![]()
FDA accepts Kythera’s ATX-101 new drug application
Kythera Biopharmaceuticals’ new drug application (NDA) for its ATX-101, a submental contouring injectable drug, has been accepted for filing by the US Food and Drug Administration (FDA).
According to Kythera Biopharmaceuticals, the ATX-101 NDA will be subject to a standard review and will have a prescription drug user fee act (PDUFA) action date of 13 May 2015. The company submitted the NDA in May 2014.
http://www.pharmaceutical-technology.com/news/newsfda-accepts-kytheras-atx-101-new-drug-application-4316052?WT.mc_id=DN_News
![]()
cas 83-44-3, C24 H40 O4
cas of Na salt….302-95-4
NSC-681065 , NSC 8797
| NAMES |
Cholan-24-oic acid, 3,12-dihydroxy-, (3α,5β,12α)- |
- OTHERS
- 5β-Cholan-24-oic acid, 3α,12α-dihydroxy- (8CI); 17β-[1-Methyl-3-carboxypropyl]-etiocholane-3α,12α-diol;
- 3α,12α-Dihydroxy-5β-cholan-24-oic acid;
- 3α,12α-Dihydroxy-5β-cholanic acid;
- 3α,12α-Dihydroxy-5β-cholanoic acid; 3α,12α-Dihydroxycholanic acid;
- 5β-Cholanic acid-3α,12α-diol;
- 5β-Deoxycholic acid; 7-Deoxycholic acid; ATX 101;
- Cholerebic; Cholic acid, deoxy-; Cholorebic; Degalol; Deoxycholatic acid; Deoxycholic acid; Desoxycholic acid; Droxolan; NSC 8797; Pyrochol; Septochol
- Deleted CAS Registry Numbers: 728917-93-9
- University of California, Oakland (Originator)
LA BioMed (Originator)
- LICENSE….
Rapid removal of body fat is an age-old ideal, and many substances have been claimed to accomplish such results, although few have shown results. ”Mesotherapy”, or the use of injectables for the removal of fat. is not widely accepted among medical practitioners due to safety and efficacy concerns, although homeopathic and cosmetic claims have been made since the 1950′s. Mesotherapy was originally conceived in Europe as a method of utilizing cutaneous injections containing a mixture of compounds for the treatment of local medical and cosmetic conditions. Although mesotherapy was traditionally employed for pain relief, its cosmetic applications, particularly fat and cellulite removal, have recently received attention in the United States. One such reported treatment for localized fat reduction, which was popularized in Brazil and uses injections of phosphatidylcholine, has been erroneously considered synonymous with mesotherapy. Despite its attraction as a purported “fat-dissolving” injection, there is little safety and efficacy data of these cosmetic treatments. See, Rotunda, A.M. and M.
olodney, Dermatologic Surgery 32:, 465-480 (2006) (“Mesotherapy and
Phosphatidy lcholine Injections: Historical Clarification and Review**).
Recently published literature reports that the bile acid, DCA, and salts thereof, have fat removing properties when injected into fatty deposits in vivo. See, WO
2005/1 17900 and WO 2005/1 12942, as well as US2005/0261258; US2005/0267080; US2006/127468; and US20060154906, each of which is incorporated herein by reference in its entirety). Deoxycholate injected into fat tissue degrades fat cells via a cytolytic mechanism. Because deoxycholate injected into fat is rapidly inactivated by exposure to protein and then rapidly returns to the intestinal contents, its effects are spatially contained. As a result of this attenuation effect that confers clinical safety, fat removal typically require 4 – 6 sessions. This localized fat removal without the need for surgery is beneficial not only for therapeutic treatment relating to pathological localized fat deposits (e.g., dyslipidemias incident to medical intervention in the treatment of HIV), but also for cosmetic fat removal without the attendant risk inherent in surgery (e.g., liposuction). See, Rotunda et ai, Dermatol. Surgery 30: 1001-1008 (2004) (“Detergent effects of sodium deoxycholate are a major feature of an injectable phosphatidylcholine formulation used for localized fat dissolution”) and Rotunda et al, J. Am. Acad. Dermatol. (2005 : 973-978) (“”Lipomas treated with subcutaneous deoxycholate injections”), both incorporated herein by reference in their entirety. US Patent Nos. 7,622,130 and
7,754,230 describe using DCA for fat removal.
In addition, many important steroids have a 12- -hydroxy-substituent on the C- ring of the steroid. Such compounds include, by way of example, bile acids such as DCA, cholic acid, lithocholic acid, and the like. Heretofore, such compounds were typically- recovered from bovine and ovine sources which provided a ready source of bile acids on a cost effective basis. However, with the recent discovery that pathogens such as prions can contaminate such sources, alternative methods for the synthesis of bile acids from plant sources or synthetic starting materials have become increasingly important. For example, DCA from animals in New Zealand are a source of bile acids for human use under US regulatory regimes, as long as the animals continue to remain isolated and otherwise free of observable pathogens. Such stringent conditions impose a limitation on the amount of suitable mammalian sourced bile acids and does not preclude the possibility that the bile acid will be free of such pathogens. US Patent Publication No.
8,242,294 relates to DCA containing less than 1 ppt 14C.
ATX-101, sodium deoxycholate for injection, is awaiting for approval in the U.S. for the reduction of localized submental fat. Phase II trials for the treatment of superficial lipomas have been completed at Kythera Biopharmaceuticals and Intendis. Treatment with ATX-101 is expected to significantly reduce the size of or eliminate lipomas and provide an effective non-surgical, minimally invasive treatment option for patients.
Licensed to Kythera from Los Angeles Biomedical Institute at Harbor-UCLA Medical Center in 2007, ATX-101 is also being evaluated by the company for aesthetic applications. Specifically, phase II trials are under way for the reduction of submental fat. In 2010, ATX-101 was licensed to Intendis by Kythera Biopharmaceuticals outside of the U.S. and Canada for the treatment of dermatological disorders. In 2010, the product was licensed by Kythera Biopharmaceuticals to Bayer outside Canada and the U.S., and in 2014, Kythera acquired those same rights from Bayer.
………………………………..
WO 2011075701
http://www.google.com/patents/WO2011075701A2?cl=en
Scheme 2
![Figure imgf000077_0001]()
Conversion of Compound 24 to Compound 33:
The hydrogenation of compound 24 on 10.0 g scale using dry 10 % Pd/C (15 wt %) in ethyl acetate (20 parts) was added and applied about 50 psi hydrogen pressure and temperature raised to 70 °C. After reaching temperature 70 °C, observed increase of hydrogen pressure to about 60 psi, at these conditions maintained for 60 h. After 60 hours 0.6% of compound 24 and 2.75% of allylic alcohol were still observed, so further stirred for additional 12 h (observed 0.16% of allylic alcohol and 0.05% of compound 24). After work-up, the reaction provided 9.5g of residue.
Anther hydrogenation reaction on 25 g of compound 24 with above conditions for 76 h provided 24.5 g of residue.
Method A
10% Pd/C (900 mg) was added to a solution of compound 24 (2.0 g, 4.5 mmol) in EtOAc (150 mL) and the resulting slurry was hydrogenated in a Parr apparatus (50 psi) at 50 °C for 16 h. At this point the reaction was determined to be complete by TLC. The mixture was filtered through a small plug of Celite® and the solvent was removed under vacuum, providing compound 33 (1.6 g, 80% yield) as a white solid.
TLC: p-anisaldehyde charring, Rf for 33 = 0.36 and Rf for 25 = 0.32.
