Image may be NSFW. Clik here to view.Originally posted on lyranara.me:
Image may be NSFW. Clik here to view.
In this artist’s representation of the adult subependymal neurogenic niche (viewed from underneath the ependyma), electrical signals generated by the ChAT+ neuron give rise to newborn migrating neuroblasts, seen moving over the underside of ependymal cells. Credit: O’Reilly Science Art
Duke researchers have found a new type of neuron in the adult brain that is capable of telling stem cells to make more new neurons. Though the experiments are in their early stages, the finding opens the tantalizing possibility that the brain may be able to repair itself from within.
Neuroscientists have suspected for some time that the brain has some capacity to direct the manufacturing of new neurons, but it was difficult to determine where these instructions are coming from, explains Chay Kuo, M.D. Ph.D., an assistant professor of cell biology, neurobiology and pediatrics.
In a study with mice, his team found a previously unknown population of neurons within…
Image may be NSFW. Clik here to view.Originally posted on lyranara.me:
A team of University of South Carolina researchers led by Mitzi Nagarkatti, Prakash Nagarkatti and Xiaoming Yang have discovered a novel pathway through which marijuana can suppress the body’s immune functions. Their research has been published online in the Journal of Biological Chemistry.
Marijuana is the most frequently used illicit drug in the United States, but as more states legalize the drug for medical and even recreational purposes, research studies like this one are discovering new and innovative potential health applications for the federal Schedule I drug.
Marijuana is now regularly and successfully used to alleviate the nausea and vomiting many cancer patients experience as side effects to chemotherapy, combat the wasting syndrome that causes some AIDS patients to lose significant amounts of weight and muscle mass and ease chronic pain that is unresponsive to opioids, among other applications.
The university study has uncovered yet another potential application for
Image may be NSFW. Clik here to view.Originally posted on lyranara.me:
Image may be NSFW. Clik here to view.
Toll-like receptor 2 is normally localized to the cell membrane (green outlines, left panel). However, a KSHV protein affects the normal distribution (diffuse green, right panel). Endoplasmic reticulum is shown in red.
Pathogens entering our body only remain unnoticed for a short period. Within minutes our immune cells detect the invader and trigger an immune response. However, some viruses have developed strategies to avoid detection and elimination by our immune system. Researchers from the Helmholtz Centre for Infection Research (HZI) in Braunschweig have now been able to show how the herpesviruses achieve this.
The Kaposi’s sarcoma-associated herpesvirus (KSHV), a gammaherpesvirus that can cause multiple forms of cancer, establishes lifelong infections within the body. To do so the virus has to find a way to modulate the immune system of its host.
“Intruders are usually fought off immediately by an antiviral immune response that is triggered by sensors including the toll-like…
Image may be NSFW. Clik here to view.Originally posted on lyranara.me:
There are a number of reasons to avoid taking aspirin or ibuprofen often. You may be seeking alternatives because you experience pain but like to stay away from conventional medicines. Alternatively, you could be someone who just learned about the potential dangers that come with taking aspirin and ibuprofen regularly and are ready for something different. Ibuprofen and Aspirin have been linked to anemia, DNA damage, heart disease, hearing loss, hypertension, miscarriage and even influenza mortality (these are just 7 of the over 24 adverse health effects its been connected with.)
“Long-term high-dose use of painkillers such as ibuprofen or diclofenac is ‘equally hazardous’ in terms of heart attack risk as use of the drug Vioxx, which was withdrawn due to its potential dangers.” – Reuters
Although there are a number of adverse health effects that go along with Ibuprofen, we continue to take it in vast amounts because we…
Image may be NSFW. Clik here to view.Originally posted on lyranara.me:
Image may be NSFW. Clik here to view.
Capsule endoscopes with built-in cameras for imaging the GI tract are not new anymore, providing the ability to scan areas otherwise difficult to access and potentially replacing traditional upper and lower endoscopies with a less invasive, more dignified approach. Yet, they still require bowel prep cleansing and are limited to the visual spectrum. A new electronic pill from Check-Cap(Isfiya, Israel) has an X-ray source that can provide 3D visualization of GI tract. It’s currently an investigational device, but a study, recently presented at theDigestive Disease Week annual meeting in Chicago, showed safety and feasibility of colon imaging using the pill without bowel cleansing.
It’s 30 mm in length and 11 mm in diameter and delivers a radiation dose similar to a dental X-ray. Moreover, unlike virtual CT colonoscopy, the radiation from the pill doesn’t have to penetrate the rest of the body to get to the colon, and…
Image may be NSFW. Clik here to view.Originally posted on lyranara.me:
Image may be NSFW. Clik here to view.
As everyone knows, in life often the whole is more than the sum of its parts. Sometimes, by bringing already available technologies together, a brand new capability presents itself. In cancer research, drugs, X-rays, lasers, and gold nanoparticles have all been used in different ways, often complementing each other to improve the effectiveness of a treatment. Now researchers at Rice University have developed a new approach that combines all four technologies to effectively kill aggressive cancer cells by literally exploding them. They dubbed the technology quadrapeutics, which significantly amplifies the killing effect of anti-cancer drugs and chemo, but only in cancer cells.
The technique harnesses plasmonic nanobubbles, tiny droplets of vapor that form around plasmonic gold nanoparticles, which can then pop and try to destroy the cell from within. If it doesn’t, the explosion causes the delivered drug to be spread through the cells and the effect of the chemotherapy also…
Image may be NSFW. Clik here to view.Originally posted on lyranara.me:
Levels of a small molecule found only in humans and in other primates are lower in the brains of depressed individuals, according to researchers at McGill University and the Douglas Institute. This discovery may hold a key to improving treatment options for those who suffer from depression.
Depression is a common cause of disability, and while viable medications exist to treat it, finding the right medication for individual patients often amounts to trial and error for the physician. In a new study published in the journal Nature Medicine, Dr. Gustavo Turecki, a psychiatrist at the Douglas and professor in the Faculty of Medicine, Department of Psychiatry at McGill, together with his team, discovered that the levels of a tiny molecule, miR-1202, may provide a marker for depression and help detect individuals who are likely to respond to antidepressant treatment.
“Using samples from the Douglas Bell-Canada Brain Bank…
Image may be NSFW. Clik here to view.Originally posted on lyranara.me:
A team of University of South Carolina researchers led by Mitzi Nagarkatti, Prakash Nagarkatti and Xiaoming Yang have discovered a novel pathway through which marijuana can suppress the body’s immune functions. Their research has been published online in the Journal of Biological Chemistry.
Marijuana is the most frequently used illicit drug in the United States, but as more states legalize the drug for medical and even recreational purposes, research studies like this one are discovering new and innovative potential health applications for the federal Schedule I drug.
Marijuana is now regularly and successfully used to alleviate the nausea and vomiting many cancer patients experience as side effects to chemotherapy, combat the wasting syndrome that causes some AIDS patients to lose significant amounts of weight and muscle mass and ease chronic pain that is unresponsive to opioids, among other applications.
The university study has uncovered yet another potential application for
ScinoPharm to Provide Active Pharmaceutical Ingredient to F*TaiGen for Novel Stem Cell Drug
MarketWatch
The drug has received a Clinical Trial Application from China’s FDA for the initiation of … In addition, six products have entered Phase III clinical trials.
TAINAN, June 8, 2014 — ScinoPharm Taiwan, Ltd. (twse:1789) specializing in the development and manufacture of active pharmaceutical ingredients, and TaiGen Biotechnology (4157.TW; F*TaiGen) jointly announced today the signing of a manufacturing contract for the clinical supply of the API of Burixafor, a new chemical entity discovered and developed by TaiGen. The API will be manufactured in ScinoPharm’s plant in Changshu, China. This cooperation not only demonstrates Taiwan’s international competitive strength in new drug development, but also sees the beginning of a domestic pharmaceutical specialization and cooperation mechanisms, thus establishing a groundbreaking milestone for Taiwan’s pharmaceutical industry.
Dr. Jo Shen, President and CEO of ScinoPharm said, “This cooperation with TaiGen is of representative significance in the domestic pharmaceutical companies’ upstream and downstream cooperation and self-development of new drugs, and indicates the Taiwanese pharmaceutical industry’s cumulative research and development momentum is paving the way forward.” Dr. Jo Shen emphasized, “ScinoPharm’s Changshu Plant provides high-quality API R&D and manufacturing services through its fast, flexible, reliable competitive advantages, effectively assisting clients of new drugs in gaining entry into China, Europe, the United States, and other international markets.”
According to Dr. Ming-Chu Hsu, Chairman and CEO of TaiGen, “R&D is the foundation of the pharmaceutical industry. Once a drug is successfully developed, players at all levels of the value chain could reap the benefit. Burixafor is a 100% in-house developed product that can be used in the treatment of various intractable diseases. The cooperation between TaiGen and ScinoPharm will not only be a win-win for both sides, but will also provide high-quality novel dug for patients from around the world.”
Burixafor is a novel stem cell mobilizer that can efficiently mobilize bone marrow stem cells and tissue precursor cells to the peripheral blood. It can be used in hematopoietic stem cell transplantation, chemotherapy sensitization and other ischemic diseases. The results of the ongoing Phase II clinical trial in the United States are very impressive. The drug has received a Clinical Trial Application from China’s FDA for the initiation of a Phase II clinical trial in chemotherapy sensitization under the 1.1 category. According to the pharmaceutical consultancy company JSB, with only stem cell transplant and chemotherapy sensitizer as the indicator, Burixafor’s annual sales are estimated at USD1.1 billion.
ScinoPharm currently has accepted over 80 new drug API process research and development plans, of which five new drugs have been launched in the market. In addition, six products have entered Phase III clinical trials. Through the Changshu Plant’s operation in line with the latest international cGMP plant equipment and quality management standards, the company provides customers with one stop shopping services in professional R&D, manufacturing, and outsourcing, thereby shortening the customer development cycle of customers’ products and accelerating the launch of new products to the market.
TaiGen’s focus is on the research and development of novel drugs. Besides Burixafor, the products also include anti-infective, Taigexyn®, and an anti-hepatitis C drug, TG-2349. Taigexyn® is the first in-house developed novel drug that received new drug application approval from Taiwan’s FDA. TG-2349 is intended for the 160 million global patients with hepatitis C with huge market potential. TaiGen hopes to file one IND with the US FDA every 3-4 years to expand TaiGen’s product line.
