heated to reflux under reduced pressure. Approx. 500 mL of distillate were collected and discarded while being continuously replaced with a solution of toluene in tert-BuOH. Vacuum was removed and distillate was switched to percolate through a column filled with molecular sieves and allowed to return to the vessel. After drying was complete, DPPA (52.4mL; dissolved in 60 mL toluene) was added slowly to the slurry at 80 0C. Upon complete conversion (HPLC), tert- BuOH was removed by vacuum distillation and continuously replaced with toluene. The mixture was cooled to room temperature and washed twice with 10% aqueous K2HPO4 (lx800mL, 1×400 mL) and water (40OmL). The organic phase was heated and concentrated in vacuo to approx. 27OmL. Vacuum was removed and heptane (1.1 L) was added slowly at approx. 80 0C, followed by seeds of 9 (~lg). The slurry was slowly cooled to room temperature and benzyl {(S)-l-[(lS,2R,4R)-2- tert- butoxycarbonylamino-4-(isopropyl-methyl-amino)-cyclo-hexyl]-2-oxo-pyrrolidin-3- yl} -carbamate 9 was isolated by filtration as a white solid (86.76g, 78% yield).
![Figure imgf000090_0001]()
[00234] Example 1, Alternative Preparation, Step 5: The tert-Butyl carbamate 9 (5Og) was dissolved in Toluene (50OmL) and /-PrOH (15OmL). The resulting solution was then heated to 6O0C. Methanesulfonic acid (19.6mL) was added below 65°C. Upon reaction completion (HPLC), the mixture was cooled to RT and triethylamine (69.4mL) added slowly below 25°C. Acetic anhydride was then added below 25°C. After Ih acetic acid (25OmL) was added below 25°C. The toluene phase was discarded and 2-methyl-THF (50OmL) was added to the aqueous phase. The mixture was stirred vigorously and basified with NaOH (25% aqueous solution) to pH 12. The aqueous phase was discarded and the organic layer was washed with brine (25OmL). The organic layer was concentrated under reduced pressure and continuously replaced with /-PrOH. The solution was cooled and filtered to provide benzyl {(5′)-l-[(15r,2R,4R)-2-acetylamino-4-(isopropyl-methyl-amino)-cyclohexyl]-2- oxo-pyrrolidin-3-yl} -carbamate 10 in /-PrOH solution which was used directly in the hydrogenation.
[00235] Example 1, Alternative Preparation, Step 6: To a solution containing acetamide 10 (~61g) in /-PrOH (-625 mL) was added 10% Pd/C wet catalyst (2.5 g) and the suspension was hydrogenated at 30 psig and approx. 25 0C for at least 2 h. Upon completion (HPLC), the catalyst was removed by filtration and the filtrate was concentrated to approx. 550 mL. Water (8.8 mL) was added, followed by 5.6 N hydrochloric acid in /-PrOH solution (69.5 mL). The resulting slurry was held at room temperature overnight. The product was isolated by filtration and the cake was rinsed with /-PrOH (2×100 mL) and dried in vacuo to constant weight at -50 0C to give N-[(li?,25r,5R)-2-((5′)-3-amino-2-oxo-pyrrolidin-l-yl)-5-(isopropyl-methyl- amino)-cyclohexyl]-acetamide 11 (55.6 g, 97% yield) as its hydrochloric acid salt (73.6% free base assay, HPLC).
NH,
CL,
Example 1
![Figure imgf000091_0001]()
[00236] Example 1, Alternative Preparation, Step 7: To 6-trifluoromethyl- quinazolin-4-ol 12 (20.1 g) in MeCN (400 mL) was added 5.5 M solution of sodium methoxide in methanol (17.0 mL). The resulting suspension was distilled under reduced pressure and continuously replaced by MeCN to remove methanol. To the slurry was added DMF (1.4 g), followed by oxalyl chloride (13.0 mL) below 50 0C. Upon reaction completion (HPLC), excess reagent was removed under reduced pressure to give -400 mL of slurry. The mixture was cooled to room temperature and washed with 10 % aqueous K2HPO4 (lxl.O L, 1×0.5 L) to afford 4-chloro-6- trifluoromethyl-quinazoline 13 (-21.2 g) in approx. 450 mL of wet MeCN solution, which was used directly in the subsequent coupling reaction (HPLC purity 99.8 %). [00237] Example 1, Alternative Preparation, Step 8: To a mixture of acetamide 11 (28.5 g, HCl salt, 73.6% free base assay), acetonitrile (100 mL), N,N,-di-isopropyl- N-ethylamine (61 mL) at room temperature was added a solution of 13 (-21.2 g) in MeCN (-450 mL). The homogeneous mixture was held overnight. Upon reaction completion (HPLC), the mixture was concentrated in vacuo to approx. 125 mL. A 9.5% aqueous solution of acetic acid (240 mL) was added and the aqueous phase was extracted with methylene chloride. The aqueous phase was separated and methyl tert- butyl ether (450 mL) was added, followed by 2N aqueous lithium hydroxide solution to adjust to pH >11.5. The organic layer was separated, washed with water and filtered. Approx. half of the ether phase was diluted with methyl tert-bvAyl ether (-250 mL) and concentrated in vacuo. Heptane (45 mL) was added slowly below 60 0C, followed by seed crystals of Example 1 (0.4 g). Additional heptane (125 mL) was added and the mixture was slowly cooled to room temperature and the resulting slurry was held overnight. The product was isolated by filtration, the cake was washed with heptane and dried in vacuo to constant weight to give N-((lR,25′,5R)-5- (isopropylamino)-2-((5′)-2-oxo-3-(6-(trifluoromethyl)-quin-azolin-4- ylamino)pyrrolidin-l-yl)cyclohexyl)acetamide 14 (15.0 g, 85% yield).
Crystallization Procedures for Example 1
[00238] Example 1, Production of bis-BSA salt and purification: The entirety of the amorphous free base from Example 1, Step 11 was dissolved in methanol (600 mL). The resultant solution was heated at 60 0C and charged with benzenesulfonic acid (2.5 eq). The mixture was cooled to room temperature and the resultant white solid was collected by filtration to yield the bis-benzene sulfonic acid salt of the title compound (95 g, 86%). This material was >99% pure by HPLC. This material was further purified by re-crystallization from 80/20 EtOH/H2θ, which provided the salt free from any residual methanol. HPLC purity = 99.8%. 1H ΝMR (500 MHz, D2O) δ ppm 8.75 (1 H, s), 8.66 (1 H, s), 8.25 (1 H, d, J=8.80 Hz), 7.90 (1 H, d, J=8.80 Hz), 7.75 (4 H, d, J=8.25 Hz), 7.43 – 7.57 (6 H, m), 5.42 (1 H, t), 4.33 – 4.44 (1 H, m), 4.09 – 4.19 (1 H, m), 3.83 – 3.91 (1 H, m), 3.74 – 3.83 (2 H, m), 3.61 (1 H, t, J=I 1.55 Hz), 2.75 (3 H, d, J=6.60 Hz), 2.61 – 2.70 (1 H, m), 2.31 – 2.44 (1 H, m), 2.20 – 2.27 (1 H, m), 2.17 (2 H, d, J=12.10 Hz), 1.94 – 2.04 (1 H, m, J=12.65 Hz), 1.90 – 1.95 (3 H, m), 1.72 – 1.91 (2 H, m), 1.37 (3 H, d, J=6.05 Hz), 1.29 (3 H, d, J=6.60 Hz). Differential scanning calorimetry utilized a heating rate of 10 °C/min and revealed a melting / decomposition endotherm with an onset temperature of 297.6 0C and a peak temperature at 299.1 0C. [00239] Example 1, Crystallization of the Free Base: A sample of the amorphous free base of N-((lR,25r,5R)-5-(isopropyl(methyl)amino)-2-((5′)-2-oxo-3- (6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin- 1 -yl)cyclohexyl)acetamide ( 1 g) was dissolved in dichloromethane (5 mL). The solution was charged with heptane (30 mL) and then warmed to distill the dichloromethane. The solution was cooled to 40 0C; a white solid precipitated. The suspension was heated to 90 0C and stirred for 2 h. The suspension was cooled to room temperature and filtered to provide the pure free base of the title compound. No residual solvent was apparent by 1H-NMR.