TLC mobile phase: 20% – EtOAc in hexanes. 1H NMR (500 MHz, CDC13): δ = 4.67-4.71 (m, 1H), 3.66 (s, 3H), 2.45-2.50 (t, J = 15 Hz, 2H), 2.22-2.40 (m, 1H), 2.01 (s, 3H), 1.69-1.96 (m, 9H), 1.55 (s, 4H), 1.25-1.50 (m, 8H), 1.07-1.19 (m, 2H), 1.01 (s, 6H), 0.84-0.85 (d, J= 7.0 Hz, 3H).
13C NMR (125 MHz, CDC13): δ = 214.4, 174.5, 170.4, 73.6, 58.5, 57.4, 51.3, 46.4, 43.9, 41.2, 38.0, 35.6, 35.5, 35.2, 34.8, 32.0, 31.2, 30.4, 27.4, 26.8, 26.2, 25.9, 24.2, 22.6,
21.2, 18.5,1 1.6,
Mass (m/z) = 447.0 [M+ + 1], 464.0 [M+ + 18].
IR (KBr) = 3445, 2953, 2868, 1731, 1698, 1257, 1029 cm-1.
m.p. =142.2-144.4 °C (from EtOAc/hexanes mixture).
[α]D = +92 (c = 1 % in CHCl3).
ELSD Purity: 96.6%: Retention time = 9.93 (Inertsil ODS 3 V, 250 χ 4.6 mm, 5um, ACN:
0.1 % TFA in water (90: 10)
Method B
A slurry of 10% Pd/C (9 g in 180 mL of ethyl acetate) was added to a solution of compound 24 (36 g, 81 mmol) in EtOAc (720 mL) and the resulting slurry was treated with hydrogen gas (50 psi) at 45-50 °C for 16 h. (A total of 1080 mL of solvent may be used). At this point the reaction was determined to be complete by HPLC (NMT 1% of compound 24). The mixture was filtered through Celite® (10 g) and washed with ethyl acetate (900 mL). The filtrate was concentrated to 50% of its volume via vacuum distillation below 50 °C. To the concentrated solution was added pyridinium
chlorochromate (20.8 g) at 25-35 °C and the mixture was stirred for 2 h at 25-35 °C, when the reaction completed by HPLC (allylic alcohol content is NMT 1%).
The following process can be conducted if compound 24 content is more than 5%. Filter the reaction mass through Celite® (10 g) and wash with ethyl acetate (360 mL). Wash the filtrate with water (3 x 460 mL) and then with saturated brine (360 mL). Dry the organic phase over sodium sulphate (180 g), filter and wash with ethyl acetate (180 mL). Concentrate the volume by 50% via vacuum distillation below 50 °C. Transfer the solution to a clean and dry autoclave. Add slurry of 10% palladium on carbon (9 g in 180 mL of ethyl acetate). Pressurize to 50 psi with hydrogen and stir the reaction mixture at 45-50 °C for 16 h. Upon complete consumption of compound 24 by HPLC (the content of compound 24 being NMT 1%), the reaction mixture was filtered through Celite® (10 g) and the cake was washed with ethyl acetate (900 mL). The solvent was concentrated to dryness via vacuum distillation below 50 °C. Methanol (150 mL) was added and concentrated to dryness via vacuum distillation below 50 °C. Methanol (72 mL) was added to the residue and the mixture was stirred for 15-20 min at 10-15 °C, filtered and the cake was washed with methanol (36 mL). The white solid was dried in a hot air drier at 45-50 °C for 8 h to LOD being NMT 1 % to provide compound 33 (30 g, 83.1 % yield).
Conversion of Compound 33 to Compound 34:
Method A
A THF solution of lithium tri-tert-butoxyaluminum hydride (1 M, 22.4 mL, 22.4 mmol) was added drop wise to a solution of compound 33 (2.5 g, 5.6 mmol) in THF (25 mL) at ambient temperature. After stirring for an additional 4-5 h, the reaction was determined to be complete by TLC. The reaction was quenched by adding aqueous HCl (1 M, 10 mL) and the mixture was diluted with EtOAc (30 mL). The phases were separated and the organic phase was washed sequentially with water (15 mL) and saturated brine solution (10 mL). The organic phase was then dried over anhydrous Na2S04 (3 g) and filtered. The filtrate was concentrated under vacuum and the resulting solid was purified by column chromatography [29 mm (W) x 500 mm (L), 60-120 mesh silica, 50 g], eluting with EtOAc/hexane (2:8) [5 mL fractions, monitored by TLC with p- anisaldehyde charring]. The fractions containing the product were combined and concentrated under vacuum to provide compound 34 (2.3 g, 91%) as a white solid.
TLC: p-anisaldehyde charring, Rf for 34 = 0.45 and Rf for 33 = 0.55.
TLC mobile phase: 30% – EtOAc in hexanes.
1H NMR (500 MHz, CDC13): δ = 4.68-4.73 (m, 1H), 3.98 (s, 1H), 3.66 (s, 3H), 2.34-2.40 (m, 1H), 2.21-2.26 (m, 1H), 2.01 (s, 3H), 1.75-1.89 (m, 6H), 1.39-1.68 (m, 16H), 1.00-1.38 (m, 3H), 0.96-0.97 (d, J= 5.5 Hz, 3H), 0.93 (s, 3H), 0.68 (s, 3H).
13C NMR (125 MHz, CDCI3): δ = 174.5, 170.5, 74.1, 72.9, 51.3, 48.1, 47.2, 46.4, 41.7, 35.8, 34.9, 34.7, 34.0, 33.5, 32.0, 30.9, 30.8, 28.6, 27.3, 26.8, 26.3, 25.9, 23.4, 22.9, 21.3, 17.2, 12.6 Mass (m/z) = 449.0 [M+ + 1], 466.0 [M + 18].
IR ( Br) = 3621, 2938, 2866, 1742, 1730, 1262, 1 162, 1041, cm-1.
m.p = 104.2-107.7 °C (from EtOAc).
[α]D = +56 (c = 1% in CHCl3).
ELSD Purity: 97.0%: Retention time = 12.75 (Inertsil ODS 3V, 250 χ 4.6 mm, 5um, ACN: Water (60:40)
Method B
A THF solution of lithium tri-rert-butoxyaluminum hydride (1 M, 107.6 mL, 107.6 mmol) was added over 1 h to a solution of compound 33 (30.0 g, 67 mmol) in dry THF (300 mL) at 0-5 °C. After stirring for an additional 4 h at 5-10 °C, the reaction was determined to be complete by HPLC (NMT 1% of compound 33). The reaction was cooled to 0-5 °C and quenched by adding 4N HCl (473 mL). The phases were separated. The aqueous layer was extracted with DCM (2 x 225 mL) and the combined organic phase was washed sequentially with water (300 mL) and saturated brine solution (300 mL). The organic phase was then was concentrated to dryness by vacuum distillation below 50 °C. Methanol (150 mL) was added to the residue and concentrated to dryness by vacuum distillation below 50 °C. Water (450 mL) was then added to the residue and the mixture was stirred for 15-20 min., filtered and the cake was washed with water (240 mL). The white solid was dried in a hot air drier at 35-40 °C for 6 h to provide compound 34 (30 g, 99.6%).
Conversion of Compound 34 to crude DCA:
Method A
A solution of LiOH (187 mg, 4.4 mmol) in H20 (2.0 mL) was added to a solution of compound 34 (500 mg, 1.1 1 mmol) in THF (8 mL) and MeOH (8 mL). The resulting mixture was stirred for 3-4 h at 50 °C. Upon complete disappearance of the starting material by TLC, the reaction mixture was concentrated under vacuum. A mixture of water (10 mL) and 3 N HCl (1 mL) were combined and cooled to 0 °C and then added to the crude product. After stirring for 1 h at 0 °C, the precipitated solids were filtered and then washed with water (10 mL) and hexane (20 mL). Drying under vacuum at room temperature provided deoxycholic acid (DCA, 400 mg, 91% yield) as a white solid. TLC: -anisaldehyde charring, Rf for DC A = 0.32 and Rf for 2.1a = 0.82.
