
- C24H27FN2O
- Average mass: 378.482391 Da

Neuropathic pain
Neuropathic pain is caused when peripheral nerves are damaged by mechanical, metabolic or inflammatory way. The pain occurring images are mainly due to the occurrence of spontaneous pain, hyperalgesia and allodynia (pain is already triggered by non-noxious stimuli) in. As a result, the lesions to increased expression of Na + channels and thus to spontaneous activity in the damaged axons and their Nachbaraxonen (England et al., Neurology, 1996, 47, 272-276).The excitability of the neurons is increased and they react to incoming stimuli with an increased discharge frequency. This results in an increased sensitivity to pain, which contributes to the development of hyperalgesia and spontaneous pain (Baron, Clin J Pain 2000;. 16 (2 Suppl), 12-20). The causes and manifestations, and therefore the treatment needs of neuropathischerm pain are varied. They arise as a result of injury or disease of the brain, spinal cord or peripheral nerves.Causes may be operations, such as phantom pain after amputation, stroke, multiple sclerosis, spinal cord injury, alcohol or drug abuse or other toxins, cancers but also
Metabolic diseases such as diabetes, gout, kidney failure or liver cirrhosis, or infectious diseases such as mononucleosis, ehrlichiosis, typhoid, diphtheria, HIV, syphilis or Lyme disease. The pain experience is very different signs and symptoms that can change over time in number and intensity. Paradoxically, patients with neuropathic pain outline a slowdown or failure of acute pain perception and the simultaneous increase of neuropathic pain. The typical symptoms of neuropathic pain as tingling, burning, shooting or described, or radiating electrifying. Pharmacological basis for treatment of neuropathic pain include tricyclic antidepressants and anticonvulsants, which are used as monotherapy or in combination with opioids. These drugs usually provide only a certain pain relief during a pain-free but is often not achieved. The often-adjusting side effects are dose increases while the drug to achieve adequate pain relief often in the way. In fact, a higher dosage of a μ-opioid is often required as the treatment of acute pain, thereby reducing the side effects get even more important for satisfactory treatment of neuropathic pain. By the occurrence of typical μ-opioid tolerance development and the concomitant need for dose escalation of this problem is exacerbated. In summary it can be stated that neuropathic pain is difficult to treat and today is alleviated by high doses of μ-opioids only partially (Saudi Pharm J. 2002, 10 (3), 73-85). There is therefore an urgent need for medicines for the treatment of chronic pain, the dose should not be increased until the occurrence of intolerable side effects to ensure a satisfactory pain treatment.
……………
http://www.google.com/patents/US7547707
Example 24 1,1-(3-Dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole hemicitrate, More Non-polar diastereoisomer
4-Dimethylamino-4-phenylcyclohexanone (651 mg, 3 mmoles) and 2-(5-fluoro-1H-indol-3-yl)-ethanol (“5-fluorotryptophol”, 537 mg, 3 mmoles) were initially introduced into abs. MC (20 ml) under argon. Trifluoromethanesulfonic acid trimethylsilyl ester (0.6 ml, 3.1 mmoles) was then added very rapidly. The mixture was stirred at RT for 20 h. For working up, 1 M NaOH (30 ml) was added to the reaction mixture and the mixture was stirred for 30 min. The organic phase was separated, and the aqueous phase which remained was extracted with MC (3×60 ml). The combined organic phases were washed with water (2×30 ml) and dried over sodium sulfate. Methanol (30 ml) was added to the solid residue obtained after the solvent had been distilled off, and the mixture was heated, and stirred for 15 hours. The solid contained in the suspension was filtered off with suction and dried. 955 mg of the more non-polar diastereoisomer of 1,1-(3-dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole were obtained (m.p. 284-292° C.). 850 mg of this were dissolved in hot ethanol (900 ml), and a similarly hot solution of citric acid (1 g, 5.2 mmoles) in ethanol (20 ml) was added. After approx. 15 minutes, crystals precipitated out at the boiling point. After cooling to approx. 5° C., the mixture was left to stand for 2 h. The solid formed was filtered off with suction. 640 mg of the hemicitrate were obtained as a white solid (m.p. 258-282° C.).
Example 25 1,1-(3-Dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole hemicitrate, More Polar diastereoisomer
4-Dimethylamino-4-phenylcyclohexanone (217 mg, 1 mmole) and 2-(5-fluoro-1H-indol-3-yl)-ethanol (“5-fluorotryptophol”, 179 mg, 1 mmole) were dissolved in conc. acetic acid (4 ml). Phosphoric acid (1 ml, 85 wt. %) was slowly added dropwise to this mixture. The mixture was stirred at RT for 16 h. For working up, the mixture was diluted with water (20 ml), brought to pH 11 with 5 M NaOH and extracted with MC (3×20 ml). The combined organic phases were dried with sodium sulfate and evaporated. The residue (364 mg of white solid) was suspended in hot ethanol (20 ml), and a similarly hot solution of citric acid (185 mg, 0.96 mmole) in ethanol (5 ml) was added. The residue thereby dissolved completely and no longer precipitated out even on cooling to approx. 5° C. Ethanol was removed on a rotary evaporator and the hemicitrate of the more polar diastereoisomer of 1,1-(3-dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole was obtained in this way in a yield of 548 mg as a white solid (m.p. 148-155° C.).
| 24 |
 |
hemicitrate | more non-polar diastereomer |
| 25 |
 |
hemicitrate | more polar diastereomer |
(1 r,4r)-6′-fluoro-N,N- dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1 ,1 '-pyrano[3,4-b]indol]-4-amine (free base), has the following structural formula (I):
http://www.google.com/patents/WO2013113690A1?cl=en