![Image result for bristol myers squibb]()
PATENT
US 7671062
http://google.com/patents/US7671062
The present invention provides a novel antagonist or partial agonists/antagonists of MCP-1 receptor activity: N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide,
or a pharmaceutically acceptable salt, solvate or prodrug, thereof, having an unexpected combination of desirable pharmacological characteristics. Crystalline forms of the present invention are also provided. Pharmaceutical compositions containing the same and methods of using the same as agents for the treatment of inflammatory diseases, allergic, autoimmune, metabolic, cancer and/or cardiovascular diseases is also an objective of this invention. The present disclosure also provides a process for preparing compounds of Formula (I), including N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide:
wherein R1, R8, R9, R10, and
are as described herein. Compounds that are useful intermediates of the process are also provided herein.
1st embodiment, the disclosure provides a process for preparing a compound of formula IV, or a salt thereof:
![Figure US07671062-20100302-C00010]()
Example 1 N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide
![Figure US07671062-20100302-C00060]()
Example 1, Step 1: (1R,2S,5R)-tert-Butyl 2-benzyloxycarbonylamino-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate (89.6 g, 0.24 mol, see: P. H. Carter, et al. PCT application WO 2005/021500) was dissolved in ethyl acetate (1.5 L) and the resulting solution was washed with sat. NaHCO3 (2×0.45 L) and sat. NaCl (1×0.45 L). The solution was dried (Na2SO4) and then filtered directly into a 3-necked 3 L round-bottom flask. The solution was purged with direct nitrogen injection before being charged with 10% Pd/C (13.65 g) under nitrogen atmosphere. The flask was evacuated and back-filled with hydrogen; this was repeated twice more. Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated, back-filled with nitrogen, and charged with fresh catalyst (6 g of 10% Pd/C). Hydrogen was bubbled through the solution for 30 min and then the reaction was stirred under 1 atm H2 for 18 h. The flask was evacuated and back-filled with nitrogen. The mixture was filtered through Celite; the filter pad was then washed with ethyl acetate. The filtrate (˜1.6 L EtOAc volume) was diluted with acetonitrile (0.3 L) and charged sequentially with L-N-Cbz-methionine (68 g, 0.24 mol), TBTU (77 g, 0.24 mol), and N,N-diisopropylethylamine (42 mL, 0.24 mol). The reaction was stirred at room temperature for 4 h, during which time it changed from a suspension to a clear solution. The reaction was quenched with the addition of sat. NH4Cl (0.75 L) and water (0.15 L); the mixture was diluted further with EtOAc (0.75 L). The phases were mixed and separated and the organic phase was washed with sat. Na2CO3 (2×0.9 L) and sat. NaCl (1×0.75 L). The solution was dried (Na2SO4), filtered, and concentrated in vacuo to give (1R,2S,5R)-tert-butyl 2-((S)-2-(benzyloxycarbonylamino)-4-(methylthio)butanamido)-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate as an oil, which was taken into the next step without further purification. LC/MS for primary peak: [M-Boc+H]+=406.3; [M+Na]+=528.3. 1H-NMR (400 MHz, d4-MeOH): δ 7.36 (m, 5H), 5.11 (s, 2H), 4.32 (m, 1H), 4.2 (m, 1H), 4.0 (m, 1H), 2.5-2.7 (m, 3H), 2.25 (m, 1H), 2.11 (s, 3H), 2.05 (m, 4H), 1.9 (m, 1H), 1.7 (m, 2H), 1.54 (s, 9H). Also present are EtOAc [1.26 (t), 2.03 (s), 4.12 (q)] and N,N,N,N-tetramethylurea [2.83 (s)].
Example 1, Step 2: A sample of (1R,2S,5R)-tert-butyl 2-((S)-2-(benzyloxycarbonylamino)-4-(methylthio)butanamido)-7-oxo-6-aza-bicyclo[3.2.1]octane-6-carboxylate (0.24 mol assumed; see previous procedure) was dissolved in iodomethane (1,250 g) and stirred for 48 h at room temperature. The reaction was concentrated in vacuo. The residue was dissolved in dichloromethane and concentrated in vacuo. This was repeated twice more. The resultant sludge was dissolved in dichloromethane (0.4 L) and poured into a rapidly stirring solution of MTBE (4.0 L). The resultant yellow solids were collected via suction filtration and dried under high vacuum to afford the sulfonium salt (179 g). This material was taken into the next step without further purification. LC/MS for primary peak: [M-Me2S+H]+=458.4; [M]+=520.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.09 (s, 2H), 4.33 (m, 1H), 4.28 (m, 1H), 3.98 (m, 1H), 3.3-3.45 (m, 2H), 2.97 (s, 3H), 2.94 (s, 3H), 2.78 (m, 1H), 2.0-2.3 (m, 4H), 1.7 (m, 2H), 1.52 (s, 9H). Also present are MTBE [1.18 (s), 3.2 (s)] and traces of N,N,N,N-tetramethylurea [2.81 (s)].
Example 1, Step 3: All of the sulfonium salt from the previous step (0.24 mol assumed) was dissolved in DMSO (2.0 L). The resultant solution was stirred under nitrogen at room temperature and charged with cesium carbonate (216 g) portionwise. The suspension was stirred at room temperature for 3 h and then filtered to remove the solids. The solution was divided into ˜0.22 L portions and worked up as follows: the reaction mixture (˜0.22 L) was diluted with ethyl acetate (1.5 L) and washed successively with water (3×0.5 L) and brine (1×0.3 L). The organic phase was dried (Na2SO4), filtered, and concentrated in vacuo. The desired (1R,2S,5R)-tert-butyl 2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-7-oxo-6-azabicyclo[3.2.1]octane-6-carboxylate (90.8 g, 83%) was obtained as a microcrystalline foam, free from tetramethyl urea impurity. LC/MS for primary peak: [M-Boc+H]+=358.4; [M+Na]+=480.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.12 (s, 2H), 4.35 (m, 2H), 4.2 (m, 1H), 3.6 (m, 1H), 3.3 (m, 1H), 2.64 (m, 1H), 2.28-2.42 (m, 2H), 2.15 (m, 1H), 1.7-2.0 (m, 5H), 1.55 (s, 9H). If desired, this material can be isolated as a solid by dissolving in MTBE (1 volume), adding to heptane (3.3 volumes), and collecting the resultant precipitate.
Example 1, Step 4: A stirring solution of (1R,2S,5R)-tert-butyl 2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-7-oxo-6-azabicyclo[3.2.1]octane-6-carboxylate (108 g, 0.236 mol) in THF (1 L) was charged with lithium hydroxide monohydrate (21.74 g, 0.519 mol). Water (0.3 L) was added slowly, such that the temperature did not exceed 20° C. The reaction was stirred at room temperature overnight and the volatiles were removed in vacuo. The pH was adjusted to ˜4 through the addition of 1N HCl (450 mL) and NaH2PO4. The resultant white precipitates were collected by filtration and washed with water (2×1 L). The solid was dissolved in dichloromethane (1.5 L) and water (˜1 L). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was dissolved in EtOAc (0.7 L) and the resultant solution was heated at reflux for 1 h. Solids separated after cooling to RT, and were collected via filtration. These solids were purified by recrystallization in isopropanol to afford the desired (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxylic acid as a white solid (104.5 g, 93% yield). LC/MS for primary peak: [M-tBu+H]+=420.2; [M-Boc+H]+=376.2; [M+H]+=476.2. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.35 (m, 2H), 3.71 (m, 1H), 3.45-3.6 (m, 2H), 2.99 (m, 1H), 2.41 (m, 1H), 2.15 (m, 1H), 2.0 (m, 2H), 1.6-1.9 (m, 4H), 1.46 (s, 9H).