TLC mobile phase: 10% – Methanol in DCM.
1H NMR (500 MHz, DMSO): δ = 11.92 (s, 1H), 4.44 (s, 1H), 4.19 (s, 1H), 3.77 (s, 1H), 3.35-3.36 (m, 1H), 2.19-2.21 (m, 1H), 2.08-2.10 (m, 1H), 1.73-1.80 (m, 4H), 1.43- 1.63 (m, 6H), 1.15-1.35 (m, 12H), 0.98-1.05 (m, 2H), 0.89-0.90 (d, J = 6.0 Hz, 3H),
0.83 (s, 3H), 0.58 (s, 3H).
13C NMR (125 MHz, DMSO): δ =174.8, 71.0, 69.9, 47.4, 46.1, 46.0, 41.6, 36.3, 35.6, 35.1, 34.9, 33.8, 32.9, 30.8, 30.7, 30.2, 28.6, 27.1, 27.0, 26.1, 23.5, 23.0, 16.9, 12.4.
Mass (m/z) = 393 [M+, + 1].
IR = 3363, 2933, 2863, 1694, 1453, 1372, 1042, cm-1.
m.p. = 171.4-173.6 °C (from ethanol); 174-176 °C (Alfa Aesar) and 171-174 °C (Aldrich)
[<x]D = +47 (c = 1% in EtOH ), +54° (c = 2% in ethanol) [Alfa Aesar]
ELSD Purity: 99.7%: Retention time = 5.25 (Inertsil ODS 3 V, 250 χ 4.6 mm, 5um, ACN:
0.1% TFA in water (90:10).
Method B
A 20% solution of NaOH (40 g, 270 mmol) in H20 (54 mL) was added to a solution of compound 34 (30 g, 67 mmol) in THF (120 mL) and MeOH (120 mL) at 0-5 °C. The resulting mixture was stirred for 4 h at 25-35 °C. Upon completion of reaction by HPLC (NMT 0.5% of compound 34 and intermediates), the solvent was removed via vacuum distillation below 50 °C. The residue was dissolve in water (300 mL) and washed with DCM (2 x 150 mL). The pH of aqueous layer was adjusted to 1-2 with 2N HCl (~ 173 mL). The solids were filtered, washed thoroughly with water (3 L) and dried by a hot air drier at 70-75 °C until the moisture content is less than 2% to provide deoxycholic acid (DCA, 26 g, 99% yield) as a white solid.
EXAMPLE 9
Deoxycholic acid (DCA) Purification
1. Solvent Selection
Two solvent systems were explored for further purification of DCA: • 10% Hexanes in EtOAc
• DCM
The following experiments have been conducted and the experimental results tabulated below.
* The DCA to be purified was dissolved in a mixture of methanol and DCM and then the methanol was removed by azeotropic distillation. The amount of methanol required to dissolve the crude DCA depends on how pure it is to begin with.
Typical crude material was—75% pure and could be dissolved at reflux using 10% methanol-DCA (by volume) using—20 mL per gram. With purer DCA, the percentage of methanol had to be increased to 15%.
Effective purification was achieved by crystallization of the product from DCM following dissolution in a mixture of methanol and DCM and azeotropic removal of the methanol via atmospheric distillation.
2. Solvent Quantity
Experiments have been conducted using different solvent volumes and the experimental results are tabulated below.
Excellent recoveries and product quality were obtained at all solvent levels.
3. Isolation Temperature
The following experiments have been conducted by varying the isolation temperature and the results are tabulated below:
Higher quality product was obtained when isolation is done at 25-30 °C as compared to 10-15 °C. Purification of DCA in 100 g Scale
The final purification procedure for this step is given below:
Crude DCA (110 g) was dissolved in 10% methanol in DCM (2.5 L) at reflux temperature. To this clear solution 2.5 L of dichloromethane was added at reflux temperature and then about 3.0 L of solvent was distilled at atmospheric pressure (GC analysis of reaction mass supernatant revealed the presence of about 3% of methanol). The reaction slurry was cooled to 20-25 °C and then stirred for 3-4 h. The mixture was filtered and the solids were washed with DCM (300 mL). The product was dried in a hot air oven at 50-55 °C for 6-8 h.
The above dried DCA was added to water (1.0 L) and then 10% sodium hydroxide solution (122 mL) was added resulting in a clear solution. This solution was filtered through 5μ filter paper. The filtrate was diluted with water (2.0 L), and the pH was adjusted to 1— 2 with 2N HCl solution (204 mL). The precipitated solids were stirred for 1 h, filtered and the solids were washed with additional water (7.0 L). After drying in a hot air oven at 70-75 °C for 16-20 h, purified DCA (~ 66 g with more than 99% purity by HPLC RI detection) was obtained as a white solid.
TLC: 7-Anisaldehyde charring, Rf for DCA = 0.32 and Rf for compound 34 = 0.82. Eluent = 10% methanol in DCM. 1H NMR (500 MHz, DMSO): δ = 11.92(s, 1H),4.44(s, 1H), 4.19(s, 1H), 3.77 (s, 1H), 3.36-3.35 (m, 1H), 2.21-2.19 (m, 1H), 2.10-2.08 (m, 1H), 1.80-1.73 (m, 4H), 1.63- 1.43(m, 6H), 1.35-1.15(m, 12H), 1.05-0.98(m, 2H), 0.90-0.89 (d, J = 6.0 Hz, 3H), 0.83 (s, 3H), 0.58 (s, 3H).
1 C NMR (125 MHz, DMSO): δ =174.8, 71.0, 69.9, 47.4, 46.1, 46.0, 41.6, 36.3, 35.6, 35.1, 34.9, 33.8, 32.9, 30.8, 30.7, 30.2, 28.6, 27.1, 27.0, 26.1, 23.5, 23.0, 16.9, 12.4.
Mass (m/z) = 393 [M+, + 1].
IR = 3363, 2933, 2863, 1694, 1453, 1372, 1042, cm-1.
m.p. = 171.4-173.6 °C (from ethanol); 174-176 °C (Alfa Aesar) and 171-174 °C (Aldrich).
Recrystallization of Deoxycholic acid (DC A)
DCA obtained from Method B (26 g) above, was charged into a clean and dry flask. Methanol (65 mL) and DCM (585 mL) were added. The mixture was heated to reflux to obtain a clear solution. DCM (650 mL) was charged to the solution and the solvent was distilled atmospherically until 780 mL of solvent was collected. The mixture was assayed by GC to determine the solvent composition. If the methanol content is more than 2%, add DCM (200 mL) and distill atmospherically until 200 mL of distillate have been collected. (Check for the methanol content by GC). The reaction mixture was cooled over 1-2 h to 20-25 °C and stirred at this temperature for 3-4 h. The product was filtered and washed with DCM (81 mL), dried in a hot air drier at 50-55 °C for 8 h. The purity was determined by HPLC. If single max impurity is more than 0.1%, the above process is repeated.