About ScinoPharm
ScinoPharm Taiwan, Ltd. is a leading process R&D and API manufacturing service provider to the global pharmaceutical industry. With research and manufacturing facilities in both Taiwan and China, ScinoPharm offers a wide portfolio of services ranging from custom synthesis for early phase pharmaceutical activities to contract services for brand companies as well as APIs for the generic industry. For more information, please visit the Company’s website at http://www.scinopharm.com
About TaiGen Biotechnology
TaiGen Biotechnology is a leading research-based and product-driven biotechnology company in Taiwan with a wholly-owned subsidiary in Beijing, China. The company’s first product, Taigexyn®, have already received NDA approval from Taiwan’s FDA. In addition to Taigexyn®, TaiGen has two other in-house discovered NCEs in clinical development under IND with US FDA: TG-0054, a chemokine receptor antagonist for stem cell transplantation and chemosensitization, in Phase 2 and TG-2349, a HCV protease inhibitor for treatment of chronic hepatitis infection, in Phase 2. Both TG-0054 and TG-2349 are currently in clinical trials in patients in the US.
SOURCE ScinoPharm Taiwan Ltd.
Image may be NSFW. Clik here to view.
TG-0054 is a potent and selective chemokine CXCR4 (SDF-1) antagonist in phase II clinical studies at TaiGen Biotechnology for use in stem cell transplantation in cancer patients. Specifically, the compound is being developed for the treatment of stem cell transplantation in multiple myeloma, non-Hodgkin’s lymphoma, Hodgkin’s lymphoma and myocardial ischemia.
Preclinical studies had also been undertaken for the treatment of diabetic retinopathy, critical limb ischemia (CLI) and age-related macular degeneration. In a mouse model, TG-0054 efficiently mobilizes stem cells (CD34+) and endothelial progenitor cells (CD133+) from bone marrow into peripheral circulation.
BACKGROUND
Chemokines are a family of cytokines that regulate the adhesion and transendothelial migration of leukocytes during an immune or inflammatory reaction (Mackay C.R., Nat. Immunol, 2001, 2:95; Olson et al, Am. J. Physiol. Regul. Integr. Comp. Physiol, 2002, 283 :R7). Chemokines also regulate T cells and B cells trafficking and homing, and contribute to the development of lymphopoietic and hematopoietic systems (Ajuebor et al, Biochem. Pharmacol, 2002, 63:1191). Approximately 50 chemokines have been identified in humans. They can be classified into 4 subfamilies, i.e., CXC, CX3C, CC, and C chemokines, based on the positions of the conserved cysteine residues at the N-terminal (Onuffer et al, Trends Pharmacol ScI, 2002, 23:459). The biological functions of chemokines are mediated by their binding and activation of G protein-coupled receptors (GPCRs) on the cell surface.
Stromal-derived factor- 1 (SDF-I) is a member of CXC chemokines. It is originally cloned from bone marrow stromal cell lines and found to act as a growth factor for progenitor B cells (Nishikawa et al, Eur. J. Immunol, 1988, 18:1767). SDF-I plays key roles in homing and mobilization of hematopoietic stem cells and endothelial progenitor cells (Bleul et al, J. Exp. Med., 1996, 184:1101; and Gazzit et al, Stem Cells, 2004, 22:65-73). The physiological function of SDF-I is mediated by CXCR4 receptor. Mice lacking SDF-I or CXCR4 receptor show lethal abnormality in bone marrow myelopoiesis, B cell lymphopoiesis, and cerebellar development (Nagasawa et al, Nature, 1996, 382:635; Ma et al, Proc. Natl. Acad. ScI, 1998, 95:9448; Zou et al, Nature, 1998, 393:595; Lu et al, Proc. Natl. Acad. ScI, 2002, 99:7090). CXCR4 receptor is expressed broadly in a variety of tissues, particularly in immune and central nervous systems, and has been described as the major co-receptor for HIV- 1/2 on T lymphocytes. Although initial interest in CXCR4 antagonism focused on its potential application to AIDS treatment (Bleul et al, Nature, 1996, 382:829), it is now becoming clear that CXCR4 receptor and SDF-I are also involved in other pathological conditions such as rheumatoid arthritis, asthma, and tumor metastases (Buckley et al., J. Immunol., 2000, 165:3423). Recently, it has been reported that a CXCR4 antagonist and an anticancer drug act synergistically in inhibiting cancer such as acute promuelocutic leukemia (Liesveld et al., Leukemia
Research 2007, 31 : 1553). Further, the CXCR4/SDF-1 pathway has been shown to be critically involved in the regeneration of several tissue injury models. Specifically, it has been found that the SDF-I level is elevated at an injured site and CXCR4-positive cells actively participate in the tissue regenerating process.
Water (10.0 L) and (BoC)2O (3.33 kgg, 15.3 mol) were added to a solution of trans-4-aminomethyl-cyclohexanecarboxylic acid (compound 1-1, 2.0 kg, 12.7 mol) and sodium bicarbonate (2.67 kg, 31.8 mol). The reaction mixture was stirred at ambient temperature for 18 hours. The aqueous layer was acidified with concentrated hydrochloric acid (2.95 L, pH = 2) and then filtered. The resultant solid was collected, washed three times with water (15 L), and dried in a hot box (60 0C) to give trα/?5-4-(tert-butoxycarbonylamino-methyl)-cyclo-hexanecarboxylic acid (Compound l-II, 3.17 kg, 97%) as a white solid. Rf = 0.58 (EtOAc). LC-MS m/e 280 (M+Na+). 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, IH), 2.98 (t, J= 6.3 Hz, 2H), 2.25 (td, J = 12, 3.3 Hz, IH), 2.04 (d, J= 11.1 Hz, 2H), 1.83 (d, J= 11.1 Hz, 2H), 1.44 (s, 9H), 1.35-1.50 (m, 3H), 0.89-1.03 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 181.31, 156.08, 79.12, 46.41, 42.99, 37.57, 29.47, 28.29, 27.96. M.p. 134.8-135.0 0C. A suspension of compound l-II (1.0 kg, 3.89 mol) in THF (5 L) was cooled at
-10 0C and triethyl amine (1.076 L, 7.78 mol) and ethyl chloroformate (0.441 L, 4.47 mol) were added below -10 0C. The reaction mixture was stirred at ambient temperature for 3 hours. The reaction mixture was then cooled at -10 0C again and NH4OH (3.6 L, 23.34 mol) was added below -10 0C. The reaction mixture was stirred at ambient temperature for 18 hours and filtered. The solid was collected and washed three times with water (10 L) and dried in a hot box (6O0C) to give trans-4- (tert-butoxycarbonyl-amino-methyl)-cyclohexanecarboxylic acid amide (Compound l-III, 0.8 kg, 80%) as a white solid. Rf= 0.23 (EtOAc). LC-MS m/e 279, M+Na+. 1H NMR (300 MHz, CD3OD) δ 6.63 (brs, IH), 2.89 (t, J= 6.3 Hz, 2H), 2.16 (td, J = 12.2, 3.3 Hz, IH), 1.80-1.89 (m, 4H), 1.43 (s, 9H), 1.37-1.51 (m, 3H), 0.90-1.05 (m, 2H). 13C NMR (75 MHz, CD3OD) δ 182.26, 158.85, 79.97, 47.65, 46.02, 39.28, 31.11, 30.41, 28.93. M.p. 221.6-222.0 0C.
A suspension of compound l-III (1.2 kg, 4.68 mol) in CH2Cl2 (8 L) was cooled at -1O0C and triethyl amine (1.3 L, 9.36 mol) and trifluoroacetic anhydride (0.717 L, 5.16 mol) were added below -10 0C. The reaction mixture was stirred for 3 hours. After water (2.0 L) was added, the organic layer was separated and washed with water (3.0 L) twice. The organic layer was then passed through silica gel and concentrated. The resultant oil was crystallized by methylene chloride. The crystals were washed with hexane to give £rαns-(4-cyano-cyclohexylmethyl)-carbamic acid tert-butyl ester (Compound 1-IV, 0.95 kg, 85%) as a white crystal. Rf = 0.78 (EtOAc). LC-MS m/e 261, M+Na+. 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, IH), 2.96 (t, J = 6.3 Hz, 2H), 2.36 (td, J= 12, 3.3 Hz, IH), 2.12 (dd, J= 13.3, 3.3 Hz, 2H), 1.83 (dd, J = 13.8, 2.7 Hz, 2H), 1.42 (s, 9H), 1.47-1.63 (m, 3H), 0.88-1.02 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 155.96, 122.41, 79.09, 45.89, 36.92, 29.06, 28.80, 28.25, 28.00. M.p. 100.4~100.6°C.
Compound 1-IV (1.0 kg, 4.196 mol) was dissolved in a mixture of 1 ,4-dioxane (8.0 L) and water (2.0 L). To the reaction mixture were added lithium hydroxide monohydrate (0.314 kg, 4.191), Raney-nickel (0.4 kg, 2.334 mol), and 10% palladium on carbon (0.46 kg, 0.216 mol) as a 50% suspension in water. The reaction mixture was stirred under hydrogen atmosphere at 5O0C for 20 hours. After the catalysts were removed by filtration and the solvents were removed in vacuum, a mixture of water (1.0 L) and CH2Cl2 (0.3 L) was added. After phase separation, the organic phase was washed with water (1.0 L) and concentrated to give £rα/?s-(4-aminomethyl- cyclohexylmethyl)-carbamic acid tert- butyl ester (compound 1-V, 0.97 kg, 95%) as pale yellow thick oil. Rf = 0.20 (MeOH/EtOAc = 9/1). LC-MS m/e 243, M+H+. 1H NMR (300 MHz, CDCl3) δ 4.67 (brs, IH), 2.93 (t, J= 6.3 Hz, 2H), 2.48 (d, J= 6.3 Hz, 2H), 1.73-1.78 (m, 4H), 1.40 (s, 9H), 1.35 (brs, 3H), 1.19-1.21 (m, IH), 0.77-0.97 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 155.85, 78.33, 48.27, 46.38, 40.80, 38.19, 29.87, 29.76, 28.07. A solution of compound 1-V (806 g) and Et3N (1010 g, 3 eq) in 1-pentanol
(2.7 L) was treated with compound 1-VI, 540 g, 1 eq) at 900C for 15 hours. TLC showed that the reaction was completed. Ethyl acetate (1.5 L) was added to the reaction mixture at 25°C. The solution was stirred for 1 hour. The Et3NHCl salt was filtered. The filtrate was then concentrated to 1.5 L (1/6 of original volume) by vacuum at 500C. Then, diethyl ether (2.5 L) was added to the concentrated solution to afford the desired product 1-VII (841 g, 68% yield) after filtration at 250C .