Example 1, Step 5: A 3 L round bottom flask was charged with (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxylic acid (75.5 g, 0.158 mol), EDC.HCl (33.5 g, 0.175 mol), 1-hydroxybenzotriazole (23.6 g, 0.175 mol), and dichloromethane (1 L). The reaction was stirred at room temperature for 2 h, during which time it changed from a white suspension to a clear solution. Ammonia (gas) was bubbled into the solution until the pH was strongly basic (paper) and the reaction was stirred for 10 min; this ammonia addition was repeated and the reaction was stirred for an additional 10 min. Water was added. The organic phase was washed with sat. NaHCO3, NaH2PO4, and brine before being concentrated in vacuo. The residue was slurried with acetonitrile (0.5 L) and then concentrated in to give (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxamide as a white solid (75.9 g, ˜100%), which was used in the next step without further purification. LC/MS for primary peak: [M-Boc+H]+=375.3; [M+H]+=475.4; [M-tBu+H]+=419.3. 1H-NMR (400 MHz, d4-MeOH): δ 7.35 (m, 5H), 5.11 (s, 2H), 4.25 (m, 2H), 3.70 (m, 1H), 3.6 (m, 1H), 3.45 (m, 1H), 2.91 (m, 1H), 2.38 (m, 1H), 2.12 (m, 1H), 1.9-2.05 (m, 2H), 1.65-1.9 (m, 4H), 1.46 (s, 9H).
Example 1, Step 6: The reaction was run in three equal portions and combined for aqueous workup. A 5 L, 3-necked round bottom flask was charged with (1R,2S,5R)-2-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)-5-(tert-butoxycarbonylamino)cyclohexanecarboxamide (25.3 g, 53 mmol), acetonitrile (1.9 L), and 2.6 L of water/ice. The mixture was stirred and cooled to 0° C. Iodobenzene diacetate (25.77 g, 80 mmol) was added and the reaction was stirred for 2 h; another 0.5 eq of iodobenzene diacetate was added. The reaction was stirred for 9 h (reaction temp<10° C.). The mixture was charged with 8 eq N,N-diisopropylethylamine and 2 eq acetic anhydride. Over the next thirty minutes, 4 eq N,N-diisopropylethylamine and 2 eq acetic anhydride were added every ten minutes, until the reaction had proceeded to completion (HPLC). The acetonitrile was removed in vacuo; some solid separated from the residue, and this was collected by filtration. The remaining residue was extracted with dichloromethane (3 L, then 1 L). The organic phase was washed sequentially with water, sat. NaHCO3, and brine. The collected solids were added to the organic phase, along with activated carbon (15 g). The mixture was stirred for 30 minutes at 40° C. before being filtered and concentrated in vacuo. The residue was dissolved in EtOAc (1 L), and the resultant solution was stirred at 75° C. for 1 h before being allowed to cool to room temperature. A solid separated and was collected by filtration. This solid was purified further by recrystallization: it was first dissolved in 0.5 L CH2Cl2, then concentrated in vacuo, then re-crystallized from 1 L EtOAc; this was repeated three times. The solids obtained from the mother liquors of the above were recrystallized three times using the same method. The combined solids were recrystallized twice more from acetonitrile (0.7 L) to provide 66 g (84%) of tert-butyl (1R,3R,4S)-3-acetamido-4-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)cyclohexylcarbamate (purity>99.5% by HPLC). LC/MS for primary peak: [M+H]+=489.4; [M-tBu+H]+=433.3. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.11 (s, 2H), 4.35 (m, 1H), 4.15 (m, 1H), 4.04 (m, 1H), 3.8 (m, 1H), 3.6 (m, 2H), 2.44 (m, 1H), 2.12 (m, 1H), 1.87-2.05 (m, 4H), 1.87 (s, 3H), 1.55-1.7 (m, 2H), 1.46 (s, 9H). The stereochemical fidelity of the Hofmann rearrangement was confirmed through X-ray crystal structure analysis of this compound, as shown in FIG. 1.
Example 1, Step 7: A stirring solution of tert-butyl (1R,3R,4S)-3-acetamido-4-((S)-3-(benzyloxycarbonylamino)-2-oxopyrrolidin-1-yl)cyclohexylcarbamate (66 g, 0.135 mol) in dichloromethane (216 mL) was charged with trifluoroacetic acid (216 mL). The reaction was stirred for 2 h at room temperature and concentrated in vacuo. The residue was dissolved in methanol and the resultant solution was concentrated in vacuo; this was repeated once. Benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate was obtained as an oil and used directly in Step 8 below. LC/MS found [M+H]+=389.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.12 (s, 2H), 4.41 (br. s, 1H), 4.15 (m, 1H), 4.00 (t, J=9.3 Hz, 1H), 3.81 (t, J=9.1 Hz, 1H), 3.65 (q, J=8.4 Hz, 1H), 3.3-3.4 (m, 1H), 2.45 (m, 1H), 1.95-2.24 (m, 5H), 2.00 (s, 3H), 1.6-1.8 (m, 2H).
Example 1, Step 8: A stirring solution of benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-aminocyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (˜0.135 mol) in methanol (675 mL) was charged sequentially with acetone (37.8 g, 4 eq), sodium acetate (33.2 g, 3 eq), and sodium cyanoborohydride (16.9 g, 2 eq). The mixture was stirred at room temperature for 6 h and filtered. The filtrate was dissolved in dichloromethane (1 L); this solution was washed with 1N NaOH (1 L). The solids collected in the filtration were dissolved in 1N NaOH (1 L) at 0° C. and then extracted with dichloromethane (1 L). The organic extracts were combined and extracted with aqueous HCl (200 mL 1N HCl+800 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then 1N NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate as an oil. LC/MS found [M+H]+=431.45. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.12 (s, 2H), 4.31 (m, 1H), 4.24 (t, J=9.4 Hz, 1H), 4.11 (m, 1H), 3.61 (t, J=9.1 Hz, 1H), 3.52 (q, J=8.6 Hz, 1H), 3.04 (br. s, 1H), 2.96 (sep, J=6.3 Hz, 1H), 2.40 (m, 1H), 2.15 (m, 1H), 1.92 (s, 3H), 1.7-1.9 (m, 5H), 1.65 (m, 1H), 1.12 (app. dd, J=6.3, 1.1 Hz, 6H).
Example 1, Step 9 (See Alternative Step 9, below): A stirring solution of benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropylamino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (˜115 mmol) in dichloromethane (600 mL) was cooled to 0° C. and charged sequentially with formaldehyde (18.6 g, 37 wt % solution), triethylamine (23 mL), and sodium triacetoxyborohydride (28.7 g). The mixture was stirred at room temperature for 30 minutes and diluted with dichloromethane (up to 1.2 L). This solution was washed thrice with 500 mL sat. NaHCO3+NaOH (sat. NaHCO3, pH to 11 w/1N NaOH). The organic layer was extracted with aq. HCl (200 mL 1N HCl+600 mL water). The aqueous phase was basified with sat. NaHCO3 (500 mL) and then 1N NaOH (100 mL) until pH 11. The aqueous phase was extracted with dichloromethane (1.2 L). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated in vacuo to give benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate as an oil, which was used directly in Step 10 below. LC/MS found [M+H]+=445.4. 1H-NMR (400 MHz, d4-MeOH): δ 7.3-7.4 (m, 5H), 5.12 (s, 2H), 4.33 (br s, 1H), 4.25 (t, J=9.2 Hz, 1H), 4.11 (br s, 1H), 3.5-3.6 (m, 2H), 2.77 (v br s, 2H), 2.41 (m, 1H), 2.26 (s, 3H), 2.0-2.1 (m, 2H), 1.92 (s, 3H), 1.7-1.9 (m, 5H), 1.10 (app. dd, J=17, 6.4 Hz, 6H).