The dried material from the above was charged in to a clean flask. Water (190 mL) was added and followed by 10% aqueous NaOH (3.18 g in 31.8 mL of water). The solution was filtered through 5μ filter paper and the filtrate was diluted with additional water (380 mL). The pH was adjusted to 1-2 with 2 N HCl (53 mL). The resulting solids was filtered, washed thoroughly with water (1.9 L), and dried in a hot air drier at 70-75 °C until the water content is below 1% to give DCA as a white solid (17 g, % of recovery: 65). EXAMPLE 10
Alternate method of Synthesis and purification of DCA from compound 33
Step la— Hydrogenation of methyl 3a-acetoxy-12-oxo—5fi-chol-9(ll)-en-24-oate (24)
Dry Pd/C (75.0 g, 25 wt %) was added to 24 (300.0 g, 0.7 mol) in EtOAc (7.5 L, 25 vol). The reaction mixture was heated to 45°— 50°C and pressurized to 50 psi of H2. HPLC analysis after 21 hours indicated < 1.0% area under the curve (AUC) of 24 remained; 4.6% AUC of the allylic alcohol impurity 86 and 1 1.1% AUC of the 87 formed. The reaction mixture was cooled to 30° – 35°C, filtered over Hyflo® (300 g) and washed with EtOAc (7.5 L) to remove the catalyst. The resulting filtrate was
concentrated to about 6 L and taken forward without further manipulation (67.8% AUC by HPLC, 5.5% AUC of the allylic alcohol impurity 86 and 13.0% AUC of 87).
Step lb/c – Oxidation of allylic alcohol 86 and 87 and rehydrogenation of 24 to methyl 3a-acetoxy-12-oxo-5fi-cholan-24-oate (33)
Step lb – PCC oxidation of allylic alcohol 86 and 87
A slurry of PCC (149.1 g, 1.03 equiv.) in EtOAc (1.5 L) was added to the 33 solution from above at 20°— 25°C. The reaction was allowed to proceed for 3.5 hours where HPLC analysis showed that < 1% AUC of the allylic alcohol 86 and < 1% AUC of 87 remained. The reaction mixture was filtered over Hyflo® (300 g) and washed with EtOAc (3.0 L). The EtOAc filtrate was washed with deionized (DI) water (2 x 3.6 L) and brine (3.6 L), filtered over Hyflo® (300 g) and washed with EtOAc (3.0 L). The resulting filtrate was concentrated to -7.5 L and taken forward without further manipulation (77.7% AUC by HPLC containing 5.3% AUC of 24).
Step lc— Rehydrogenation of 24 to 33
Powder activated carbon DARCO (60 g, 20 wt %) was added to the crude 33 solution from above containing 24. The resulting slurry was heated to 45°— 50°C for 4 hours, cooled to 30°— 35°C and filtered over Celite®. The filter cake was washed with EtOAc (7.5 L), concentrated to -7.5 L and added to dry Pd/C (60.0 g, 20 wt %). The reaction mixture was heated to 45° – 50°C and pressurized to 50 psi of H2 for 6 hours. HPLC analysis indicated < 1.0% AUC of 24 remained; 1.1% AUC of 86 impurity and < 1.0% AUC of 87 formed. The reaction was deemed complete and cooled to 30° – 35°C, filtered over Celite® and washed with EtOAc (7.5 L). The EtOAc filtrate was concentrated to—5 volumes and azeotroped with MeOH (2 x 4.5 L) back down to—5 volumes. The resulting slurry was diluted with DI water (2.4 L) and maintained at 20-25 °C. The slurry was filtered, washed with DI water (2 x 600 mL) and dried under vacuum at 40° – 50°C to yield 266 g (88%) of 33 (66.2% AUC by HPLC).
Step 2— Synthesis of 34
A solution of 33 (245 g, 0.5 mol) in THF (2.5 L) was cooled to 0° – 5°C and 1 M solution of Li(t-BuO)3A1H (822.9 niL, 1.5 equiv.) was added while maintaining the temperature below 5°C. The reaction mixture was stirred at 5° – 10°C for 22 hours. Reaction may be complete in 2-4 hours. HPLC analysis indicated that the reaction was complete with < 1% of 33 remaining. The reaction was quenched with 4 M HCl (3.7 L) while maintaining the temperature below 20°C. The reaction mixture was extracted with CH2CI2 (2 x 2.5 L) and the combined organic phases were washed with DI water (2 x 2.5 L). The CH2C12 phase was concentrated to afford 300 g (122%) of 34 (73.5% AUC by HPLC). 1H NMPv analysis indicated that 9.7 wt % of THF and 0.8 wt % of CH2C12 remained.
Step 3 – Synthesis of DCA
A NaOH solution (87.6 g, 4 equiv.) in DI water (438.6 mL) was added to a solution of 34 (245 g, 0.5 mol) in MeOH (980 mL) and THF (475 mL) at 0° – 5°C. The reaction mixture was allowed to warm to 20° – 25°C. HPLC analysis showed that the reaction was complete after 1 hour with < 0.5% 34 and < 0.5% of the hydrolysis intermediates remaining. The reaction was diluted with DI water (2.5 L) and
concentrated to—10 volumes. The aqueous solution was washed with CH2C12 (2 x 1.3 L) and adjusted to pH 1.7— 2.0 using 2 M HCl (1.6 L). A white slurry formed and was stirred at 20° – 25 °C for 1 hour. The slurry was filtered, washed with DI water (7 x 1 L) and dried under vacuum to yield 195 g (91%) of DCA (82.2% AUC by HPLC).
Step 4 – Purification of DCA
A solution of DCA obtained above (190 g, 0.48 mol) in MeOH (475 mL) and CH2C12 (4275 mL) was heated to 35° – 40°C. The MeOH/CH2Cl2 was distilled out of the mixture while CH2CI2 (4740 mL) was added matching the rate of distillation. Analysis of the solvent composition by Ή NMR indicated 4.5 mol % of MeOH remained relative to CH2C12. The slurry was allowed to cool to 20°— 25°C and held for 16 hours. The solids were isolated by filtration, washed with CH2Cl2 (600 mL) and dried under vacuum to yield 104 g (55%) of DCA (> 99% AUC by HPLC-RID and 98.7% AUC by HPLC- CAD).
The recrystallization was repeated by heating a mixture of DCA (103 g, 0.3 mol) in MeOH (359 mL) and CH2C12 (1751 mL) to 35° – 40°C. The MeOH/CH2Cl2 was distilled out of the mixture while CH2CI2 (3760 mL) was added matching the rate of distillation. Analysis of the solvent composition by 1H NMR indicated 4.7 mol % of MeOH remained relative to CH2C12. The slurry was allowed to cool to 20°— 25°C. After 1 hour, the solids were isolated by filtration, washed with CH2CI2 (309 mL) and dried under vacuum to afford 82 g (79%) of DCA (> 99% AUC by HPLC-RID and 99.3% AUC by HPLC-C AD).
To assess the effect of additional purification and reprocessing, the product was recrystallized a third time prior to the normal final water isolation step. The above sample of DCA (80 g, 0.2 mol) in MeOH (240 mL) and CH2C12 (1400 mL) was heated to 35° – 40°C. The MeOH/CH2Cl2 was distilled out of the mixture while CH2C12 (2000 mL) was added matching the rate of distillation. Analysis of the solvent composition by !H NMR indicated 6.7 mol % of MeOH remained relative to CH2C12. The slurry was allowed to cool to 20° – 25°C. After 1 hour, the solids were isolated by filtration, washed with CH2CI2 (240 mL) and dried under vacuum to afford 72 g (89%) of DCA (99.7% AUC by HPLC-CAD).
The sample was slurried in DI water (840 mL) and diluted with a solution of
NaOH (14.0 g) in DI water (140 mL). The resulting solution was filtered over Celite® and washed with DI water (1.4 L). The filtrate was adjusted to pH 1.6 with 2 M HCl (—300 mL) resulting in a white precipitate which was held for 1 hour at 20°— 25°C. The product was isolated by filtration, washed with DI water (9.0°L) and dried under vacuum to afford 63 g (87%) of DCA (99.7% AUC by HPLC-CAD).
SEE MORE IN PATENT
………………………………..