A solution of intermediate 1-VII (841 g) was treated with 4 N HCl/dioxane (2.7 L) in MeOH (8.1 L) and stirred at 25°C for 15 hours. TLC showed that the reaction was completed. The mixture was concentrated to 1.5 L (1/7 of original volume) by vacuum at 500C. Then, diethyl ether (5 L) was added to the solution slowly, and HCl salt of 1-VIII (774 g) was formed, filtered, and dried under vacuum (<10 torr). For neutralization, K2CO3 (2.5 kg, 8 eq) was added to the solution of HCl salt of 1-VIII in MeOH (17 L) at 25°C. The mixture was stirred at the same temperature for 3 hours (pH > 12) and filtered (estimated amount of 1-VIII in the filtrate is 504 g). Aldehyde 1-IX (581 g, 1.0 eq based on mole of 1-VII) was added to the filtrate of 1-VIII at 0-100C. The reaction was stirred at 0-100C for 3 hours. TLC showed that the reaction was completed. Then, NaBH4 (81 g, 1.0 eq based on mole of 1-VII) was added at less than 100C and the solution was stirred at 10-150C for Ih. The solution was concentrated to get a residue, which then treated with CH2Cl2 (15 L). The mixture was washed with saturated aq. NH4Cl solution (300 mL) diluted with H2O (1.2 L). The CH2Cl2 layer was concentrated and the residue was purified by chromatography on silica gel (short column, EtOAc as mobile phase for removing other components; MeOH/28% NH4OH = 97/3 as mobile phase for collecting 1-X) afforded crude 1-X (841 g). Then Et3N (167 g, leq) and BoC2O (360 g, leq) were added to the solution of
1-X (841 g) in CH2Cl2 (8.4 L) at 25°C. The mixture was stirred at 25°C for 15 hours. After the reaction was completed as evidenced by TLC, the solution was concentrated and EtOAc (5 L) was added to the resultant residue. The solution was concentrated to 3L (1/2 of the original volume) under low pressure at 500C. Then, n-hexane (3 L) was added to the concentrated solution. The solid product formed at 500C by seeding to afford the desired crude product 1-XI (600 g, 60% yield) after filtration and evaporation. To compound 1-XI (120.0 g) and piperazine (1-XII, 50.0 g, 3 eq) in 1- pentanol (360 niL) was added Et3N (60.0 g, 3.0 eq) at 25°C. The mixture was stirred at 1200C for 8 hours. Ethyl acetate (480 mL) was added to the reaction mixture at 25°C. The solution was stirred for Ih. The Et3NHCl salt was filtered and the solution was concentrated and purified by silica gel (EtOAc/MeOH = 2:8) to afforded 1-XIII (96 g) in a 74% yield.
A solution of intermediate 1-XIII (100 mg) was treated with 4 N HCl/dioxane (2 mL) in CH2Cl2 (1 mL) and stirred at 25°C for 15 hours. The mixture was concentrated to give hydrochloride salt of compound 1 (51 mg). CI-MS (M+ + 1): 459.4
Compound 2 Intermediate 1-XIII was prepared as described in Example 1.
To a solution of 1-XIII (120 g) in MeOH (2.4 L) were added diethyl vinyl phosphonate (2-1, 45 g, 1.5 eq) at 25°C. The mixture was stirred under 65°C for 24 hours. TLC and HPLC showed that the reaction was completed. The solution was concentrated and purified by silica gel (MeOH/CH2Cl2 = 8/92) to get 87 g of 2-11 (53% yield, purity > 98%, each single impurity <1%) after analyzing the purity of the product by HPLC.
A solution of 20% TFA/CH2C12 (36 mL) was added to a solution of intermediate 2-11 (1.8 g) in CH2Cl2 (5 mL). The reaction mixture was stirred for 15 hours at room temperature and concentrated by removing the solvent to afford trifluoracetic acid salt of compound 2 (1.3 g). CI-MS (M+ + 1): 623.1
Intermediate 2-11 was prepared as described in Example 2. To a solution of 2-11 (300 g) in CH2Cl2 (1800 mL) was added TMSBr (450 g, 8 eq) at 10-150C for 1 hour. The mixture was stirred at 25°C for 15 hours. The solution was concentrated to remove TMSBr and solvent under vacuum at 400C.
CH2Cl2 was added to the mixture to dissolve the residue. TMSBr and solvent were removed under vacuum again to obtain 36O g crude solid after drying under vacuum (<1 torr) for 3 hours. Then, the crude solid was washed with 7.5 L IPA/MeOH (9/1) to afford compound 3 (280 g) after filtration and drying at 25°C under vacuum (<1 torr) for 3 hours. Crystallization by EtOH gave hydrobromide salt of compound 3 (19Og). CI-MS (M+ + 1): 567.0.
The hydrobromide salt of compound 3 (5.27 g) was dissolved in 20 mL water and treated with concentrated aqueous ammonia (pH=9-10), and the mixture was evaporated in vacuo. The residue in water (30 mL) was applied onto a column (100 mL, 4.5x8 cm) of Dowex 50WX8 (H+ form, 100-200 mesh) and eluted (elution rate, 6 mL/min). Elution was performed with water (2000 mL) and then with 0.2 M aqueous ammonia. The UV-absorbing ammonia eluate was evaporated to dryness to afford ammonia salt of compound 3 (2.41 g). CI-MS (M+ + 1): 567.3.
The ammonia salt of compound 3 (1.5 g) was dissolved in water (8 mL) and alkalified with concentrated aqueous ammonia (pH=l 1), and the mixture solution was applied onto a column (75 mL, 3x14 cm) of Dowex 1X2 (acetate form, 100-200 mesh) and eluted (elution rate, 3 mL/min). Elution was performed with water (900 mL) and then with 0.1 M acetic acid. The UV-absorbing acetic acid eluate was evaporated, and the residue was codistilled with water (5x50 mL) to afford compound 3 (1.44 g). CI-MS (M+ + 1): 567.4. Example 4: Preparation of Compound 4
Intermediate 1-XIII was obtained during the preparation of compound 1. To a solution of diethyl vinyl phosphonate (4-1, 4 g) in CH2Cl2 (120 mL) was added oxalyl chloride (15.5 g, 5 eq) and the mixture was stirred at 300C for 36 hours. The mixture were concentrated under vacuum on a rotatory evaporated to give quantitatively the corresponding phosphochloridate, which was added to a mixture of cyclohexyl amine (4-II, 5.3 g, 2.2 eq), CH2Cl2 (40 mL), and Et3N (6.2 g, 2.5 eq). The mixture was stirred at 35°C for 36 hours, and then was washed with water. The organic layer was dried (MgSO4), filtered, and evaporated to afford 4-III (4.7 g, 85% yield) as brown oil.
Compound 4-III (505 mg) was added to a solution of intermediate 1-XIII (500 mg) in MeOH (4 mL). The solution was stirred at 45°C for 24 hours. The solution was concentrated and the residue was purified by column chromatography on silica gel (EtOAc/ MeOH = 4: 1) to afford intermediate 4-IV (420 mg) in a 63% yield.
A solution of HCl in ether (5 mL) was added to a solution of intermediate 4- IV (420 mg) in CH2Cl2 (1.0 mL). The reaction mixture was stirred for 12 hours at room temperature and concentrated by removing the solvent. The resultant residue was washed with ether to afford hydrochloride salt of compound 4 (214 mg). CI-MS (M+ + 1): 595.1
Intermediate l-II was prepared as described in Example 1. To a suspension of the intermediate l-II (31.9 g) in toluene (150 mL) were added phosphorazidic acid diphenyl ester (51-1, 32.4 g) and Et3N (11.9 g) at 25°C for 1 hour. The reaction mixture was stirred at 800C for 3 hours and then cooled to 25°C. After benzyl alcohol (51-11, 20 g) was added, the reaction mixture was stirred at 800C for additional 3 hours and then warmed to 1200C overnight. It was then concentrated and dissolved again in EtOAc and H2O. The organic layer was collected. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with 2.5 N HCl, saturated aqueous NaHCO3 and brine, dried over anhydrous MgSO4, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (EtOAc/Hexane = 1 :2) to give Intermediate 51-111 (35 g) in a 79% yield. A solution of intermediate 51-111 (35 g) treated with 4 N HCl/dioxane (210 rnL) in MeOH (350 mL) was stirred at room temperature overnight. After ether (700 mL) was added, the solution was filtered. The solid was dried under vacuum. K2CO3 was added to a suspension of this solid in CH3CN and ώo-propanol at room temperature for 10 minutes. After water was added, the reaction mixture was stirred at room temperature for 2 hours, filtered, dried over anhydrous MgSO4, and concentrated. The resultant residue was purified by column chromatography on silica gel (using CH2Cl2 and MeOH as an eluant) to give intermediate 51-IV (19 g) in a 76% yield. Intermediate 1-IX (21 g) was added to a solution of intermediate 51-IV (19 g) in CH2Cl2 (570 mL). The mixture was stirred at 25°C for 2 hours. NaBH(OAc)3 (23 g) was then added at 25°C overnight. After the solution was concentrated, a saturated aqueous NaHCO3 solution was added to the resultant residue. The mixture was then extracted with CH2Cl2. The solution was concentrated and the residue was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 51-V (23.9 g) in a 66% yield.
A solution of intermediate 51-V (23.9 g) and BoC2O (11.4 g) in CH2Cl2 (200 mL) was added to Et3N (5.8 mL) at 25°C for overnight. The solution was then concentrated and the resultant residue was purified by column chromatography on silica gel (using EtOAc and Hexane as an eluant) to give intermediate 51-VI (22 g) in a 77% yield.
10% Pd/C (2.2 g) was added to a suspension of intermediate 51-VI (22 g) in MeOH (44 mL). The mixture was stirred at ambient temperature under hydrogen atmosphere overnight, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 51-VII (16.5 g) in a 97% yield.
Intermediate 51-VII (16.5 g) and Et3N (4.4 mL) in 1-pentanol (75 mL) was allowed to react with 2,4-dichloro-6-aminopyrimidine (1-VI, 21 g) at 1200C overnight. The solvent was then removed and the residue was purified by column chromatography on silica gel (using EtOAc and hexane as an eluant) to afford intermediate 51-VIII (16.2 g) in a 77% yield.