Example 1, Step 10: To a solution of benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropyl(methyl)amino)-cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate (0.115 mol) in methanol (600 mL) was added 10% Pd/C (6 g of 50% wet catalyst). The flask was evacuated and back-filled with hydrogen. The mixture was stirred under 1 atm H2 for 2 h and the catalyst was removed by filtration through Celite. The filtrate was concentrated in vacuo to provide N-((1R,2S,5R)-2-((S)-3-amino-2-oxopyrrolidin-1-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide as an oil, which was taken on to the next step without further purification. LC/MS found [M+H]+=311.47. 1H-NMR (400 MHz, d4-MeOH): δ 4.39 (br s, 1H), 4.00 (m, 1H), 3.3-3.5 (m, 4H), 2.73 (m, 1H), 2.38 (m, 1H), 2.25 (s, 3H), 2.0-2.2 (m, 3H), 1.94 (s, 3H), 1.6-1.75 (m, 4H), 1.07 (app. dd, J=21, 6.4 Hz, 6H).
Example 1, Step 11: To a solution of N-((1R,2S,5R)-2-((S)-3-amino-2-oxopyrrolidin-1-yl)-5-(isopropyl(methyl)amino)cyclohexyl)acetamide (˜35 g, 0.115 mol) in isopropanol (600 mL) was added 4-chloro-6-(trifluoromethyl)quinazoline (32 g, 0.138 mol, 1.2 eq, see: P. H. Carter et al., PCT application WO 2005/021500). The mixture was stirred at room temperature overnight before being charged with triethylamine (46 g, 0.46 mol, 4 eq). The mixture was stirred at 60° C. for 10 h. The solvent was removed under reduced pressure to give an oil. Azeotropic distillation with isopropanol was performed twice. The residue was dissolved in dichloromethane (600 mL) and extracted with water (250 mL, containing 4 eq acetic acid). Dichloromethane (600 mL) was added to the combined aqueous washes, and the mixture was cooled to 0° C. Aqueous NaOH (50% by weight) was added with stirring until the pH reached 11. The water layer was extracted with dichloromethane twice (2×600 mL). The combined organic extracts were dried (Na2SO4), filtered, and concentrated in vacuo to give the amorphous free base of the title compound (99% purity by HPLC). LC/MS found [M+H]+=507.3. 1H-NMR (400 MHz, d4-MeOH): δ 8.82 (s, 1H), 8.59 (s, 1H), 8.05 (dd, J=8.8, 1.8 Hz, 1H), 7.9 (d, J=8.7 Hz, 1H), 5.28 (t, J=8.6 Hz, 1H), 4.58 (br s, 1H), 4.06 (m, 1H), 3.52-3.68 (m, 2H), 3.43 (m, 1H), 2.76 (br s, 1H), 2.55 (m, 1H), 2.28 (s, 3H), 2.1-2.3 (m, 3H), 2.0 (s, 3H), 2.0 (m, 1H), 1.65-1.8 (m, 3H), 1.09 (app. dd, J=24, 6.4 Hz, 6 H).
Example 1 Alternative Step 9
![Figure US07671062-20100302-C00061]()
Example 1, Alternative step 9ai: To a hydrogenator were charged ethyl (7R,8S)-8-((S)-1-phenyl-ethylamino)-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1A (1417 g, 2.8 moles, c.f.: WO2004098516, prepared analogous to U.S. Pat. No. 6,835,841), ethanol (200 proof, 11.4 L), and 10% Pd/C catalyst (50% wet, 284 g). The mixture was inerted with nitrogen, then pressurized with hydrogen gas (45 psig) and agitated vigorously at approx. 40° C. until starting material was consumed (HPLC). The suspension was cooled, purged with nitrogen gas and the catalyst was removed by filtration while inerted. The spent catalyst was washed with ethanol (4.3 L). The filtrate and washings were combined and concentrated under vacuum to a volume of 2-3 L while maintaining the batch between 40°-60° C. Isopropyl acetate (5 L) was charged and the mixture was concentrated to a volume of ˜2 L until most ethanol was removed (<0.5%) and residual moisture content was <1,000 ppm. Batch volume was adjusted to ˜7.5 L by the addition of isopropyl acetate. The mixture was heated to 80° C. until clear, then cooled 65°-70° C. Seed crystals of 1 (5 g) were added and the batch was cooled to 50° C. over 2 hours, then further cooled to 20° C. over 4 hours and held for ˜10 hours. The resulting slurry was filtered and the cake was washed with isopropyl acetate (2 L). The product was dried under vaccum at ˜35° C. until volatiles were reduced below ˜1% (LOD). Ethyl (7R,8S)-8-amino-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 was obtained as a white, crystalline solid (936 g, 83% yield; HPLC purity: 99.8%). 1H-NMR: (300 MHz, CDCl3) 8.14-7.89 (brs, 3H), 7.75 (d, J 9.0 Hz, 2H), 7.15 (d, J 8.0 Hz, 2H), 4.22-4.04 (m, 2H), 4.01-3.77 (m, 4H), 3.55-3.43 (m, 1H,), 3.20-3.13 (m, 1H), 2.40-2.27 (m, 4H), 2.21-1.94 (m, 2H), 1.81-1.51 (m, 3H), 1.23 (t, J 7.0 Hz, 3H); HPLC: Waters Xterra MS C18 4.6 mm×150 mm i.d., 3.5 μm particle size, 0.05% NH4OH (5% ACN, 95% H2O, solvent A), to 0.05% NH4OH (95% ACN, 5% H2O, solvent B), 5% B to 20% B in 10 minutes, changed to 95% B in 25 minutes, and then changed to 5% B in 1 minute; 11.1 minutes (aminoester 1).
![Figure US07671062-20100302-C00062]()
Example 1, Alternative Step 9aii: Aminoester 1 (63 g, 0.16M, 1 eq.; the product of reductive deprotection of a known compound—(See e.g. R. J. Cherney, WO 2004/098516 and G. V. Delucca & S. S. Ko, WO 2004/110993) was placed in a round bottom flask and MeCN (500 mL) was added. EDAC (33.1 g, 0.17M, 1.1 eq), HOBt.H2O (21.2 g, 0.16M, 1.0 eq) and N-Cbz-L-methionine (46.7 g, 0.17M, 1.05 eq) were then added followed by TEA (48.0 mL, 0.35M, 2.2 eq). An exotherm to 38° C. was observed. The reaction mass was left to stir at RT. After 30 mins, HPLC indicated complete conversion. The reaction mass was diluted with EtOAc (2.5 L) and washed with KHCO3 (4×500 mL, 20 wt % aq. solution) and brine (500 mL). The organic phase was separated, dried over MgSO4 and concentrated. The residue was dissolved in TBME and reconcentrated to give ethyl (7R,8S)-8-{(2S)-2-benzyloxycarbonylamino-4-methylsulfanyl-butyr-yl-amino}-1,4-dioxa-spiro[4.5]decane-7-carboxylate 2 as a sticky semi-solid (76.2 g, 98% yield, 93 AP purity). 1H-NMR: (300 MHz, CDCl3) δ 7.36-7.30 (m, 5H), 7.03 (d, J 9.0 Hz, 1H), 5.66 (d, J 8.0 Hz, 1H), 5.10 (s, 2H), 4.35-4.25 (m, 2H), 4.19-4.04 (m, 2H,), 3.98-3.86 (m, 4H), 2.87-2.80 (m, 1H), 2.55-2.45 (m, 2H), 2.18 (dd, J 14.0 Hz, 7.0 Hz, 1H), 2.08 (s, 3H), 2.05-1.67 (m, 6H), 1.26 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 10.01 min (Compound 2, 93.1 AP). HRMS: m/z 495.2166 [Calc: C24H35N2O7S 495.2165].