WO 2013044119
http://www.google.com/patents/WO2013044119A1?cl=en
![Figure imgf000019_0001]()
Scheme 10
Example 4: Converting Compound 129 To DCA
[0125| In Scheme 1 below, there is provided a scheme for the synthesis and purification of DCA from compound 1.
Scheme 10
A. Conversion of Compound 129 to Compound 130:
Method Al
[0126] 10% Pd/C (900 mg) was added to a solution of compound 129 (2.0 g, 4.5 mmol) in EtOAc (150 mL) and the resulting slurry was hydrogenated in a Parr apparatus (50 psi) at 50 °C for 16 h. At this point the reaction was determined to be complete by TLC. The mixture was filtered through a small plug of Celite® and the solvent was removed under vacuum, providing compound 130 (1.6 g, 80% yield) as a white solid.
TLC: -anisaldehyde charring, Rt for 130 = 0.36. TLC mobile phase: 20% – EtOAc in hexanes.
Ή NMR (500 MHz, CDCL): δ = 4.67-4.71 (m, 1 H), 3.66 (s, 3H), 2.45-2.50 (t, J = 15 Hz, 2H ), 2.22-2,40 (m, 1H), 2.01 (s, 3H). 1 ,69- 1 .96 (m, 9H), 1 ,55 (s, 4H), 1 ,25- 1.50 (m, 8H)5 1.07-1 . 19 (m. 2H), 1 .01 (s, 6H), 0.84-0.85 (d, J = 7.0 Hz, 3H).
13C NMR (125 MHz, CDC13): δ = 214.4, 174.5, 170.4, 73.6, 58,5, 57.4, 51.3, 46,4, 43.9, 41.2, 38.0, 35.6, 35.5, 35.2, 34.8, 32.0, 31 .2, 30.4, 27.4. 26.8, 26.2, 25.9, 24.2, 22.6, 21 .2, 18.5, 1 1.6,.
Mass (m/z) = 447.0 | \! + 1 ], 464.0 [Mf + 18]. IR ( Br) = 3445, 2953, 2868, 1731 , 1698, 1257, 1029 cm“1 , m.p. = 142,2- 144.4 °C (from EtOAc/hexanes mixture). [a]D = +92 (c = l % in CHCl3).
ELSD Purity: 96.6%: Retention time = 9.93 (Inertsil ODS 3V, 250 * 4.6 mm, 5 urn, ACN: 0.1 % TFA in water (90: 10)
Method A2
[0127J A slurry of 10% Pd/C (9 g in 180 mL of ethyl acetate) was added to a solution of compound 129 (36 g, 81 mmol) in EtOAc (720 mL) and the resulting slurry was treated with hydrogen gas (50 psi) at 45-50 °C for 16 h. (A total of 1080 mL of solvent may be used). At this point the reaction was determined to be complete by HPLC (NMT 1 % of compound 129). The mixture was filtered through Cclite® (10 g) and washed with ethyl acetate (900 mL). The filtrate was concentrated to 50% of its volume via vacuum distillation below 50 °C. To the concentrated solution was added pyridinium
chlorochromate (20.8 g) at 25-35 °C and the mixture was stirred for 2 h at 25-35 °C, when the reaction completed by LIPLC (allylic alcohol content is NMT 1 %).
[0128] The following process can be conducted if compound 129 content is more than 5%. Filter the reaction mass through Celite® (10 g) and wash with ethyl acetate (360 mL). Wash the filtrate with water (3 x 460 mL) and then with saturated brine (360 mL). Dry the organic phase over sodium sulphate (180 g), filter and wash with ethyl acetate ( 180 mL). Concentrate the volume by 50% via vacuum distillation below 50 °C. Transfer the solution to a clean and dry autoclave. Add slurry of 10% palladium on carbon (9 g in 1 80 mL of ethyl acetate). Pressurize to 50 psi with hydrogen and stir the reaction mixture at 45-50 °C for 16 h.
[0129] Upon complete consumption of compound 129 by HPLC ( the content of compound 129 being NMT 1 %), the reaction mixture was filtered through Celite® ( 10 g) and the cake was washed with ethyl acetate (900 mL). The solvent was concentrated to dryness via vacuum distillation below 50 °C. Methanol (150 mL) was added and concentrated to dryness via vacuum distillation below 50 °C. Methanol (72 mL) was added to the residue and the mixture was stirred for 15-20 min at 10- 15 °C, filtered and the cake was washed with methanol (36 mL). The white solid was dried in a hot air drier at 45-50 °C for 8 h to LOD being NMT 1% to provide compound 230 (30 g, 83.1 % yield).
B. Conversion of Compound 130 to Compound 1 1.a
Method Bl
[0130J A THF solution of lithium tri-te -butoxyaluminum hydride (1 M. 22.4 mL, 22.4 mmol) was added drop wise to a solution of compound 130 (2.5 g, 5.6 mmol) in THF (25 mL) at ambient temperature. After stirring for an additional 4-5 h, the reaction was determined to be complete by TLC. The reaction was quenched by adding aqueous HQ (1 M, 10 mL) and the mixture was diluted with EtOAc (30 mL). The phases were separated and the organic phase was washed sequentially with water (15 mL) and saturated brine solution (10 mL). The organic phase was then dried over anhydrous Na2SO-i (3 g) and filtered. The filtrate was concentrated under vacuum and the resulting solid was purified by column chromatography [29 mm (W) x 500 mm (L), 60-120 mesh silica, 50 g], eluting with EtOAc/hexane (2:8) [5 mL fractions, monitored by TLC with p- anisaldehyde charring]. The fractions containing the product were combined and concentrated under vacuum to provide compound 131. a (2.3 g, 91 %) as a white solid.
TLC: /7-anisaldehyde charring, Rf for 131. a = 0.45 and Rt for 130 = 0.55. TLC mobile phase: 30% – EtOAc in hexanes.
Ή NMR (500 MHz, CDC13): δ = 4.68-4.73 (m, 1 H), 3.98 (s, 1 H), 3.66 (s, 3H), 2.34-2.40 (m, 1H), 2.21-2.26 (m, 1H), 2.01 (s, 3H), 1.75-1.89 (m, 6H), 1.39-1.68 (m, 16H), 1.00-1.38 (m, 3H), 0.96-0.97 (d, J = 5.5 Hz, 3H), 0.93 (s, 3H), 0.68 (s, 3H).
13C NMR (125 MHz, CDCI3): δ = 174.5, 170.5, 74.1 , 72.9, 51.3, 48.1 , 47.2, 46.4, 41.7, 35.8, 34.9, 34.7, 34.0, 33.5, 32.0, 30.9, 30.8, 28.6, 27.3, 26.8, 26.3, 25.9, 23.4. 22.9, 21.3. 17.2, 12.6
Mass (m/z) = 449.0 [M+ + 1 ], 466.0 [M+ + 18].
IR (KBr) = 3621 , 2938, 2866, 1742, 1730, 1262, 1 162, 1041 , cm4. m.p = 104.2-107.7 °C (from EtOAc).
[<x]D = +56 (c = 1% in CHCI3). ELSD Purity: 97.0%: Retention time = 12.75 (Inertsil ODS 3V, 250 χ 4.6 mm, 5 urn, ACN: Water (60:40)
Method B2
[0131 ] A THF solution of lithium tri-/er?-butoxyaluminum hydride (1 M, 107.6 mL, 107.6 mmol) was added over 1 h to a solution of compound 130 (30.0 g, 67 mmol) in dry THF (300 mL) at 0-5 °C. After stirring for an additional 4 h at 5-10 °C, the reaction was determined to be complete by HPLC (NMT 1% of compound 130). The reaction was cooled to 0-5 °C and quenched by adding 4N HC1 (473 mL). The phases were separated. The aqueous layer was extracted with DCM (2 x 225 mL) and the combined organic phase was washed sequentially with water (300 mL) and saturated brine solution (300 mL). The organic phase was then was concentrated to dryness by vacuum distillation below 50 °C. Methanol (150 mL) was added to the residue and concentrated to dryness by vacuum distillation below 50 °C. Water (450 mL) was then added to the residue and the mixture was stirred for 15-20 min., filtered and the cake was washed with water (240 mL). The white solid was dried in a hot air drier at 35-40 °C for 6 h to provide compound 131.a (30 g, 99.6%).