A solution of intermediate 51-VIII (16.2 g) and piperazine (1-XII, 11.7 g) in 1-pentanol (32 mL) was added to Et3N (3.3 mL) at 1200C overnight. After the solution was concentrated, the residue was treated with water and extracted with CH2Cl2. The organic layer was collected and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc/ MeOH to 28% NH40H/Me0H as an eluant) to afford Intermediate 51-IX (13.2 g) in a 75% yield. Diethyl vinyl phosphonate (2-1) was treated with 51-IX as described in
Example 3 to afford hydrobromide salt of compound 51. CI-MS (M+ + 1): 553.3
Water (10.0 L) and (Boc)2O (3.33 kgg, 15.3 mol) were added to a solution of trans-4-aminomethyl-cyclohexanecarboxylic acid (compound 1-I, 2.0 kg, 12.7 mol) and sodium bicarbonate (2.67 kg, 31.8 mol). The reaction mixture was stirred at ambient temperature for 18 hours. The aqueous layer was acidified with concentrated hydrochloric acid (2.95 L, pH=2) and then filtered. The resultant solid was collected, washed three times with water (15 L), and dried in a hot box (60° C.) to give trans-4-(tert-butoxycarbonylamino-methyl)-cyclo-hexanecarboxylic acid (Compound 1-II, 3.17 kg, 97%) as a white solid. Rf=0.58 (EtOAc). LC-MS m/e 280 (M+Na+). 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, 1H), 2.98 (t, J=6.3 Hz, 2H), 2.25 (td, J=12, 3.3 Hz, 1H), 2.04 (d, J=11.1 Hz, 2H), 1.83 (d, J=11.1 Hz, 2H), 1.44 (s, 9H), 1.35˜1.50 (m, 3H), 0.89˜1.03 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 181.31, 156.08, 79.12, 46.41, 42.99, 37.57, 29.47, 28.29, 27.96. M.p. 134.8˜135.0° C.
A suspension of compound 1-II (1.0 kg, 3.89 mol) in THF (5 L) was cooled at 10° C. and triethyl amine (1.076 L, 7.78 mol) and ethyl chloroformate (0.441 L, 4.47 mol) were added below 10° C. The reaction mixture was stirred at ambient temperature for 3 hours. The reaction mixture was then cooled at 10° C. again and NH4OH (3.6 L, 23.34 mol) was added below 10° C. The reaction mixture was stirred at ambient temperature for 18 hours and filtered. The solid was collected and washed three times with water (10 L) and dried in a hot box (60° C.) to give trans-4-(tert-butoxycarbonyl-amino-methyl)-cyclohexanecarboxylic acid amide (Compound 1-III, 0.8 kg, 80%) as a white solid. Rf=0.23 (EtOAc). LC-MS m/e 279, M+Na+. 1H NMR (300 MHz, CD3OD) δ 6.63 (brs, 1H), 2.89 (t, J=6.3 Hz, 2H), 2.16 (td, J=12.2, 3.3 Hz, 1H), 1.80˜1.89 (m, 4H), 1.43 (s, 9H), 1.37˜1.51 (m, 3H), 0.90˜1.05 (m, 2H). 13C NMR (75 MHz, CD3OD) δ 182.26, 158.85, 79.97, 47.65, 46.02, 39.28, 31.11, 30.41, 28.93. M.p. 221.6˜222.0° C.
A suspension of compound 1-III (1.2 kg, 4.68 mol) in CH2Cl2 (8 L) was cooled at 10° C. and triethyl amine (1.3 L, 9.36 mol) and trifluoroacetic anhydride (0.717 L, 5.16 mol) were added below 10° C. The reaction mixture was stirred for 3 hours. After water (2.0 L) was added, the organic layer was separated and washed with water (3.0 L) twice. The organic layer was then passed through silica gel and concentrated. The resultant oil was crystallized by methylene chloride. The crystals were washed with hexane to give trans-(4-cyano-cyclohexylmethyl)-carbamic acid tent-butyl ester (Compound 1-IV, 0.95 kg, 85%) as a white crystal. Rf=0.78 (EtOAc). LC-MS m/e 261, M+Na+. 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, 1H), 2.96 (t, J=6.3 Hz, 2H), 2.36 (td, J=12, 3.3 Hz, 1H), 2.12 (dd, J=13.3, 3.3 Hz, 2H), 1.83 (dd, J=13.8, 2.7 Hz, 2H), 1.42 (s, 9H), 1.47˜1.63 (m, 3H), 0.88˜1.02 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 155.96, 122.41, 79.09, 45.89, 36.92, 29.06, 28.80, 28.25, 28.00. M.p. 100.4˜100.6° C.
Compound 1-IV (1.0 kg, 4.196 mol) was dissolved in a mixture of 1,4-dioxane (8.0 L) and water (2.0 L). To the reaction mixture were added lithium hydroxide monohydrate (0.314 kg, 4.191), Raney-nickel (0.4 kg, 2.334 mol), and 10% palladium on carbon (0.46 kg, 0.216 mol) as a 50% suspension in water. The reaction mixture was stirred under hydrogen atmosphere at 50° C. for 20 hours. After the catalysts were removed by filtration and the solvents were removed in vacuum, a mixture of water (1.0 L) and CH2Cl2 (0.3 L) was added. After phase separation, the organic phase was washed with water (1.0 L) and concentrated to give trans-(4-aminomethyl-cyclohexylmethyl)-carbamic acid tert-butyl ester (compound 1-V, 0.97 kg, 95%) as pale yellow thick oil. Rf=0.20 (MeOH/EtOAc=9/1). LC-MS m/e 243, M+H+. 1H NMR (300 MHz, CDCl3) δ 4.67 (brs, 1H), 2.93 (t, J=6.3 Hz, 2H), 2.48 (d, J=6.3 Hz, 2H), 1.73˜1.78 (m, 4H), 1.40 (s, 9H), 1.35 (brs, 3H), 1.19˜1.21 (m, 1H), 0.77˜0.97 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 155.85, 78.33, 48.27, 46.38, 40.80, 38.19, 29.87, 29.76, 28.07.
A solution of compound 1-V (806 g) and Et3N (1010 g, 3 eq) in 1-pentanol (2.7 L) was treated with compound 1-VI, 540 g, 1 eq) at 90° C. for 15 hours. TLC showed that the reaction was completed.
Ethyl acetate (1.5 L) was added to the reaction mixture at 25° C. The solution was stirred for 1 hour. The Et3NHCl salt was filtered. The filtrate was then concentrated to 1.5 L (1/6 of original volume) by vacuum at 50° C. Then, diethyl ether (2.5 L) was added to the concentrated solution to afford the desired product 1-VII (841 g, 68% yield) after filtration at 25° C.
A solution of intermediate 1-VII (841 g) was treated with 4 N HCl/dioxane (2.7 L) in MeOH (8.1 L) and stirred at 25° C. for 15 hours. TLC showed that the reaction was completed. The mixture was concentrated to 1.5 L (1/7 of original volume) by vacuum at 50° C. Then, diethyl ether (5 L) was added to the solution slowly, and HCl salt of 1-VIII (774 g) was formed, filtered, and dried under vacuum (<10 ton). For neutralization, K2CO3 (2.5 kg, 8 eq) was added to the solution of HCl salt of 1-VIII in MeOH (17 L) at 25° C. The mixture was stirred at the same temperature for 3 hours (pH>12) and filtered (estimated amount of 1-VIII in the filtrate is 504 g).
Aldehyde 1-IX (581 g, 1.0 eq based on mole of 1-VII) was added to the filtrate of 1-VIII at 0-10° C. The reaction was stirred at 0-10° C. for 3 hours. TLC showed that the reaction was completed. Then, NaBH4 (81 g, 1.0 eq based on mole of 1-VII) was added at less than 10° C. and the solution was stirred at 10-15° C. for 1 h. The solution was concentrated to get a residue, which then treated with CH2Cl2 (15 L). The mixture was washed with saturated aq. NH4Cl solution (300 mL) diluted with H2O (1.2 L). The CH2Cl2 layer was concentrated and the residue was purified by chromatography on silica gel (short column, EtOAc as mobile phase for removing other components; MeOH/28% NH4OH=97/3 as mobile phase for collecting 1-X) afforded crude 1-X (841 g).
Then Et3N (167 g, 1 eq) and Boc2O (360 g, 1 eq) were added to the solution of 1-X (841 g) in CH2Cl2 (8.4 L) at 25° C. The mixture was stirred at 25° C. for 15 hours. After the reaction was completed as evidenced by TLC, the solution was concentrated and EtOAc (5 L) was added to the resultant residue. The solution was concentrated to 3 L (1/2 of the original volume) under low pressure at 50° C. Then, n-hexane (3 L) was added to the concentrated solution. The solid product formed at 50° C. by seeding to afford the desired crude product 1-XI (600 g, 60% yield) after filtration and evaporation.
To compound 1-XI (120.0 g) and piperazine (1-XII, 50.0 g, 3 eq) in 1-pentanol (360 mL) was added Et3N (60.0 g, 3.0 eq) at 25° C. The mixture was stirred at 120° C. for 8 hours. Ethyl acetate (480 mL) was added to the reaction mixture at 25° C. The solution was stirred for 1 h. The Et3NHCl salt was filtered and the solution was concentrated and purified by silica gel (EtOAc/MeOH=2:8) to afforded 1-XIII (96 g) in a 74% yield.
To a solution of 1-XIII (120 g) in MeOH (2.4 L) were added diethyl vinyl phosphonate (1-XIV, 45 g, 1.5 eq) at 25° C. The mixture was stirred under 65° C. for 24 hours. TLC and HPLC showed that the reaction was completed. The solution was concentrated and purified by silica gel (MeOH/CH2Cl2=8/92) to get 87 g of 1-XV (53% yield, purity>98%, each single impurity<1%) after analyzing the purity of the product by HPLC.
A solution of 20% TFA/CH2Cl2 (36 mL) was added to a solution of intermediate 1-XV (1.8 g) in CH2Cl2 (5 mL). The reaction mixture was stirred for 15 hours at room temperature and concentrated by removing the solvent to afford trifluoracetic acid salt of compound 1 (1.3 g).
Intermediate 1-XV was prepared as described in Example 1.
To a solution of 1-XV (300 g) in CH2Cl2 (1800 mL) was added TMSBr (450 g, 8 eq) at 10-15° C. for 1 hour. The mixture was stirred at 25° C. for 15 hours. The solution was concentrated to remove TMSBr and solvent under vacuum at 40° C. CH2Cl2 was added to the mixture to dissolve the residue. TMSBr and solvent were removed under vacuum again to obtain 360 g crude solid after drying under vacuum (<1 torr) for 3 hours. Then, the crude solid was washed with 7.5 L IPA/MeOH (9/1) to afford compound 2 (280 g) after filtration and drying at 25° C. under vacuum (<1 ton) for 3 hours. Crystallization by EtOH gave hydrobromide salt of compound 2 (190 g). CI-MS (M++1): 567.0.