![Figure US07671062-20100302-C00063]()
Example 1, Alternative Step 9b: Methionine amide 2 (75.0 g, 0.15M) was dissolved in MeI (225 mL, 3 mL/g)—some off gassing was noted but no exotherm. The reaction mass was left to stir in the dark for 16.5 h. After this time a thick light yellow precipitate had formed. The flask was then evacuated to 200 mmHg and some of the MeI removed. The remaining material was slurried in TBMF (500 mL), after a 30 min stir-out the slurry was filtered, the cake washed with TBMF (500 mL). NMR analysis of this material indicated a small amount of MeI remaining. The cake was re-slurried in TBMF (500 mL), filtered, washed with TBMF (500 mL) and dried under vacuum to give [(3S)-3-benzyloxycarbonylamino-3-{(7R,8S)-7-ethoxycarbonyl-1,4-di-oxa-spiro[4.5]dec-8-ylcarbamoyl}-propyl]-dimethylsulfonium iodide 3 as a free flowing off-white solid (93.5 g, 97%, 99 area % purity). 1H-NMR: (300 MHz, CDCl3) δ 7.75 (d, J 9.0 Hz, 1H), 7.38-7.27 (m, 5H), 6.40 (d, J 7.0 Hz, 1H), 5.10 (s, 2H), 4.76-4.65 (m, 1H), 4.48-4.39 (m, 1H), 4.14-3.85 (m, 6H), 3.84-7.73 (m, 1H), 3.68-3.55 (m, 1H), 3.21 (s, 3H), 3.12 (s, 3H), 2.90-2.83 (s, 1H), 2.52-1.55 (m, 8H), 1.24 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 2.45 min (I−), 8.14 min (Compound 3, 43.6 AP, I−54.6 AP). HRMS: m/z 509.2341 [Calc: C25H37N2O7S 509.2321].
![Figure US07671062-20100302-C00064]()
Example 1, Alternative Step 9c: Cs2CO3 (61.5 g, 0.19M, 1.5 eq) was placed in an round bottom flask and anhydrous DMSO (2.4 L) was added. Sulfonium salt 3 (80.0 g, 0.13M, 1.0 eq) was then added portionwise. Once the addition was complete the reaction mass was left to stir in the dark for 20 h. The reaction mass was then split in half and each half worked up separately: the reaction mass was diluted with EtOAc (2.0 L) and washed with brine (2 L), the organic phase was washed with brine (500 mL). The combined aq. layers were then washed EtOAc (500 mL). The combined organic phases were then washed with brine (3×750 mL). The second half of the reaction mass was treated in an identical manner and the combined organics dried over MgSO4 and concentrated to give ethyl (7R,8S)-8-{(3S)-3-Benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl}-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4 as a light colored oil (56.5 g, 0.13M, ˜100 area-% purity) pure by NMR analysis. 1H-NMR: (300 MHz, CDCl3) δ 7.38-7.30 (m, 5H), 5.37 (br d, J 4.0 Hz, 1H), 5.11 (s, 2H), 4.27-4.18 (m, 1H), 4.17-3.82 (m, 8H), 3.32 (td, J 10.0Hz, 60.0 Hz, 1H), 3.23 (q, J 5.0 Hz, 1H), 2.63-2.57 (m, 1H), 2.42-2.25 (m, 2H), 1.94-1.68 (m, 5H), 1.25 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 8.99 min (Compound 5, produced on column, 4.2 AP), 9.48 (Compound 4, 74.3 AP). HRMS: m/z 447.2127 [Calc: C23H31N2O7 447.2131].
![Figure US07671062-20100302-C00065]()
Example 1, Alternative Step 9d: Pyrrolidinone 4 (50.0 g, 0.11M) was dissolved in acetone (500 mL) and 1N HCl (500 mL) was added. The reaction mass was then heated to 65° C. After 20 mins HPLC indicated complete reaction. The reaction mass was allowed to cool to RT and the acetone was removed on a rotary evaporator. During this distillation the product precipitated from solution as a white solid. This was isolated by filtration and the cake washed with water. The cake was then dried azeotropically with toluene (3×300 mL) to give ethyl (1R,2S)-2-((3S)-3-Benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-oxo-cyclohexanecarboxylate 5 as a white solid (39.8 g, 88%, 97 area-% purity). 1H-NMR: (300 MHz, CDCl3) δ 7.37-7.32 (m, 5H), 6.65 (br d, J 4.0 Hz, 1H), 5.12 (s, 2H), 4.54-4.47 (m, 1H), 4.34-4.26 (m, 1H), 4.18 (dq, J 11.0 Hz, 7.0 Hz, 1H), 4.09 (dq, J 11.0 Hz, 7.0 Hz, 1H), 3.36-3.20 (m, 3H), 2.70-2.35 (m, 6H), 2.05-1.96 (m, 1H), 1.81 (quin., J 11.0 Hz, 1H), 1.24 (t, J 7.0 Hz, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 8.95 min (Compound 5). HRMS: m/z 403.1864 [Calc: C21H27N2O6403.1869].
![Figure US07671062-20100302-C00066]()
Example 1, Alternative Step 9e: Cyclohexanone 5 (22.5 g, 0.06M, 1 eq), DMSO (30 mL) and Ti(O-iPr)4 (33.7 mL, 0.11M, 2.04 eq) were placed in a round bottom flask. N-isopropyl-N-methylamine (11.6 mL, 0.11M, 2.0 eq) was then added in one portion. The mixture was left to stir for 30 mins at room temperature before being cooled to <3° C. in ice/water. MeOH (30 mL) was then added followed by the portionwise addition of NaBH4 (4.33 g, 0.11M, 2.04 eq)—temperature kept <8° C. 30 mins after the addition was completed the reaction mass was diluted with methylene chloride (300 mL) and then NaOH (1N, 40 mL). The resulting slurry was filtered through Celite, and the cake washed with methylene chloride (100 mL). The resulting liquor was concentrated under reduced pressure and the residue dissolved in EtOAc (500 mL). This solution was extracted with 1N HCl (2×400 mL), the combined aqueous layers were then basified with Na2CO3. Extraction with EtOAc (4×250 mL) provided a clear and colorless organic phase which was dried over Na2SO4 and concentrated to give a white powder (24.6 g, 96%, 7:1 d.r.). This material was then slurried overnight in hexane (670 mL). The solid was isolated by filtration and dried under reduced pressure to give ethyl (1R,2S,5R)-2-((3S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylate 6 as a while solid (20.9 g, 81%, 24:1 d.r.). 1H-NMR: (300 MHz, CDCl3) δ 7.37-7.28 (m, 5H), 5.55 (d, J 4.5, 1H), 5.10 (s, 2H), 4.42 (q, J 4.5, 1H), 4.23-4.12 (m, 1H), 4.08 (dq, J 10.5, 7.0, 1H), 4.02 (dq, J 10.5, 7.0, 1H), 3.84 (t, J 9.0, 1H), 3.46-3.36 (m, 1H), 3.04 (septet, J 6.5, 1H), 2.86-2.80 (m, 1H), 2.63-2.48 (m, 2H), 2.17 (s, 3H, Me), 2.10-1.63 (m, 7H), 1.22 (t, J 7.0, 3H), 1.00 (d, J 6.5, 3H), 0.97 (d, J 6.5, 3H). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.01M NH4OAc (MeOH:water 20:80) to 0.01M NH4OAc (MeOH:water:MeCN 20:5:75) 10 to 100% 15 min gradient. 8.23 (Compound 6), 8.88 (5-epi-Compound 6). HRMS: 460.2798 [Calc: C25H38N3O5 460.2811].