C. Conversion of Compound 131.a to crude DCA:
[01321 To a solution of 131. a in MeOH (4 vol) and THF (4 vol) was added a solution of NaOH (4.0 equiv) in DI water (5 M) maintaining the temperature below 20 °C. HPLC analysis after 20 hours at 20-25 °C indicated <0.5% AUC of 131.a and the two
intermediates remained. The reaction was deemed complete, diluted with DI water (10 vol) and concentrated to -10 volumes. The sample was azeotroped with 2-MeTHF (2 x 10 vol) and assayed by Ή NMR to indicate MeOH was no longer present. The rich aqueous phase was washed with 2-MeTHF (2 x 10 vol) and assayed by HPLC to indicate 0.3% AUC of the alcohol impurity remained. The aqueous phase was diluted with 2- MeTHF (10 vol ) and adjusted to pH = 1 .7-2.0 using 2 M HC1 (~4 vol ). The phases were separated and the 2-MeTHF phase was washed with DI water (2 x 10 vol). The 2- MeTHF phase was filtered over Celite and the filter cake was washed with 2-MeTHF (2 vol). The 2-MeTHF filtrate was distillated to -10 volumes and azeotroped with ^-heptane containing Statsafe™ 5000 (3 x 10 vol) down to -10 vol. The mixture was assayed by Ή N MR to indicate <5 mol% of 2-MeTHF remained relative to o-heptane. The slurry was held for a minimum of 2 hours at 20-25 °C and filtered. The filter cake was washed with //-heptane (2 x 10 vol) and conditioned under vacuum on the Niitsche filter with N2 for a minimum of 1 hour to afford DCA-crude as white solids. Purity = 94.6% (by HPLC). HPLC analysis for DS-DCA (NMT 5% AUC).
D. Recrystallization of DCA
|0133] DCA-crude was diluted with 2 mol% MeOH in CH2C12 (25 vol) and heated to 35—37 °C for 1 hour. The slurry was allowed to cool to 28-30 °C and filtered. The filter cake was washed with CITC (5 vol) and dried under vacuum at 40 °C to afford DCA. HPLC analysis for DS-DCA (NMT 0.15% AUC).
[0134] DCA was dissolved in 10% DI water/ EtOH (12 vol), polish filtered over Celite and washed with 10% DI water/ EtOH (3 vol). The resulting 15 volume filtrate was added to DI water (30 vol) and a thin white slurry was afforded. The slurry was held for 24 hours, filtered, washed with DI water (20 vol) and dried under vacuum at 40 °C to afford pure DCA. OVI analysis for CH2C12. EtOH. ^-heptane, MeOH and MeTHF was conducted to ensure each solvent was below ICH guideline. KF analysis conducted (NMT 2.0%). Purity = 99.75% (by HPLC). Yield from DCA-crude = 59%.
……………………………
WO 2012174229
http://www.google.com/patents/WO2012174229A2?cl=en
In Scheme 1 below, there is provided a scheme for the synthesis and purification of deoxycholic acid from compound 1.
Scheme 1
Conversion of Compound 1 to Compound 2:
[0043] The hydrogenation of compound 1 on 10.0 g scale using dry 10 % Pd/C (15 wt %) in ethyl acetate (20 parts) was added and applied about 50 psi hydrogen pressure and temperature raised to 70 °C. After reaching temperature 70 °C, observed increase of hydrogen pressure to about 60 psi, at these conditions maintained for 60 h. After 60 hours 0.6% of compound 2 and 2.75%> of allylic alcohol were still observed, so further stirred for additional 12 h (observed 0.16% of allylic alcohol and 0.05% of compound 2). After work-up, the reaction provided 9.5 g of residue.
[0044] Anther hydrogenation reaction on 25 g of compound 1 with above conditions for 76 h provided 24.5 g of residue.
Method A
[0045] 10% Pd/C (900 mg) was added to a solution of compound 1 (2.0 g, 4.5 mmol) in EtOAc (150 mL) and the resulting slurry was hydrogenated in a Parr apparatus (50 psi) at 50 °C for 16 h. At this point the reaction was determined to be complete by TLC. The mixture was filtered through a small plug of Celite® and the solvent was removed under vacuum, providing compound 2 (1.6 g, 80%> yield) as a white solid.
TLC: /?-anisaldehyde charring, Rf for 2 TLC mobile phase: 20% – EtOAc in hexanes.
1H NMR (500 MHz, CDC13): δ = 4.67-4.71 (m, 1H), 3.66 (s, 3H), 2.45-2.50 (t, J = 15 Hz, 2H), 2.22-2.40 (m, 1H), 2.01 (s, 3H), 1.69-1.96 (m, 9H), 1.55 (s, 4H), 1.25-1.50 (m, 8H), 1.07-1.19 (m, 2H), 1.01 (s, 6H), 0.84-0.85 (d, J= 7.0 Hz, 3H).
13C NMR (125 MHz, CDC13): δ = 214.4, 174.5, 170.4, 73.6, 58.5, 57.4, 51.3, 46.4, 43.9, 41.2, 38.0, 35.6, 35.5, 35.2, 34.8, 32.0, 31.2, 30.4, 27.4, 26.8, 26.2, 25.9, 24.2, 22.6, 21.2, 18.5,11.6,.
Mass (m/z) = 447.0 [M+ + 1], 464.0 [M+ + 18].
IR (KBr) = 3445, 2953, 2868, 1731, 1698, 1257, 1029 cm“1.
m.p. =142.2-144.4 °C (from EtO Ac/hex anes mixture).
[a]D = +92 (c = 1% in CHC13).
ELSD Purity: 96.6%: Retention time = 9.93 (Inertsil ODS 3V, 250 4.6 mm, 5 urn, ACN: 0.1% TFA in water (90: 10)
Method B
[0046] A slurry of 10%> Pd/C (9 g in 180 mL of ethyl acetate) was added to a solution of compound 1 (36 g, 81 mmol) in EtO Ac (720 mL) and the resulting slurry was treated with hydrogen gas (50 psi) at 45-50 °C for 16 h. (A total of 1080 mL of solvent may be used). At this point the reaction was determined to be complete by HPLC (NMT 1% of compound 1). The mixture was filtered through C elite® (10 g) and washed with ethyl acetate (900 mL). The filtrate was concentrated to 50% of its volume via vacuum distillation below 50 °C. To the concentrated solution was added pyridinium
chlorochromate (20.8 g) at 25-35 °C and the mixture was stirred for 2 h at 25-35 °C, when the reaction completed by HPLC (allylic alcohol content is NMT 1%).
[0047] The following process can be conducted if compound 1 content is more than 5%>. Filter the reaction mass through Celite® (10 g) and wash with ethyl acetate (360 mL). Wash the filtrate with water (3 x 460 mL) and then with saturated brine (360 mL). Dry the organic phase over sodium sulphate (180 g), filter and wash with ethyl acetate (180 mL). Concentrate the volume by 50% via vacuum distillation below 50 °C. Transfer the solution to a clean and dry autoclave. Add slurry of 10% palladium on carbon (9 g in 180 mL of ethyl acetate). Pressurize to 50 psi with hydrogen and stir the reaction mixture at 45-50 °C for 16 h.