The hydrobromide salt of compound 2 (5.27 g) was dissolved in 20 mL water and treated with concentrated aqueous ammonia (pH=9-10), and the mixture was evaporated in vacuo. The residue in water (30 mL) was applied onto a column (100 mL, 4.5×8 cm) of Dowex 50WX8 (H+ form, 100-200 mesh) and eluted (elution rate, 6 mL/min). Elution was performed with water (2000 mL) and then with 0.2 M aqueous ammonia. The UV-absorbing ammonia eluate was evaporated to dryness to afford ammonia salt of compound 2 (2.41 g). CI-MS (M++1): 567.3.
The ammonia salt of compound 2 (1.5 g) was dissolved in water (8 mL) and alkalified with concentrated aqueous ammonia (pH=11), and the mixture solution was applied onto a column (75 mL, 3×14 cm) of Dowex 1×2 (acetate form, 100-200 mesh) and eluted (elution rate, 3 mL/min). Elution was performed with water (900 mL) and then with 0.1 M acetic acid. The UV-absorbing acetic acid eluate was evaporated, and the residue was codistilled with water (5×50 mL) to afford compound 2 (1.44 g). CI-MS (M++1): 567.4.
Intermediate 1-XIII was obtained during the preparation of compound 1.
To a solution of diethyl vinyl phosphonate (3-I, 4 g) in CH2Cl2 (120 mL) was added oxalyl chloride (15.5 g, 5 eq) and the mixture was stirred at 30° C. for 36 hours. The mixture were concentrated under vacuum on a rotatory evaporated to give quantitatively the corresponding phosphochloridate, which was added to a mixture of cyclohexyl amine (3-II, 5.3 g, 2.2 eq), CH2Cl2 (40 mL), and Et3N (6.2 g, 2.5 eq). The mixture was stirred at 35° C. for 36 hours, and then was washed with water. The organic layer was dried (MgSO4), filtered, and evaporated to afford 3-III (4.7 g, 85% yield) as brown oil.
Compound 3-III (505 mg) was added to a solution of intermediate 1-XIII (500 mg) in MeOH (4 mL). The solution was stirred at 45° C. for 24 hours. The solution was concentrated and the residue was purified by column chromatography on silica gel (EtOAc/MeOH=4:1) to afford intermediate 3-IV (420 mg) in a 63% yield.
A solution of HCl in ether (5 mL) was added to a solution of intermediate 3-IV (420 mg) in CH2Cl2 (1.0 mL). The reaction mixture was stirred for 12 hours at room temperature and concentrated by removing the solvent. The resultant residue was washed with ether to afford hydrochloride salt of compound 3 (214 mg).
Compound 4 was prepared in the same manner as that described in Example 2 except that sodium 2-bromoethanesulfonate in the presence of Et3N in DMF at 45° C. was used instead of diethyl vinyl phosphonate. Deportations of amino-protecting group by hydrochloride to afford hydrochloride salt of compound 4.
Compound 5 was prepared in the same manner as that described in Example 2 except that diethyl-1-bromopropylphosphonate in the presence of K2CO3 in CH3CN was used instead of diethyl vinyl phosphonate.
Compound 6 was prepared in the same manner as that described in Example 5 except that 1,4-diaza-spiro[5.5]undecane dihydrochloride was used instead of piperazine.
Intermediate 1-II was prepared as described in Example 1.
To a suspension of the intermediate 1-II (31.9 g) in toluene (150 mL) were added phosphorazidic acid diphenyl ester (7-I, 32.4 g) and Et3N (11.9 g) at 25° C. for 1 hour. The reaction mixture was stirred at 80° C. for 3 hours and then cooled to 25° C. After benzyl alcohol (7-II, 20 g) was added, the reaction mixture was stirred at 80° C. for additional 3 hours and then warmed to 120° C. overnight. It was then concentrated and dissolved again in EtOAc and H2O. The organic layer was collected. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with 2.5 N HCl, saturated aqueous NaHCO3 and brine, dried over anhydrous MgSO4, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (EtOAc/Hexane=1:2) to give Intermediate 7-III (35 g) in a 79% yield.
A solution of intermediate 7-III (35 g) treated with 4 N HCl/dioxane (210 mL) in MeOH (350 mL) was stirred at room temperature overnight. After ether (700 mL) was added, the solution was filtered. The solid was dried under vacuum. K2CO3 was added to a suspension of this solid in CH3CN and iso-propanol at room temperature for 10 minutes. After water was added, the reaction mixture was stirred at room temperature for 2 hours, filtered, dried over anhydrous MgSO4, and concentrated. The resultant residue was purified by column chromatography on silica gel (using CH2Cl2 and MeOH as an eluant) to give intermediate 7-IV (19 g) in a 76% yield.
Intermediate 1-IX (21 g) was added to a solution of intermediate 7-IV (19 g) in CH2Cl2 (570 mL). The mixture was stirred at 25° C. for 2 hours. NaBH(OAc)3 (23 g) was then added at 25° C. overnight. After the solution was concentrated, a saturated aqueous NaHCO3 solution was added to the resultant residue. The mixture was then extracted with CH2Cl2. The solution was concentrated and the residue was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 7-V (23.9 g) in a 66% yield.
A solution of intermediate 7-V (23.9 g) and Boc2O (11.4 g) in CH2Cl2 (200 mL) was added to Et3N (5.8 mL) at 25° C. for overnight. The solution was then concentrated and the resultant residue was purified by column chromatography on silica gel (using EtOAc and Hexane as an eluant) to give intermediate 7-VI (22 g) in a 77% yield. 10% Pd/C (2.2 g) was added to a suspension of intermediate 7-VI (22 g) in MeOH (44 mL). The mixture was stirred at ambient temperature under hydrogen atmosphere overnight, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 7-VII (16.5 g) in a 97% yield.
Intermediate 7-VII (16.5 g) and Et3N (4.4 mL) in 1-pentanol (75 mL) was allowed to react with 2,4-dichloro-6-aminopyrimidine (1-VI, 21 g) at 120° C. overnight. The solvent was then removed and the residue was purified by column chromatography on silica gel (using EtOAc and hexane as an eluant) to afford intermediate 7-VIII (16.2 g) in a 77% yield.
A solution of intermediate 7-VIII (16.2 g) and piperazine (1-XII, 11.7 g) in 1-pentanol (32 mL) was added to Et3N (3.3 mL) at 120° C. overnight. After the solution was concentrated, the residue was treated with water and extracted with CH2Cl2. The organic layer was collected and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc/MeOH to 28% NH4OH/MeOH as an eluant) to afford Intermediate 7-IX (13.2 g) in a 75% yield.
Diethyl vinyl phosphonate (2-I) was treated with 7-IX as described in Example 3 to afford hydrobromide salt of compound 7.
Cis-1,4-cyclohexanedicarboxylic acid (8-I, 10 g) in THF (100 ml) was added oxalyl chloride (8-II, 15.5 g) at 0° C. and then DMF (few drops). The mixture was stirred at room temperature for 15 hours. The solution was concentrated and the residue was dissolved in THF (100 ml). The mixture solution was added to ammonium hydroxide (80 ml) and stirred for 1 hour. The solution was concentrated and filtration to afford crude product 8-III (7.7 g).
Compound 8-III (7.7 g) in THF (200 ml) was slowly added to LiAlH4 (8.6 g) in THF (200 ml) solution at 0° C. The mixture solution was stirred at 65° C. for 15 hours. NaSO4.10H2O was added at room temperature and stirred for 1 hours. The resultant mixture was filtered to get filtrate and concentrated. The residue was dissolved in CH2Cl2 (100 ml). Et3N (27 g) and (Boc)2O (10 g) were added at room temperature. The solution was stirred for 15 h, and then concentrated to get resultant residue. Ether was added to the resultant residue. Filtration and drying under vacuum afforded solid crude product 8-IV (8.8 g).
A solution of compound 8-IV (1.1 g) and Et3N (1.7 g) in 1-pentanol (10 ml) was reacted with 2,4-dichloro-6-aminopyrimidine (1-VI, 910 mg) at 90° C. for 15 hours. TLC showed that the reaction was completed. Ethyl acetate (10 mL) was added to the reaction mixture at 25° C. The solution was stirred for 1 hour. The Et3NHCl salt was removed. The filtrate was concentrated and purified by silica gel (EtOAc/Hex=1:2) to afford the desired product 8-V (1.1 g, 65% yield).
A solution of intermediate 8-V (1.1 g) was treated with 4 N HCl/dioxane (10 ml) in MeOH (10 ml) and stirred at 25° C. for 15 hours. TLC showed that the reaction was completed. The mixture was concentrated, filtered, and dried under vacuum (<10 ton). For neutralization, K2CO3 (3.2 g) was added to the solution of HCl salt in MeOH (20 ml) at 25° C. The mixture was stirred at the same temperature for 3 hours (pH>12) and filtered. Aldehyde 1-IX (759 mg) was added to the filtrate at 0-10° C. The reaction was stirred at 0-10° C. for 3 hours. TLC showed that the reaction was completed. Then, NaBH4 (112 mg) was added at less than 10° C. and the solution was stirred at 10-15° C. for 1 hour. The solution was concentrated to get a residue, which was then treated with CH2Cl2 (10 mL). The mixture was washed with saturated NH4Cl (aq) solution. The CH2Cl2 layer was concentrated and the residue was purified by chromatography on silica gel (MeOH/28% NH4OH=97/3) to afford intermediate 8-VI (1.0 g, 66% yield).
Et3N (600 mg) and Boc2O (428 mg) were added to the solution of 8-VI (1.0 g) in CH2Cl2 (10 ml) at 25° C. The mixture was stirred at 25° C. for 15 hours. TLC showed that the reaction was completed. The solution was concentrated and purified by chromatography on silica gel (EtOAc/Hex=1:1) to afford intermediate 8-VII (720 mg, 60% yield).
To a solution compound 8-VII (720 mg) and piperazine (1-XII, 1.22 g) in 1-pentanol (10 mL) was added Et3N (1.43 g) at 25° C. The mixture was stirred at 120° C. for 24 hours. TLC showed that the reaction was completed. Ethyl acetate (20 mL) was added at 25° C. The solution was stirred for 1 hour. The Et3NHCl salt was removed and the solution was concentrated and purified by silica gel (EtOAc/MeOH=2:8) to afford 8-VIII (537 mg) in 69% yield.
To a solution of 8-VIII (537 mg) in MeOH (11 ml) was added diethyl vinyl phosphonate (2-I, 201 mg) at 25° C. The mixture was stirred under 65° C. for 24 hours. TLC and HPLC showed that the reaction was completed. The solution was concentrated and purified by silica gel (MeOH/CH2Cl2=1:9) to get 8-IX (380 mg) in a 57% yield.