![Figure US07671062-20100302-C00067]()
Example 1, Alternative Step 9f: The aminoester 6 (9.76 g, 2.12 mmol) was dissolved in 2N HCl (80 mL), then heated to ˜55° C. under inert atmosphere. The reaction was stirred for 20 h, then cooled to room temperature. The reaction solution was washed twice with toluene (25 mL portions), neutralized to pH 6-7 by the addition of KOH pellets, then extracted eight times with methylene chloride (100 mL portions). The combined extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure to 50 mL total volume. The concentrated solution was then slowly added to methyl tert-butyl ether (300 mL) over 15 min in an addition funnel with vigorous stirring. The resulting white slurry was stirred at ambient temperature for Ih, then cooled to 0° C. and stirred for 1 h. The product was filtered, and washed twice with methyl tert-butyl ether (25 mL portions). Water from the wet cake was removed by azeotropic distillation with acetonitrile (300 mL). The product was dried under reduced pressure to provide (1R,2S,5R)-2-((3S)-3-Benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylic acid 7, (7.69 g, 84% yield) as a white foam. 1H-NMR: (400 MHz, 50° C., CDCl3) δ 7.44-7.32 (m, 5H), 6.10 (broad s, 1H), 5.19 (app s, 2H), 4.42 (dd, J=15.6, 7.8 Hz, 1H), 4.29-4.23 (m, 1H), 3.68-3.60 (m, 2H), 3.33-3.27 (m, 2H), 3.20 (broad s, 1H), 2.99 (broad s, 1H), 2.51 (s, 3H), 2.49-2.45 (m, 3H), 2.33-2.31 (m, 1H), 2.00 (ddd, J=9.0, 8.6, 3.9 1H), 1.95-1.78 (m, 2H), 1.36-1.21 (m, 6H). LCMS: m/z 432.20 [Calc: C23H34N3O5 432.25].
![Figure US07671062-20100302-C00068]()
Example 1, Alternative Step 9g: Amino acid 7 (6.3 g, 14.7 mmol, 1.0 eq) was dissolved in THF (80 mL) under N2 and NaH (584 mg, 14.7 mmol, 1.0 eq, 60 wt % dispersion in mineral oil) was added portionwise. When the addition was complete, and the evolution of gas had ceased, the reaction mass was concentrated under reduced pressure and the resulting solid azeotroped with toluene (50 mL) to give a white solid (KF 0.59 wt %). This solid was slurried in toluene (100 mL) under N2and heated to 90° C. DPPA (3.32 mL, 15.3 mmol, 1.05 eq) was added dropwise over ˜2 min. After ˜5 min all the solids had dissolved, after 10 mins precipitation of a white solid was observed. After 30 mins HPLC analysis indicated complete reaction. The reaction mass was allowed to cool to RT before being filtered, the cake was washed with toluene. The liquors where then slowly added into AcOH/Ac2O (80/20, 168 mL) solution at 90° C. After 45 mins HPLC still indicated some isocyanate. At 1.15 h, the reaction mass was cooled to RT and diluted with toluene (100 mL) and water (100 mL). The organic layer was removed and the toluene washed with 1N HCl (100 mL). The combined aq. phases were then basified with K2CO3(s) and brought to pH 12 with NaOH (10N), keeping the temperature below 20° C. The aq layer was then extracted with methylene chloride (4×150 mL), the combined organic layers dried over K2CO3 and concentrated to give benzyl (S)-1-((1S,2R,4R)-2-acetamido-4-(isopropyl(methyl)amino)cyclohexyl)-2-oxopyrrolidin-3-ylcarbamate 8 as a white foam (4.5 g, 70%, 94AP purity). The 1H-NMR was identical to material obtained from the route described above (Example 1, Step 9). HPLC: YMC-Pack Pro C18 5 μm 4.6×150 mm, 0.05% TFA (20% MeOH, 80% H2O), to 0.05% TFA (20% MeOH, 80% MeCN), 0-100% 10 min gradient. 7.20 min (Compound 8), 7.85 min (urea dimer). HRMS: 445.2809 [Calc: C24H37N4O4445.2815].
Alternative Preparation of Example 1
![Figure US07671062-20100302-C00069]()
Example 1, Alternative Preparation, Step 1: Ethyl (7R,8S)-8-amino-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4-toluenesulfonate salt 1 (450.1 g), was combined with 1-ethyl-3-(3-dimethyl-amino-propyl)carbo-diimide hydrochloride (236.3 g), 1-hydroxy benzotriazole hydrate (171.9 g), N-carbobenzyloxy-L-methionine (333.4 g) and acetonitrile (3.1 L). To the stirred mixture was added triethylamine (249.5 g) below 30° C. Upon reaction completion (HPLC), the mixture was diluted with ethyl acetate (8.2 L) and washed with aqueous 25% potassium bicarbonate solution (2×4.5 L) followed by water (4.5 L). The organic phase was separated and concentrated under reduced pressure to obtain a solution of ethyl (7R,8S)-8-((S)-2-benzyloxycarbonylamino-4-methylsulfanyl-butyrylamino)-1,4-dioxa-spiro[4.5]decane-7-carboxylate 2 (1.4 L). Methyl iodide (2.39 kg) was added, the vessel was shielded from light and the mixture was held under slow agitation for approx. 24 h. To the thick yellow precipitate was added methyl tert-butyl ether (2.7 L) and the mixture was held for approx. 1 h. The product was isolated by filtration and the cake was washed with methyl tert-butyl ether (2×1.4 L), then dried under vacuum, yielding [(S)-3-benzyloxy-carbonylamino-3-((7R,8S)-7-ethoxycarbonyl-1,4-dioxa-spiro[4.5]dec-8-ylcarbamoyl)-propyl]-dimethylsulfonium iodide 3 (671.4 g, ˜94% yield) as an off-white solid (HPLC purity 99.9%).
![Figure US07671062-20100302-C00070]()
Example 1, Alternative Preparation, Step 2: Sulfonium salt 3 (619.4 g), and cesium carbonate (416.8 g) and anhydrous dimethyl sulfoxide (6.2 L) were combined in a reactor equipped with a scrubber to neutralize volatile sulfides. Vigorous agitation was maintained until complete conversion was obtained (HPLC). Ethyl acetate (12.4 L) was added, followed by 20% brine (3 L). The organic phase was separated, washed twice with brine (2×3 L) and evaporated to obtain a solution of ethyl (7R,8S)-8-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-1,4-dioxa-spiro[4.5]decane-7-carboxylate 4 in ethyl acetate (˜0.8 L). Acetone (2.55 L) was added, followed by aqueous 0.5 M hydrochloric acid solution (2.3 L). With good mixing, the solution was heated to 50 to 60° C. until conversion of 4 to ethyl (1R,2S)-2-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-oxo-cyclohexanecarboxylate 5 was complete (HPLC). The mixture was concentrated under reduced pressure while below 40° C., cooled to ˜30° C., and water (4.1 L) was added. The resulting slurry was cooled to 5 to 10° C. and agitated for ˜1 hour. The product was filtered and the cake was washed with water (2×2.5 L). Upon deliquoring, the cake was dried to a constant weight below 40° C. in a vacuum oven. Cyclohexanone 5 (272 g, 70% yield) was obtained (HPLC purity 98.7%).