[0048] Upon complete consumption of compound 1 by HPLC (the content of compound 1 being NMT 1%), the reaction mixture was filtered through Celite® (10 g) and the cake was washed with ethyl acetate (900 mL). The solvent was concentrated to dryness via vacuum distillation below 50 °C. Methanol (150 mL) was added and concentrated to dryness via vacuum distillation below 50 °C. Methanol (72 mL) was added to the residue and the mixture was stirred for 15-20 min at 10-15 °C, filtered and the cake was washed with methanol (36 mL). The white solid was dried in a hot air drier at 45-50 °C for 8 h to LOD being NMT 1% to provide compound 2 (30 g, 83.1 % yield).
Conversion of Compound 2 to Compound 3:
Method A
[0049] A THF solution of lithium tri-tert-butoxyaluminum hydride (1 M, 22.4 mL, 22.4 mmol) was added drop wise to a solution of compound 2 (2.5 g, 5.6 mmol) in THF (25 mL) at ambient temperature. After stirring for an additional 4-5 h, the reaction was determined to be complete by TLC. The reaction was quenched by adding aqueous HCl (1 M, 10 mL) and the mixture was diluted with EtO Ac (30 mL). The phases were separated and the organic phase was washed sequentially with water (15 mL) and saturated brine solution (10 mL). The organic phase was then dried over anhydrous Na2S04 (3 g) and filtered. The filtrate was concentrated under vacuum and the resulting solid was purified by column chromatography [29 mm (W) x 500 mm (L), 60-120 mesh silica, 50 g], eluting with EtO Ac/hex ane (2:8) [5 mL fractions, monitored by TLC with p- anisaldehyde charring]. The fractions containing the product were combined and concentrated under vacuum to provide compound 3 (2.3 g, 91%) as a white solid.
TLC: /?-anisaldehyde charring, Rf for 3 = 0.45 and Rf for 2 = 0.55.
TLC mobile phase: 30% – EtO Ac in hexanes.
1H NMR (500 MHz, CDC13): δ = 4.68-4.73 (m, 1H), 3.98 (s, 1H), 3.66 (s, 3H), 2.34-2.40 (m, 1H), 2.21-2.26 (m, 1H), 2.01 (s, 3H), 1.75-1.89 (m, 6H), 1.39-1.68 (m, 16H), 1.00-1.38 (m, 3H), 0.96-0.97 (d, J= 5.5 Hz, 3H), 0.93 (s, 3H), 0.68 (s, 3H). ljC NMR (125 MHz, CDC13): δ = 174.5, 170.5, 74.1, 72.9, 51.3, 48.1, 47.2, 46.4, 41.7, 35.8, 34.9, 34.7, 34.0, 33.5, 32.0, 30.9, 30.8, 28.6, 27.3, 26.8, 26.3, 25.9, 23.4, 22.9, 21.3, 17.2, 12.6
Mass (m/z) = 449.0 [M+ + 1], 466.0 [M+ + 18].
IR (KBr) = 3621, 2938, 2866, 1742, 1730, 1262, 1162, 1041, cm“1.
m.p = 104.2-107.7 °C (from EtOAc).
[a]D = +56 (c = 1% in CHC13).
ELSD Purity: 97.0%: Retention time = 12.75 (Inertsil ODS 3V, 250 4.6 mm, 5 urn, ACN: Water (60:40)
Method B
[0050] A THF solution of lithium tri-tert-butoxyaluminum hydride (1 M, 107.6 mL, 107.6 mmol) was added over 1 h to a solution of compound 2 (30.0 g, 67 mmol) in dry THF (300 mL) at 0-5 °C. After stirring for an additional 4 h at 5-10 °C, the reaction was determined to be complete by HPLC (NMT 1% of compound 2). The reaction was cooled to 0-5 °C and quenched by adding 4N HC1 (473 mL). The phases were separated. The aqueous layer was extracted with DCM (2 x 225 mL) and the combined organic phase was washed sequentially with water (300 mL) and saturated brine solution (300 mL). The organic phase was then was concentrated to dryness by vacuum distillation below 50 °C. Methanol (150 mL) was added to the residue and concentrated to dryness by vacuum distillation below 50 °C. Water (450 mL) was then added to the residue and the mixture was stirred for 15-20 min., filtered and the cake was washed with water (240 mL). The white solid was dried in a hot air drier at 35-40 °C for 6 h to provide compound 3 (30 g, 99.6%).
Conversion of Compound 3 to crude DCA:
[0051] To a solution of 3 in MeOH (4 vol) and THF (4 vol) was added a solution of NaOH (4.0 equiv) in DI water (5 M) maintaining the temperature below 20 °C. HPLC analysis after 20 hours at 20-25 °C indicated <0.5% AUC of 3 and the two intermediates remained. The reaction was deemed complete, diluted with DI water (10 vol) and concentrated to ~10 volumes. The sample was azeotroped with 2-MeTHF (2 x 10 vol) and assayed by 1H NMR to indicate MeOH was no longer present. The rich aqueous phase was washed with 2-MeTHF (2 x 10 vol) and assayed by HPLC to indicate 0.3% AUC of the alcohol impurity remained. The aqueous phase was diluted with 2-MeTHF (10 vol) and adjusted to pH = 1.7-2.0 using 2 M HC1 (~4 vol). The phases were separated and the 2-MeTHF phase was washed with DI water (2 x 10 vol). The 2- MeTHF phase was filtered over Celite and the filter cake was washed with 2-MeTHF (2 vol). The 2-MeTHF filtrate was distillated to ~10 volumes and azeotroped with n-heptane containing Statsafe™ 5000 (3 x 10 vol) down to ~10 vol. The mixture was assayed by 1H NMR to indicate <5 mol% of 2-MeTHF remained relative to n-heptane. The slurry was held for a minimum of 2 hours at 20-25 °C and filtered. The filter cake was washed with n-heptane (2 x 10 vol) and conditioned under vacuum on the Nutsche filter with N2 for a minimum of 1 hour to afford DCA-crude as white solids. Purity = 94.6% (by HPLC). HPLC analysis for DS-DCA (NMT 5% AUC).
Recrystallization of Deoxycholic acid (DCA)
[0052] DCA-crude was diluted with 2 mol% MeOH in CH2C12 (25 vol) and heated to 35-37 °C for 1 hour. The slurry was allowed to cool to 28-30 °C and filtered. The filter cake was washed with CH2C12 (5 vol) and dried under vacuum at 40 °C to afford DCA. HPLC analysis for DS-DCA (NMT 0.15% AUC).
[0053] DCA was dissolved in 10% DI water/ EtOH ( 12 vol), polish filtered over Celite and washed with 10% DI water/ EtOH (3 vol). The resulting 15 volume filtrate was added to DI water (30 vol) and a thin white slurry was afforded. The slurry was held for 24 hours, filtered, washed with DI water (20 vol) and dried under vacuum at 40 °C to afford pure DCA. OVI analysis for CH2C12, EtOH, n-heptane, MeOH and MeTHF was conducted to ensure each solvent was below ICH guideline. KF analysis conducted (NMT 2.0%). Purity = 99.75% (by HPLC). Yield from DCA-crude = 59%.