To a solution of 8-IX (210 mg) in CH2Cl2 (5 ml) was added TMSBr (312 mg) at 10-15° C. for 1 hour. The mixture was stirred at 25° C. for 15 hours. The solution was concentrated to remove TMSBr and solvent under vacuum at 40° C., then, CH2Cl2 was added to dissolve the residue. Then TMSBr and solvent were further removed under vacuum and CH2Cl2 was added for four times repeatedly. The solution was concentrated to get hydrobromide salt of compound 8 (190 mg).
Inhibitors of human immunodeficiency virus (HIV) protease have been approved for use in the treatment of HIV infection for several years. A particularly effective and recently approved HIV protease inhibitor is (2S,3S,5S)-2-(-2,6- dimethylphenoxyacetyl)-amino-3-hydroxy-5-(2-(1-tetrahydropyrimid-2-onyl)-3- methylbutanoyl)amino-1 ,6-diphenylhexane (also known as lopinavir).
Lopinavir is known to have utility for the inhibition of HIV protease and the inhibition of HIV infection. Lopinavir is particularly effective for the inhibition of HIV protease and for the inhibition of HIV infection when coadministered with ritonavir. Lopinavir, when combined with ritonavir, is also particularly effective for the inhibition of HIV infection when used in combination with one or more reverse transcriptase inhibitors and/or one or more other HIV protease inhibitors.
Lopinavir and processes for its preparation are disclosed in U.S. Patent No. 5,914,332, issued June 22, 1999, which is hereby incorporated herein by reference. This patent also discloses processes for preparing amorphous lopinavir.
Pharmaceutical compositions comprising lopinavir or a pharmaceutically acceptable salt thereof are disclosed in U.S. Patent No. 5,914,332, issued June 22, 1999; U.S. Patent Application No. 08/966,495, filed November 7, 1997; U.S. Provisional Application for Patent No. 60/177,020, filed January 19, 2000 and U.S. Patent Application No. 09/487,739, filed January 19, 2000, all of which are hereby incorporated herein by reference.
Lopinavir (ABT-378) is an antiretroviral of the protease inhibitor class. It is marketed by Abbott as Kaletra, a co-formulation with a sub-therapeutic dose of ritonavir, as a component of combination therapy to treat HIV/AIDS.
Retroviruses are those viruses which utilize a ribonucleic acid (RNA) intermediate and a RNA-dependent deoxyribonucleic acid (DNA) polymerase, reverse transcriptase, during their life cycle. Retroviruses include, but are not limited to, the RNA viruses of the Retroviridae family, and also the DNA viruses of the Hepadnavirus and Caulimovirus families. Retroviruses cause a variety of disease states in man, animals and plants. Some of the more important retroviruses from a pathological standpoint include human immunodeficiency viruses (HIV-1 and HIV-2), which cause acquired immune deficiency syndrome (AIDS) in man, human T-cell lymphotrophic viruses I, II, IV and V, which cause human acute cell leukemia, and bovine and feline leukemia viruses which cause leukemia in domestic animals.
Proteases are enzymes which cleave proteins at specific peptide bonds. Many biological functions are controlled or mediated by proteases and their complementary protease inhibitors. For example, the protease renin cleaves the peptide angiotensinogen to produce the peptide angiotensin I. Angiotensin I is further cleaved by the protease angiotensin converting enzyme (ACE) to form the hypotensive peptide angiotensin II. Inhibitors of renin and ACE are known to reduce high blood pressure in vivo. An inhibitor of a retroviral protease will provide a therapeutic agent for diseases caused by the retrovirus.
The genomes of retroviruses encode a protease that is responsible for the proteolytic processing of one or more polyprotein precursors such as the pol and gag gene products. See Wellink, Arch. Virol. 981 (1988). Retroviral proteases most commonly process the gag precursor into core proteins, and also process the pol precursor into reverse transciptase and retroviral protease. In addition, retroviral proteases are sequence specific. See Pearl, Nature 328 482 (1987).
The correct processing of the precursor polyproteins by the retroviral protease is necessary for the assembly of infectious virions. It has been shown that in vitro mutagenesis that produces protease-defective virus leads to the production of immature core forms which lack infectivity. See Crawford, J. Virol. 53 899 (1985); Katoh, et al., Virology 145 280 (1985). Therefore, retroviral protease inhibition provides an attractive target for antiviral therapy. See Mitsuya, Nature 325 775 (1987).
Current treatments for viral diseases usually involve administration of compounds that inhibit viral DNA synthesis. Current treatments for AIDS involve administration of compounds such as 3′-azido-3′-deoxythymidine (AZT), 2′,3′-dideoxycytidine (DDC), 2′,3′-dideoxyinosine (DDI), d4T and 3TC and compounds which treat the opportunistic infections caused by the immunosuppression resulting from HIV infection. None of the current AIDS treatments have proven to be totally effective in treating and/or reversing the disease. In addition, many of the compounds currently used to treat AIDS cause adverse side effects including low platelet count, renal toxicity and bone marrow cytopenia.
Recently the HIV protease inhibitors ritonavir, saquinavir and indinavir have been approved in the U.S. for treatment of HIV infections. However, there is a continuing need for improved HIV protease inhibitors.
Pharmacology
Lopinavir is highly bound to plasma proteins (98–99%).[2]
Reports are contradictory regarding lopinavir penetration into the cerebrospinal fluid (CSF). Anecdotal reports state that lopinavir cannot be detected in the CSF; however, a study of paired CSF-plasma samples from 26 patients receiving lopinavir/ritonavir found lopinavir CSF levels above the IC50 in 77% of samples.[3]
Clinical properties
Side effects, interactions, and contraindications have only been evaluated in the drug combination lopinavir/ritonavir.
Research
A 2014 study indicates that lopinavir is effective against the
human papilloma virus (HPV). The study used the equivalent of one tablet twice a day applied topically to the cervixes of women with high grade and low grade pre-cancerous conditions. After three months of treatment, 82.6% of the women who had high-grade disease had normal cervical conditions, confirmed by smears and biopsies.[4]
Lopinavir of Formula I is chemically [1S-[1R*,(R*),3R*,4R*]]-N-[4-[[(2,6-dimethyl-phenoxy)acetyl]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]tetrahydro-alpha-(1-methylethyl)-2-oxo-1(2H)-pyrimidineacetamide and is indicated in combination with other antiretroviral agents for the treatment of HIV-infection.
U.S. Pat. No. 5,914,332 provides a process for preparing amorphous lopinavir which involves dissolving lopinavir in an organic solvent (for example, ethanol, isopropanol, acetone, or acetonitrile) and then adding the solution to water. For example, lopinavir is dissolved in ethanol (from about 2 to about 4 mL/g) and the ethanolic solution is added with stirring to water (from about 10 about 100 mL/g) to provide amorphous lopinavir. However, this process for the preparation of amorphous lopinavir is not effective on the kilogram scale and thus is not commercially suitable.
PCT Publication No. WO 01/074787 provides various crystalline Forms (Types I, II, III, IV) of solvated and non-solvated lopinavir. It further provides a process for the preparation of amorphous lopinavir which involves dehydration/desolvation of Type I hydrated crystal form/Type II solvated crystal forms.
PCT Publication Nos WO 2006/100552 and WO 2006/090264 provide process for the preparation of crystalline lopinavir.
Organic Process Research & Development, 3, 145-148 (1999), and Organic Process Research & Development, 4, 264-269 (2000); provide a crystallization process for the preparation of crystalline lopinavir which involves recrystallization from mixtures of ethyl acetate and heptane. However, the crystalline lopinavir obtained contains small amounts of solvents and removal of the final traces of solvents proved exceedingly difficult, and even extensive drying after milling (to reduce particle size) did not facilitate its complete removal. It further provides the crystallized product obtained contains appromixately 2% residual ethyl acetate which cannot be removed by further drying.
Thionyl chloride (18 ml) was added to the mixture of 2S-(1-tetrahydropyrimid-2-onyl)-3-methylbutanoic acid (25 gm), tetrahydrofuran (370 ml) and dimethylformamide (2 ml) at 0-10 deg C. and the mass was stirred for 1 hour 15 minutes. The mass was subjected to distillation under reduced pressure to remove excess thionyl chloride, n-heptane (45 ml) was added to the residue obtained and the solvent was distilled off. The reaction mass was slurried in dimethylformamide (105 ml). (2S,3S,5S)-2-(2,6-dimethylphenoxyacetyl)amino-3-hydroxy-5-amino-1,6-diphenylhexane (41 gm), imidazole (25 gm) and 4-(dimethylamino)pyridine (1.5 gm) were dissolved in ethyl acetate (420 ml). To the solution was added above slurried product at 0-10 deg C. The reaction mass was maintained for 14 hours and then ethyl acetate (165 ml) and water (250 ml) were added. The layers were separated, water (250 ml) was added to the organic layer and the pH was adjusted to 2.0-3.0 with dilute hydrochloric acid (6N HCl). The layers were separated, the organic layer was washed with aqueous sodium bicarbonate and then with water. The ethyl acetate was distilled off from the mass. The reaction mass was dissolved in ethyl acetate (80 ml) and n-heptane (80 ml) was added to the solution. The separated solid was stirred with ethyl acetate (290 ml) for 8 hours, filtered and dried the solid to obtain 33 gm of lopinavir ethyl acetate solvate
A large scale process for the synthesis of HIV protease inhibitor candidate ABT-378 has been developed which utilizes an intermediate common to the synthesis of ritonavir, Abbott’s first generation compound. The synthesis relies on the sequential acylation of this intermediate which is carried through as a mixture of diastereomers until the penultimate step. A synthesis of acid 5, derived from l-valine, is also reported.
A 500-mL, three-necked, round-bottomed flask equipped with mechanical stirring, ……………………..DELETED…………………The solid product was washed with 30 mL of 1:1 EtOAc/heptane and dried in vacuo at 70 °C for 60 h, affording 18.8 g (89% yield) of ABT-378 2 as a colorless solid. Before crystallization crude 2 assayed as >93% pure by HPLC; after crystallization >99% purity was achieved.
Anal. Calcd for C37H48N4O5: C, 70.66; H, 7.69; N 8.91. Found: C, 70.26; H, 7.73; N 8.79.
[α]d20 = − 22.85 (c 0.4 MeOH).
Crystallographic studies have shown, to our surprise, that 2 isolated by this crystallization method is not a solvate.