One particular drug that is of great interest for use in treating cancer pain (and other acute, visceral, neuropathic and chronic pain pain disorders) is (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4b]indol]-4-amine. This drug is depicted below as the compound of formula (I).

The solid forms of (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4b]indol]-4-amine that are known so far are not satisfactory in every respect and there is a demand for advantageous solid forms

In a previous communication, our efforts leading from 1 to the identification of spiro[cyclohexane-dihydropyrano[3,4-b]indole]-amine 2a as analgesic NOP and opioid receptor agonist were disclosed and their favorable in vitro and in vivo pharmacological properties revealed. We herein report our efforts to further optimize lead 2a, toward trans-6′-fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-spiro[cyclohexane-1,1′(3′H)-pyrano[3,4-b]indol]-4-amine (cebranopadol, 3a), which is currently in clinical development for the treatment of severe chronic nociceptive and neuropathic pain.
Discovery of a Potent Analgesic NOP and Opioid Receptor Agonist: Cebranopadol
http://pubs.acs.org/doi/full/10.1021/ml500117c
b]indol]-4-amine, trans-, 2-hydroxy-1,2,3-propanetricarboxylate (2:1)
2.76 (m,6 H); 3.88 (t, 2 H); 6.86 (dt, 1 H); 7.10 (dd, 1 H); 7.30-7.43 (m, 6 H); 10.91 (br
s, 1 H).
overlap); 71.5; 72.2; 102.3 (2JC,F = 23 Hz); 105.6 (3JC,F = 4 Hz); 108.3 (2JC,F = 26 Hz);
156,7 (1JC,F = 231 Hz); 171.3 (2 C), 175.3.HPLC-MS: m/z 378.9 [M + H]+
| US20120034297 * | Aug 4, 2011 | Feb 9, 2012 | Gruenenthal Gmbh | Pharmaceutical dosage forms comprising 6′-fluoro-(N-methyl- or N,N-dimethyl-)-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1'-pyrano[3,4,b]indol]-4-amine |
| US20130012563 * | Jul 6, 2012 | Jan 10, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-n,n-dimethyl-4-phenyl-4′,9′-dihydro-3′h-spiro[cyclohexane-1,1'-pyrano[3,4,b]indol]-4-amine |
| WO2004043967A1 | Nov 5, 2003 | May 27, 2004 | Otto Aulenbacher | Spirocyclic cyclohexane derivatives |
| WO2008040481A1 | Sep 26, 2007 | Apr 10, 2008 | Gruenenthal Gmbh | MIXED ORL 1/μ AGONISTS FOR TREATING PAIN |
-
CORAL - Cebranopadol Versus Morphine Prolonged-release in Patients With Chronic Moderate to Severe Pain Related to Cancer
Efficacy, Safety, and Tolerability of Oral Cebranopadol Versus Morphine Sulfate PR in Subjects With Chronic Moderate to Severe Pain Related to Cancer.Average amount of daily rescue medication at the end of the maintenance period.
UK Clinical Trials Gateway, 07 October 2013
-
CORAL XT – Open-label Extension Trial of the CORAL Trial