……………………………..
| WO2011075701A2 * |
Dec 17, 2010 |
Jun 23, 2011 |
Kythera Biopharmaceuticals, Inc. |
Methods for the purification of deoxycholic acid |
| EP0336521B1 * |
Apr 7, 1989 |
Apr 1, 1992 |
Roussel-Uclaf |
9-alpha-hydroxy-17-methylene steroids, process for their preparation and their use in the preparation of corticosteroids |
| US20100179337 * |
May 16, 2008 |
Jul 15, 2010 |
Kythera Biopharmaceuticals, Inc. |
Preparation of bile acids and intermediates thereof |
old cut paste
![]()
http://clinicaltrials.gov/ct2/show/NCT01426373
The drug is sodium deoxycholate for injection, code-named ATX-101 was developed for the treatment of lipomas – benign tumors of subcutaneous adipose tissue, as well as other unwanted fatty growths, such as a double chin. This substance, which is a salt of one of the bile acids, emulsifies fats, destroying their excess deposits
![]()
![]()
ATX-101 (a first-in-class injectable drug being studied for the reduction of localized fat. ATX-101 is a proprietary formulation of deoxycholate a well-studied endogenous compound that is present in the body), a facial injectable drug for the reduction of unwanted fat under the chin, or submental fat. V. Leroy Young, MD, FACS, presented the initial results at the American Society for Aesthetic Plastic Surgery (ASAPS) 45th Annual Aesthetic Meeting in Vancouver, British Columbia, on May 4, 2012.
In August 2010 Bayer Consumer Care AG signed a licensing and development collaboration agreement with KYTHERA, thereby obtaining commercialization rights to ATX-101 outside the US and Canada. KYTHERA and Bayer are collaborating on the development of ATX-101 in Europe.
KYTHERA Biopharmaceuticals Inc. 02 MAR 3013, announced positive interim results from a Phase IIIb multi-center open-label study (ATX-101-11-26) to evaluate the safety and efficacy of ATX-101 an investigational injectable drug for the reduction of unwanted submental fat (SMF) commonly known as double chin. The results presented at the Late Breaking Research Symposium at the 71st American Academy of Dermatology (AAD) Annual Meeting in Miami Beach Fla. found that ATX-101 is well-tolerated and may be effective in reducing SMF by both clinician and patient reported outcome measures. The ATX-101 global clinical development program has enrolled more than 2500 total patients of which more than 1500 have been treated with ATX-101.
“In my practice patients often request a non-surgical way to treat their submental fat or undesirable double chin” said investigator Susan Weinkle MD FAAD a board certified dermatologist and affiliate clinical professor at the University of South Florida. “For these patients double chin is often resistant to diet and exercise. The results of this study suggest that microinjections of ATX-101 can reduce submental fat without worsening skin laxity.”
ATX-101 is a proprietary synthetically-derived formulation of deoxycholic acid (DCA) a naturally-occurring molecule found in the body that aids in fat metabolism. In this open-label Phase IIIb study interim results three months after the last ATX-101 treatment found:
- Reduction of submental fat
- 87 percent of patients achieved at least a one-grade improvement from baseline on the Clinician-Reported Submental Fat Rating Scale (CR-SMFRS)
- Similarly 83 percent of patients achieved at least a one-grade improvement on the Patient-Reported Submental Fat Rating Scale (PR-SMFRS)
- 96 percent of patients had unchanged or improved skin laxity based on the clinician rated Submental Skin Laxity Grading Scale (SMSLG)
- 95 percent of patients were satisfied with treatment based on the Global Post Treatment Satisfaction Scale
- Adverse events were of mild to moderate intensity transient and primarily associated with the treatment area
![]()
Topline results from this study were announced in November 2012. As previously announced 71.3 percent of subjects had at least a one-grade improvement on the CR-SMFRS / PR-SMFRS composite and 14.0 percent had at least a two-grade improvement on the same composite measure.
These results are based on a multicenter 12-month open-label Phase IIIb study conducted at 21 sites across the United States evaluating 165 adults who received injections of ATX-101 for up to six treatments at four-week intervals. Patients received ATX-101 (2 mg/cm2) by subcutaneous microinjections directly into their SMF and were evaluated three months after their last treatment. The study population includes females (77.6 percent) and males (22.4 percent) with a mean age of 47 who report at least moderate SMF and dissatisfaction with the appearance of their chin. All Fitzpatrick Skin Types an industry standard scale to categorize skin tone are represented.
“We are pleased with these ATX-101 study results” said Patricia S. Walker M.D. Ph.D. chief medical officer KYTHERA Biopharmaceuticals Inc. “These results along with efficacy analyses in double-blind placebo-controlled studies support ATX-101 entering the market as potentially the first medical aesthetic drug approved for the reduction of submental fat.”
About ATX-101
ATX-101 is a potential first-in-class injectable drug candidate under clinical investigation for the reduction of unwanted submental fat. ATX-101 is a proprietary formulation of synthetic deoxycholic acid a well-characterized endogenous compound that is present in the body to promote the natural breakdown of dietary fat. ATX-101 is designed to be a locally-injected drug that causes proximal preferential destruction of adipocytes or fat cells with minimal effect on surrounding tissue. Based on clinical trials conducted to date ATX-101 has exhibited significant meaningful and durable results in the reduction of submental fat which commonly presents as an undesirable “double chin.” These results correspond with subject satisfaction measures demonstrating meaningful improvement in perceived chin appearance.
In August 2010 Bayer signed a licensing and collaboration development agreement with KYTHERA thereby obtaining development and commercialization rights to ATX-101 outside of the U.S. and Canada. Bayer recently completed two pivotal Phase III trials of ATX-101 in Europe for the reduction of submental fat. Topline results from these trials were reported in the second quarter of 2012. KYTHERA completed enrollment in its pivotal Phase III clinical program for ATX-101 in more than 1000 subjects randomized to ATX-101 or placebo in 70 centers across the United States and Canada in August 2012. The Company expects to release topline results in mid-2013.
About KYTHERA Biopharmaceuticals Inc.
KYTHERA Biopharmaceuticals Inc. is a clinical-stage biopharmaceutical company focused on the discovery development and commercialization of novel prescription products for the aesthetic medicine market. KYTHERA initiated its pivotal Phase III clinical program for ATX-101 in March 2012 and completed enrollment of more than 1000 patients randomized to ATX-101 or placebo in 70 centers across the U.S. and Canada in August 2012. KYTHERA also maintains an active research interest in hair and fat biology. Find more information at www.kytherabiopharma.com.
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
amcrasto@gmail.com
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
ABOUTME,
BRAND ANTHONYCRASTO,
COLLECTION OF SITE LINKS,
BRAVESITES,
GRAVATAR,
JIMDO,
SKILLPAGES, MIXXT, APNACIRCLE, ZIC ZAC, ZING ME, SCOOP IT, scribd, Stumbleupon, Delicious, pininterest, tumblr, Newsvine, SLIDESHARE, ACADEMIA.EDU, GOOGLE PLUS,
FACEBOOK,
ISSUU,
DIIGO
アンソニー 安东尼 Энтони 안토니 أنتوني
blogs are
![]()
MY CHINA, VIETNAM AND JAPAN BLOGS
http://me.zing.vn/u/amcrasto
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX,
![]()
Filed under:
NDA,
Uncategorized Tagged:
ATX-101,
Kythera's,
NSC 8797,
NSC-681065 ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()




























 Originally posted on
Originally posted on 











































































 Originally posted on
Originally posted on 































 Originally posted on
Originally posted on 














































































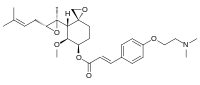
 Beloranib is an experimental drug candidate for the treatment of
Beloranib is an experimental drug candidate for the treatment of 

