The determination of the enantiomeric excess (% ee) for ABT-378 (2) can be done indirectly. Compound 17, which results from the acylation of 4 with the enantiomer of acid 5, is known to us, having been detected as an impurity in our process development.17 Compound 18 can only result from the acylation of the enantiomer of 4 (2R,3R,5R) with 5. The levels of 17/18 observed in 2 are typically <0.1%. Until there is a need for a more definitive assay, we assume this represents an upper limit to the amount of ent-2 present.
Enantiomeric excess is determined by HPLC (Chiracel OD column, elution with hexane: ethanol: trifluoroacetic acid (930: 70: 1). The desired l-isomer has a retention time of approximately 14 min; the d-isomer, 11.5 min.
Capparelli E, Holland D, Okamoto C, et al. (2005). “Lopinavir concentrations in cerebrospinal fluid exceed the 50% inhibitory concentration for HIV”. AIDS (London, England)19 (9).
Methods and compositions for the treatment or prevention of human immunodeficiency virus and related conditions using cyclooxygenase-2 selective inhibitors and antiviral agents
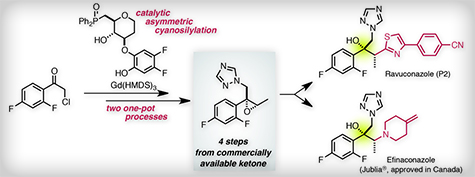
LAVAL, Quebec, June 9, 2014 /PRNewswire/ — Valeant Pharmaceuticals International, Inc. (NYSE: VRX) (TSX: VRX) today announced that that its wholly owned subsidiary, Valeant Pharmaceuticals North America LLC, received notice that the U.S. Food and Drug Administration (FDA) has approved the New Drug Application (NDA) for Jublia® (efinaconazole 10% topical solution), the first topical triazole approved for the treatment of onychomycosis of the toenails
Method for producing butanol derivatives – 1 – 2 – triazole Is a (compound described in Example 1 of Patent Document 1) a compound of formula 1 to be effective against fungal diseases of humans and animals are known, the present invention, (2R, 3R) – 2 – (2,4 – difluorophenyl) -3 – (4 – methylene piperidin-1 – yl) -1 – (1H-1, 2,4 – triazol-1 – yl) butan-2 – (generic name ol ( The present invention relates to preparation of their salts that Fina et Kona zole (Efinaconazole)), hereinafter abbreviated as “KP-103″ or even) in: INN).
The method for obtaining the amino alcohol by ring-opening addition reaction of the amine to the epoxide, in general, using a large excess of amine, and is performed for a long time at a high temperature. In the conventional method, in order to use the amine of the large excess of byproducts is large and requires a recovery step of an amine, also in terms of production costs, it is desirable as a production method on an industrial scale if the amine is expensive no. In order to increase the reactivity of the reaction, the reaction using a Lewis acid have been proposed, also, difficult to use industrially Lewis acid to be used is unstable or expensive, perchlorate, etc., are those toxic-risk is less secure high, there is a problem such as needing attention in use (Non-Patent Documents 1 and 2). It is also reported that could be the use of lithium bromide, to enhance the reactivity under solvent-free conditions at room temperature (Non-Patent Document 3). It is believed that since the liquid at normal temperature and epoxides, amines are used, the method reported in the literature, was achieved by reaction at a high concentration under solvent-free conditions starting material. Thus, a solid at room temperature and can not be applied to epoxides and amines, particularly high melting point.
On the other hand, as described in Patent Document 1, formula 1 compound is produced by ring-opening addition reaction of the amine to the epoxide. In this production process, as the epoxide (2R, 3S) -2 – (2,4 – difluorophenyl) -3 – methyl -2 – [(1H-1, 2,4 - triazol-1 - yl) methyl] oxirane and ( used that methylene piperidine (hereinafter, abbreviated as “4-MP” also) some – is used will hereinafter be abbreviated as “epoxy triazole” also) some, 4 as the amine. In this manufacturing process, it has the disadvantage for heating under reflux for a long time by using the 4-MP solution in large excess in the ring-opening addition reaction, and it is necessary to by-products are produced much in the reaction step to remove them. Furthermore, 4 – methylene-piperidine is prepared by the method described in Patent Document 2, but the purity is low because it is obtained in an aqueous solution, and also affects the reactivity when the distillation isolation there is a problem of impurities by heat generated.
The purpose of the present invention, (2R, 3S) -2 – to oxirane (2,4 – difluorophenyl) -3 – methyl-2 – [methyl-(yl 1H-1, 2,4 - - -1 triazole)] that without using methylene piperidine may yield a compound of Formula 1 under mild conditions to provide a manufacturing method with a reduced formation of by-product – 4 large excess of ring-opening addition reaction of methylene piperidine – 4 some.
As a result of intensive investigations, 4 the present inventors found that – if the acid addition salt of methylene-piperidine, 4 – impurities incorporated in the acquisition phase of the methylene piperidine has been removed, will be isolated as a solid high purity It can, therefore, four of the starting raw material in the ring-opening addition reaction of the amine to epoxy triazole – and that is able to increase the purity of methylene piperidine, the reaction solvent, the ring-opening addition reaction of amines to the epoxy triazole medium, is performed in the presence of hydroxides of alkali metals or alkaline earth metals in particular, 4 – there is no need to use excess methylene piperidine, high yield, by-products and a compound of Formula 1 under mild conditions to discover that it can be produced by reducing things, and have completed the present invention.
I will explain in detail the methods of the present invention are described below. As indicated by the following reaction formula, the present invention, (2R, 3S) -2 – (2,4 – difluorophenyl) -3 – methyl -2 – [(1H-1, 2,4 - triazol - hydrate thereof or a hydroxide of an alkali metal or alkaline earth metal is selected from the group the reaction solvent, consisting of strontium lithium, sodium, and calcium, and a methylene piperidine acid addition salt and 4 - yl) methyl] oxirane including the presence of a, is reacted relates to a manufacturing method of the formula (1) compound.
(Wherein HX represents an acid addition salt of the acid)
Starting material of the process of the present invention It can be also performed by using a starting compound of any amount ranging ton level from the g level, the method of the present invention may be determined the amount of solvent depending on the amount of the starting compound used.
(2R, 3S) -2 – (2,4 – difluorophenyl) -3 – methyl -2 – [(1H-1, 2,4 - -1 triazole - yl) methyl] oxirane, JP 2-191262 issue It can be obtained by methods described in the Gazette. 4 – is represented by the following formula methylenepiperidine acid addition salts:
Wherein an acid of the acid addition salts with HX, 4 – The acid forming the methylene piperidine acid addition salts may, for example, hydrochloric, hydrobromic, if an acid that forms a salt with an amine is basically inorganic acid acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, boric acid, chloric acid, carbonic acid, etc.; formic acid, acetic acid, trifluoroacetic acid, propionic acid, oxalic acid, methanesulfonic acid, benzenesulfonic acid, p – organic acids such as toluenesulfonic acid and the like, but is not limited thereto. Hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid, trifluoroacetic acid and the like, more preferably, preferred examples of the acid is a hydroiodic acid or hydrobromic acid.
4 – methylene-piperidine acid addition salt, 4 – can be obtained by reacting a conventional manner with an acid corresponding to the acid addition salt with methylene piperidine.
Here, the 4 – methylenepiperidine may be preferable in terms of production on an industrial scale, prepared by the method described in WO 97/11939 pamphlet. 4 is manufactured here – methylene-piperidine, and also are obtained in the form of an aqueous solution, impurities produced by heat during the distillation isolation is included, according to the manufacturing method described below, 4 – methylene-piperidine the impurities are removed, acid addition salts can be isolated as a solid high purity.
That is, 4 – preferred method of methylenepiperidine acid addition salts, the following steps: (1) 4 – reacting with an acid corresponding to the acid addition salt, a solution of methylene-piperidine, and (2) the solvent is evaporated as necessary, washed suspension crystallization or a product obtained , it is a method comprising the step of purifying.
Here, 4 (1) Step – A solution of methylene piperidine solution in a mixed solvent of alcohol or water and aqueous alcohol solution or (such as methanol), and the like. 0.9-1.0 equivalents is preferably used amount of the methylene piperidine – 4 of the acid corresponding to the acid addition salt. Reaction conditions (1) is carried out at room temperature from 0 ℃, the reaction time is several hours 15 minutes. After the step (1), if necessary, by conventional methods, for example, under reduced pressure, the temperature is carried out by heating from room temperature solvent was evaporated. In the case of decreasing the water content of the reaction system, for example, by azeotropic toluene or use of the desiccant. How to purify washed suspension or crystallization in step (2) The method of cleaning is suspended in a solvent crystals or recrystallized after being dissolved in a solvent, obtained by filtration, or distilling off the solvent I may be mentioned.
The acid addition salts, conditions of the production method is different, for example, after the reaction of step (1), the solvent was evaporated, the case of hydrochloride and hydrobromide and acetone crystals was then obtained After washing the suspension and filtered. For p-toluenesulfonate, After the reaction of step (1), the solvent was evaporated, and dissolved in ethyl acetate (10:1) / isopropanol mixture and the residue is recrystallized. For nitrate hydroiodide, and trifluoroacetic acid salt, after the reaction of step (1) to dryness by distilling off the solvent, washed and suspended by addition of diisopropyl ether to the residue.
Reaction conditions of the process of the present invention 4 – triazole for the epoxy, the amount of methylene piperidine acid addition salt is 1 to 5 equivalents, preferably 1 to 1.5 equivalents.
As the hydroxide of alkali metal or alkaline earth metal in the reaction of the present invention, a hydrate thereof or strontium hydroxide lithium hydroxide, sodium hydroxide, calcium hydroxide and the like. More preferably, lithium hydroxide, a hydrate thereof or calcium hydroxide, more preferably a hydrate thereof or a lithium hydroxide.
The amount of the hydroxide of alkaline earth metals varies depending on the basicity and the type of compound used or the alkali metals, 4 – for methylenepiperidine acid addition salt is 1 to 5 equivalents usually preferably is 1 to 1.5 equivalents.