An Open-label, Multi-site Trial to Describe the Safety and Tolerability of Oral Cebranopadol Administered for 26 Weeks in Subjects With Cancer-related Pain Who Have Completed Treatment in the KF6005/07 Trial.Absolute…
UK Clinical Trials Gateway, 12 December 2013
| WO2004043967A1 * | Nov 5, 2003 | May 27, 2004 | Otto Aulenbacher | Spirocyclic cyclohexane derivatives |
| WO2005066183A1 * | Dec 21, 2004 | Jul 21, 2005 | Gruenenthal Gmbh | Spirocyclic cyclohexane derivatives with affinity for the orl1-receptor |
| US20050153998 * | Aug 19, 2004 | Jul 14, 2005 | Fumitaka Ito | Tetrahydroisoquinoline or isochroman compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7799931 * | Feb 17, 2009 | Sep 21, 2010 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US7951948 * | Apr 19, 2010 | May 31, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US7960404 | Aug 21, 2009 | Jun 14, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US8034936 | Nov 4, 2010 | Oct 11, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds useful to treat substance dependency |
| US8053576 | Feb 17, 2009 | Nov 8, 2011 | Gruenenthal Gmbh | Treating conditions associated with the nociceptin/ORL1 receptor system, e.g. pain, drug withdrawal, anxiety, muscle relaxants, anxiolytic agents; e.g. 1,1-[3-dimethylamino-3-(pyridin-2-yl)pentamethylene]-3,4-dihydro-1H-2,9-diazafluorene |
| US8288406 | Sep 22, 2010 | Oct 16, 2012 | Gruenenthal Gmbh | Hydroxymethylcyclohexylamines |
| US8288430 | Mar 25, 2009 | Oct 16, 2012 | Grunenthal Gmbh | Spiro(5.5)undecane derivatives |
| US8293758 * | Mar 25, 2009 | Oct 23, 2012 | Grunenthal Gmbh | Substituted spirocyclic cyclohexane derivatives |
| US8357705 | Mar 25, 2009 | Jan 22, 2013 | Gruenenthal Gmbh | Substituted cyclohexyldiamines |
| US8404740 | Aug 21, 2009 | Mar 26, 2013 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US8614245 * | Jan 8, 2013 | Dec 24, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US8618156 * | Jul 6, 2012 | Dec 31, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1'-pyrano[3,4,b]indol]-4-amine |
| US20130012563 * | Jul 6, 2012 | Jan 10, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-n,n-dimethyl-4-phenyl-4′,9′-dihydro-3′h-spiro[cyclohexane-1,1'-pyrano[3,4,b]indol]-4-amine |
Filed under: Phase2 drugs, Phase3 drugs, Uncategorized Tagged: Cebranopadol, GRT 6005, neuropathic pain








 Originally posted on
Originally posted on 















 Originally posted on
Originally posted on 























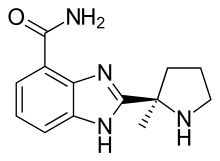




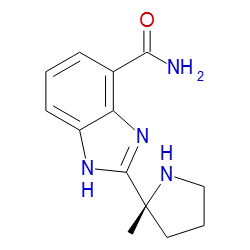











 Originally posted on
Originally posted on 












































































 Originally posted on
Originally posted on 



























