![Figure US07671062-20100302-C00071]()
Example 1, Alternative Preparation, Step 3: Cyclohexanone 5 (206 g) was dissolved in dichloromethane (1.1 L) and charged to a hydrogenator. Titanium tetraisopropoxide (218.2 g) and N-isopropyl N-methylamine (63.64 g) were added and the mixture was stirred at ambient temperature (23 to 25° C.) for at least 5 h. Platinum catalyst (5% Pt/S/C, 15 g, approx. 7.5% relative to 5) was added and hydrogenation was performed at ˜30 psig for at least 6 h, yielding a mixture of ethyl (1R,2S,5R)-2-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexanecarboxylate 6 and its 5-epi-isomer (˜7%). The catalyst was removed by filtration and the filtrate was concentrated under reduced pressure to approx. ˜600 mL. Wet ethyl acetate (˜3% water, 2.0 L) was added with vigorous agitation over a period of at least 1.5 h. Stirring was continued for at least an additional 6 h. The slurry was filtered. Filter cake was washed with ethyl acetate (1.0 L) and discarded. The combined filtrate and washings were concentrated to ˜400 mL. Toluene (2.0 L) was added and the solution was washed with 2M aqueous hydrochloric acid (2×400 mL). The aqueous layer was warmed to 50° to 60° C. for approx. 20 h or hydrolysis of 6 was deemed complete (HPLC). Aqueous sodium hydroxide solution was added to adjust to pH ˜10, and mixture was extracted with toluene (3×600 mL). The organic phase was discarded and pH was readjusted to ˜6 by addition of aqueous hydrochloric acid. The aqueous phase was concentrated to ˜600 mL under reduced pressure and extracted with methylene chloride (at least 3×2.0 L). The combined methylene chloride layers were evaporated under reduced pressure and continuously replaced with THF to obtain a solution of (1R,2S,5R)-2-((S)-3-benzyloxycarbonylamino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexane carboxylic acid 7 (˜148 g) in THF (˜4 L). Seed crystals of 8 were added, followed by 25% solution of sodium methoxide in methanol (81.24 g) below 25° C. The slurry was held for at least additional 16 h with agitation. The product was isolated by filtration and the cake was washed with THF (4×200 mL) and dried to a constant weight in vacuo below 30° C. Dry (1R,2S,5R)-2-((S)-3-benzyloxycarbonyl-amino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexane-carboxylate sodium salt 8 was obtained (139 g, ˜60% yield from 5).
![Figure US07671062-20100302-C00072]()
Example 1, Alternative Preparation, Step 4: Aminoester sodium salt 8 (100 g), diphenyl phosphate (3.86 g), tert-BuOH (1275 mL) and toluene (225 mL) were combined and heated to reflux under reduced pressure. Approx. 500 mL of distillate were collected and discarded while being continuously replaced with a solution of toluene in tert-BuOH. Vacuum was removed and distillate was switched to percolate through a column filled with molecular sieves and allowed to return to the vessel. After drying was complete, DPPA (52.4 mL; dissolved in 60 mL toluene) was added slowly to the slurry at 80° C. Upon complete conversion (HPLC), tert-BuOH was removed by vacuum distillation and continuously replaced with toluene. The mixture was cooled to room temperature and washed twice with 10% aqueous K2HPO4 (1×800 mL, 1×400 mL) and water (400 mL). The organic phase was heated and concentrated in vacuo to approx. 270 mL. Vacuum was removed and heptane (1.1 L) was added slowly at approx. 80° C., followed by seeds of 9 (˜1 g). The slurry was slowly cooled to room temperature and benzyl {(S)-1-[(1S,2R,4R)-2-tert-butoxycarbonylamino-4-(isopropyl-methyl-amino)-cyclo-hexyl]-2-oxo-pyrrolidin-3-yl}-carbamate 9 was isolated by filtration as a white solid (86.76 g, 78% yield).
![Figure US07671062-20100302-C00073]()
Example 1, Alternative Preparation, Step 5: The tert-Butyl carbamate 9 (50 g) was dissolved in Toluene (500 mL) and i-PrOH (150 mL). The resulting solution was then heated to 60° C. Methanesulfonic acid (19.6 mL) was added below 65° C. Upon reaction completion (HPLC), the mixture was cooled to RT and triethylamine (69.4 mL) added slowly below 25° C. Acetic anhydride was then added below 25° C. After 1 h acetic acid (250 mL) was added below 25° C. The toluene phase was discarded and 2-methyl-THF (500 mL) was added to the aqueous phase. The mixture was stirred vigorously and basified with NaOH (25% aqueous solution) to pH 12. The aqueous phase was discarded and the organic layer was washed with brine (250 mL). The organic layer was concentrated under reduced pressure and continuously replaced with i-PrOH. The solution was cooled and filtered to provide benzyl {(S)-1-[(1S,2R,4R)-2-acetylamino-4-(isopropyl-methyl-amino)-cyclohexyl]-2-oxo-pyrrolidin-3-yl}-carbamate 10 in i-PrOH solution which was used directly in the hydrogenation.
Example 1, Alternative Preparation, Step 6: To a solution containing acetamide 10 (˜61 g) in i-PrOH (˜625 mL) was added 10% Pd/C wet catalyst (2.5 g) and the suspension was hydrogenated at 30 psig and approx. 25° C. for at least 2 h. Upon completion (HPLC), the catalyst was removed by filtration and the filtrate was concentrated to approx. 550 mL. Water (8.8 mL) was added, followed by 5.6 N hydrochloric acid in i-PrOH solution (69.5 mL). The resulting slurry was held at room temperature overnight. The product was isolated by filtration and the cake was rinsed with i-PrOH (2×100 mL) and dried in vacuo to constant weight at ˜50° C. to give N-[(1R,2S,5R)-2-((S)-3-amino-2-oxo-pyrrolidin-1-yl)-5-(isopropyl-methyl-amino)-cyclohexyl]-acetamide 11 (55.6 g, 97% yield) as its hydrochloric acid salt (73.6% free base assay, HPLC).
![Figure US07671062-20100302-C00074]()
Example 1, Alternative Preparation, Step 7: To 6-trifluoromethyl-quinazolin-4-ol 12 (20.1 g) in MeCN (400 mL) was added 5.5 M solution of sodium methoxide in methanol (17.0 mL). The resulting suspension was distilled under reduced pressure and continuously replaced by MeCN to remove methanol. To the slurry was added DMF (1.4 g), followed by oxalyl chloride (13.0 mL) below 50° C. Upon reaction completion (HPLC), excess reagent was removed under reduced pressure to give ˜400 mL of slurry. The mixture was cooled to room temperature and washed with 10% aqueous K2HPO4 (1×1.0 L, 1×0.5 L) to afford 4-chloro-6-trifluoromethyl-quinazoline 13 (˜21.2 g) in approx. 450 mL of wet MeCN solution, which was used directly in the subsequent coupling reaction (HPLC purity 99.8%).
Example 1, Alternative Preparation, Step 8: To a mixture of acetamide 11 (28.5 g, HCl salt, 73.6% free base assay), acetonitrile (100 mL), N,N,-di-isopropyl-N-ethylamine (61 mL) at room temperature was added a solution of 13 (˜21.2 g) in MeCN (˜450 mL). The homogeneous mixture was held overnight. Upon reaction completion (HPLC), the mixture was concentrated in vacuo to approx. 125 mL. A 9.5% aqueous solution of acetic acid (240 mL) was added and the aqueous phase was extracted with methylene chloride. The aqueous phase was separated and methyl tert-butyl ether (450 mL) was added, followed by 2N aqueous lithium hydroxide solution to adjust to pH>11.5. The organic layer was separated, washed with water and filtered. Approx. half of the ether phase was diluted with methyl tert-butyl ether (˜250 mL) and concentrated in vacuo. Heptane (45 mL) was added slowly below 60° C., followed by seed crystals of Example 1 (0.4 g). Additional heptane (125 mL) was added and the mixture was slowly cooled to room temperature and the resulting slurry was held overnight. The product was isolated by filtration, the cake was washed with heptane and dried in vacuo to constant weight to give N-((1R,2S,5R)-5-(isopropylamino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)-quin-azolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide 14 (15.0 g, 85% yield).
Crystallization Procedures for Example 1Example 1, Production of bis-BSA salt and purification: The entirety of the amorphous free base from Example 1, Step 11 was dissolved in methanol (600 mL). The resultant solution was heated at 60° C. and charged with benzenesulfonic acid (2.5 eq). The mixture was cooled to room temperature and the resultant white solid was collected by filtration to yield the bis-benzene sulfonic acid salt of the title compound (95 g, 86%). This material was >99% pure by HPLC. This material was further purified by re-crystallization from 80/20 EtOH/H2O, which provided the salt free from any residual methanol. HPLC purity=99.8%. 1H NMR (500 MHz, D2O) δ ppm 8.75 (1H, s), 8.66 (1H, s), 8.25 (1H, d, J=8.80 Hz), 7.90 (1H, d, J=8.80 Hz), 7.75 (4H, d, J=8.25 Hz), 7.43-7.57 (6H, m), 5.42 (1H, t), 4.33-4.44 (1H, m), 4.09-4.19 (1H, m), 3.83-3.91 (1H, m), 3.74-3.83 (2H, m), 3.61 (1H, t, J=11.55 Hz), 2.75 (3H, d, J=6.60 Hz), 2.61-2.70 (1H, m), 2.31-2.44 (1H, m), 2.20-2.27 (1H, m), 2.17 (2H, d, J=12.10 Hz), 1.94-2.04 (1H, m, J=12.65 Hz), 1.90-1.95 (3H, m), 1.72-1.91 (2H, m), 1.37 (3H, d, J=6.05 Hz), 1.29 (3H, d, J=6.60 Hz). Differential scanning calorimetry utilized a heating rate of 10° C./min and revealed a melting/decomposition endotherm with an onset temperature of 297.6° C. and a peak temperature at 299.1° C.
Example 1, Crystallization of the Free Base: A sample of the amorphous free base of N-((1R,2S,5R)-5-(isopropyl(methyl)amino)-2-((S)-2-oxo-3-(6-(trifluoromethyl)quinazolin-4-ylamino)pyrrolidin-1-yl)cyclohexyl)acetamide (1 g) was dissolved in dichloromethane (5 mL). The solution was charged with heptane (30 mL) and then warmed to distill the dichloromethane. The solution was cooled to 40° C.; a white solid precipitated. The suspension was heated to 90° C. and stirred for 2 h. The suspension was cooled to room temperature and filtered to provide the pure free base of the title compound. No residual solvent was apparent by 1H-NMR.
![Image result for Bristol-Myers Squibb]()
PAPER
![Abstract Image]()
A concise bulk synthesis of stereochemically complex CCR2 antagonist BMS-741672 is reported. A distinct structural feature is the chiral all-cis 1,2,4-triaminocyclohexane (TACH) core, which was assembled through consecutive stereocontrolled heterogeneous hydrogenations: efficient Pt-catalyzed reduction of a β-enaminoester, directed by (S)-α-methylbenzylamine as a low-cost chiral template, and reductive amination of a 3,4-cis-disubstituted cyclohexanone over sulfided Pt/C introduced a tert-amine, setting the third stereocenter in the all-cis cyclohexane core. The heterogeneous catalysts were recycled. Ester hydrolysis produced a γ-amino acid, isolated as its Na salt. A challenging Curtius reaction to introduce the remaining C–N bond at C-2 was strongly influenced by the presence of the basic tert-amine, providing a stereoelectronically highly activated isocyanate. Detailed mechanistic and process knowledge was required to enable clean trapping with an alcohol (t-BuOH) while avoiding formation of side products, particularly an unusual carbamoyl phosphate. Deprotection, N-acetylation, and uncatalyzed SNAr coupling with known 4-chloroquinazoline provided the final product. The resulting 12-step synthesis was used to prepare 50 kg of the target compound in an average yield of 82% per step.
Stereoselective Bulk Synthesis of CCR2 Antagonist BMS-741672: Assembly of an All-cis (S,R,R)-1,2,4-Triaminocyclohexane (TACH) Core via Sequential Heterogeneous Asymmetric Hydrogenations
Joerg Deerberg*, Siva J. Prasad, Chris Sfouggatakis, Martin D. Eastgate, Yu Fan, Ramakrishnan Chidambaram†,Praveen Sharma‡, Li Li, Richard Schild, Jale Müslehiddinoğlu§, Hyei-Jha Chung∥, Simon Leung, and Victor Rosso
Chemical and Synthetic Development, Bristol-Myers Squibb Company, One Squibb Drive, New Brunswick, New Jersey 08901, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00282
Patents
| Patent ID |
Date |
Patent Title |
| US7687508 |
2010-03-30 |
CYCLIC DERIVATIVES AS MODULATORS OF CHEMOKINE RECEPTOR ACTIVITY |
| US7671062 |
2010-03-02 |
N-((IR, 2S, 5R)-5-(ISOPROPYL(METHYL)AMINO)-2-((S)-2-0XO-3-(6-TRIFLUOROMETHYL)QUINAZOLIN-4-YLAMINO)PYRROLIDIN-1-YL)CYCLOHEXYL)ACETAMIDE AND OTHER MODULATORS OF CHEMOKINE RECEPTOR ACTIVITY, CRYSTALLINE FORMS AND PROCESS. |
![Image result for Bristol-Myers Squibb]()
Bristol-Myers Squibb, Paul Biondi Senior Vice President, Head of Business Development
About Bristol-Myers Squibb
Bristol-Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information, please visit www.bms.com or follow us on Twitter at http://twitter.com/bmsnews.
////////// ////////////////BMS-741672, BMS 741672, BRISTOL MEYER SQUIB, PHASE 2, type 2 Diabetes, Neuropathic Pain, Bristol-Myers Squibb
CC(C)N(C)[C@H]1C[C@@H](NC(C)=O)[C@H](CC1)N4CC[C@H](Nc3ncnc2ccc(cc23)C(F)(F)F)C4=O
Filed under:
Phase2 drugs Tagged:
BMS-741672,
BRISTOL MEYER SQUIB,
Bristol-Myers Squibb,
neuropathic pain,
phase 2,
TYPE 2 DIABETES ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()























































































 (S)-6-Hydroxybuspirone
(S)-6-Hydroxybuspirone
























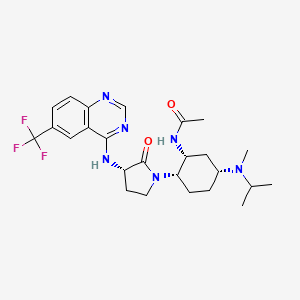



 , 1686 (s), 1635 (m), 1584 (s), 1540 (m), 1334 (m), 1307 (s), 1164 (m), 1121 (m), 870
, 1686 (s), 1635 (m), 1584 (s), 1540 (m), 1334 (m), 1307 (s), 1164 (m), 1121 (m), 870









































































































































































![[1860-5397-9-265-6]](http://i1.wp.com/www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-6.png)






















































 .
.