Production Example 1 Methanol / water mixture methylene piperidine (4-MP) – 4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 was cooled by stirring in an ice bath under a solution 0.8M 500mL (0.4mol). Thereafter, the solution is added in several portions (0.36mol) 48% hydrobromic acid 61.3g, followed by stirring for 1 hour in an ice bath. Thereafter, to precipitate white crystals by evaporating the solvent by heating under reduced pressure. Subsequently, we conducted two times operation for azeotropic water by distilling off the solvent by heating under reduced pressure and toluene was added to 50mL, and added acetone 192mL, was 2 hours under stirring ice bath. Thereafter, The crystals are filtered, washed crystals with 60mL (cooled in an ice bath) of acetone, 4-MP · HBr58g as colorless crystals (yield: 90%) After air-drying at room temperature, for 12 hours and dried under reduced pressure at 40 ℃ I got. 1 H-NMR (500MHz, CDCl 3) δ: 2.62 (4H, t, J = 6.09 Hz), 3.26 (4H, t, J = 6.09 Hz), 4.90 (2H, s), 9.18 (1H, br). Melting point (DSC): 147 ~ 147.9 ℃
Production Example 2 Water removal operation (4-MP) methylene piperidine – 4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine p-toluenesulfonic acid salt of (4-MP · PTSA) – 4 isopropanol (9.7g, 0.1mol) of (IPA) in (50mL) solution, 4-MP, which is subjected to, the resulting p-toluenesulfonic acid monohydrate (PTSA · H 2 O) (18.1g, 0.095mol) / was added (80mL) IPA, (weak exothermic) after which the mixture was stirred for 30 minutes at room temperature, evaporated under reduced pressure IPA, and was heated and dissolved in (250mL) with ethyl acetate / IPA mixture = 10:1 residue. After cooling to room temperature and allowed to stand for 20 hours at 0 ~ 5 ℃, filtered washing the precipitated crystals were obtained (91.2% yield) 4-MP · PTSA 23.34g of white crystals to dry. 1 H-NMR (400MHz, DMSO-d 6) δ: 2.29 (3H, s), 2.35 (4H, t, J = 6.4 Hz), 3.08 (4H, t, J = 6.4 Hz), 4.85 (2H, s), 7.13 (2H, d, J = 8.2 Hz ), 7.49 (2H, d, J = 8.2 Hz), 8.58 (2H, brs).
Production Example 3 4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine hydrochloride (4-MP · HCl) – - 4 subjected to moisture removal operation methylene piperidine (4-MP), obtained was cooled by stirring in an ice bath under (4.12mol) 4-MP 400g that is. Thereafter, the solution was added concentrated hydrochloric acid and 350mL (4.08mmol), and the mixture was stirred in an ice bath. After concentration under reduced pressure was performed 3 times operation for azeotropic water and concentrated under reduced pressure and toluene was added to 300mL. The washed suspension under ice-cooling and addition of acetone 300mL. The filtered crystals were washed with acetone crystals, 4 and dried under reduced pressure at room temperature – was obtained (46% yield) methylene-piperidine hydrochloride (4-MP · HCl) 336.8g. 1 H-NMR (500MHz, CDCl 3) δ: 2.58 (4H, t, J = 6.1Hz), 3.22 (4H, t, J = 6.1Hz), 4.89 (2H, s), 9.70 (1H, br s).
Preparation Example 4 Methanol / water mixture methylene piperidine (4-MP) – 4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine hydriodic acid salt of (4-MP · HI) – 4 was cooled by stirring in an ice bath under a solution 0.66M 20mL (13.19mmol). Thereafter, the solution was added 57% hydroiodic acid and 2.66g (11.84mmol), and the mixture was stirred for 15 minutes in an ice bath. After concentrated under reduced pressure, to precipitate a white solid by performing twice the operation for azeotropic water and concentrated under reduced pressure and toluene was added to 1.6mL. The washed suspension for 1 hour at room temperature by addition of diisopropyl ether 6mL. Thereafter, The crystals are filtered, washed and crystallized with diisopropyl ether, 4 and dried under reduced pressure at room temperature – was obtained (90% yield) methylene piperidine hydroiodide (4-MP · HI) 2.66g. 1 H-NMR (500MHz, CDCl 3) δ: 2.66 (4H, t, J = 6.1Hz), 3.31-3.33 (4H, m), 4.91 (2H, s), 8.34 (1H, br s).
Preparation Example 5 The reaction was carried out similarly to the above method by using trifluoroacetic acid (TFA) 1.35g and (11.87mmol) in place of hydriodic acid production 57% methylene piperidine trifluoroacetate salt of (4-MP · TFA), – 4 I got a (92% yield) methylene piperidine trifluoroacetate (4-MP · TFA) 2.55g – 4. 1 H-NMR (500MHz, CDCl 3) δ: 2.50 (4H, t, J = 6.1Hz), 3.16 (4H, t, J = 6.1Hz), 4.89 (2H, s), 9.52 (1H, br s).
Preparation Example 6 The reaction was carried out in the same manner as the above-described method using 69% nitric acid 1.08g the (11.87mmol) instead of hydroiodic acid production 57% methylene piperidine nitrate (4-MP · HNO 3), 4 – - 4 methylenepiperidine nitrate I got a (89% yield) (4-MP · HNO 3) 1.87g. 1 H-NMR (500MHz, CDCl 3) δ: 2.53 (4H, t, J = 6.1Hz), 3.28 (4H, t, J = 6.1Hz), 4.89 (2H, s), 8.85 (1H, br s).
Was stirred while addition of acetonitrile 80mL, lithium hydroxide 2.859g methylene piperidine hydrobromide (4-MP · HBr) 21.26g and (119.4mmol) and (119.4mmol) – 4 obtained in Production Example 1. Then, (2R, 3S) -2 – (2,4 – difluorophenyl) -3 – methyl -2 – [(1H-1, 2,4 - triazol-1 - yl) methyl] oxirane and 20g (79.6mmol) was added, and the mixture was heated under reflux for 14 hours at (external temperature 100 ℃) oil bath. After completion of the reaction, to precipitate the crystals by the addition of ethanol and distilled water to the reaction solution. Thereafter, the crystals were filtered, washed with ethanol / water mixture 40mL, and naturally dried at room temperature for 12 hours and dried under reduced pressure at 40 ℃, KP-103 24.2g light yellow 87.3% (yield, HPLC purity 95.3 % I got). 1 H-NMR (500MHz, CDCl 3) δ: 0.96 (3H, dd, J = 2.68, 7.08 Hz), 2.13-2.26 (4H, m), 2.35 (2H, br), 2.70 (2H, br) ,2.90-2 .94 (1H, q, J = 7.08 Hz), 4.64 (2H, s), 4.82 (1H, dd, J = 0.73, 14.39 Hz), 4.87 (1H, dd, J = 0.73, 14.39 Hz), 5.45 (1H, s), 6.72-6.81 (2H , m), 7.51 (1H, dt, J = 6.59, 9.03 Hz), 7.78 (1H, s), 8.02 (1H, s). FAB-MS m / z: 349 [M + H] + :86-89 ℃ melting point Optical rotation: [α] D25 -87 ~ -91 ° (C = 1.0, methanol)
Example 2 Epoxy triazole 0.50g (1.99mmol), 4 – in addition to acetonitrile 2mL lithium hydroxide 0.07g methylene piperidine hydrobromide (4-MP · HBr) 0.53g and (2.98mmol) and (2.96mmol), oil bath ( I was heated under reflux for 14 hours at an external temperature of 100 ℃). After the solvent was evaporated under reduced pressure of the reaction solution obtained, the solution was separated by the addition of ethyl acetate and water to the residue. The organic layer was concentrated under reduced pressure and purified by silica gel column chromatography (1:1) hexane / ethyl acetate solvent, to give (86% yield) KP-103 0.59g.
Example 3 The reaction was carried out in the same manner as in Example 2 using the calcium hydroxide 0.22g (2.97mmol) instead of lithium hydroxide, to give (82% yield) KP-103 0.57g.
Example 4 Was performed for 19 hours and the reaction in the same manner as in Example 2 using strontium hydroxide 0.36g a (2.98mmol) in place of lithium hydroxide, to give (68% yield) KP-103 0.47g.
Example 5 Was added 2mL of acetonitrile lithium hydroxide monohydrate 0.13g methylene piperidine hydrobromide (4-MP · HBr) 0.53g and (2.98mmol) and (2.96mmol) – epoxy-triazole 0.50g (1.99mmol), 4 , I was heated under reflux for 14 hours at (external temperature of 100 ℃) oil bath. Was determined to (relative area percentage of KP-103) reaction rate by HPLC measurements by sampling the reaction mixture, it was confirmed the formation of KP-103 at 81% response rate.
Example 6 The reaction was carried out in the same manner as in Example 2 using cyclopentyl methyl ether (CPME) 2mL instead of acetonitrile, to give (91% yield) KP-103 0.63g.
Example 7 1,2 instead of acetonitrile – The reaction was carried out in the same manner as in Example 2 using dimethoxyethane (DME) 2mL, was obtained (79% yield) KP-103 0.55g.
Example 8 1 in place of acetonitrile – The reaction was carried out in the same manner as in Example 2 using butanol 2mL, was obtained (72% yield) KP-103 0.59g.
Example 9 The reaction was carried out in the same manner as in Example 2 using isopropanol 2mL instead of acetonitrile, to give (86% yield) KP-103 0.50g.
Example 10 Methyl – 2 – 4 instead of acetonitrile reaction was carried out in the same manner as in Example 2 using the pentanone (MIBK) 2mL, to give (88% yield) KP-103 0.61g.
Example 11 Example Using methylene piperidine hydrochloride (4-MP · HCl) 0.40g and (2.99mmol) – 4 obtained in Production Example 3 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as 2, was obtained (67% yield) KP-103 0.47g.
Example 12 Using methylene piperidine hydroiodide (4-MP · HI) 0.67g and (2.99mmol) – 4 obtained in Production Example 4 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as in Example 2 upgrade does not give (90% yield) KP-103 0.62g.
Example 13 Using methylene piperidine trifluoroacetate (4-MP · TFA) 0.63g and (2.98mmol) – 4 obtained in Production Example 5 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as in Example 2, was obtained (78% yield) KP-103 0.54g.
Example 14 Example Using methylenepiperidine nitrate (4-MP · HNO 3) 0.48g and (3.00mmol) – 4 obtained in Production Example 6 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as 2, was obtained (71% yield) KP-103 0.49g.
Example 15 Methylenepiperidine hydroiodic acid – 4 obtained in Production Example 4 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – Sodium hydroxide 0.12g (2.98mmol), 4 instead of lithium hydroxide was performed 18 hours and the reaction in the same manner as in Example 2 using salt (4-MP · HI) 0.67g and (2.99mmol), to give (73% yield) KP-103 0.51g.
Conventional methods for producing formula 1 compound of the starting material 4 – impurity contamination at the acquisition stage of methylene piperidine, and by-products to be produced during the manufacture of the formula 1 compound was a problem. In the method of the present invention, as a starting material of the production method of the formula 1 compound, 4 – by making the acid addition salt of methylene-piperidine, 4 – impurities incorporated in the acquisition phase of the methylene piperidine has been removed, high purity it is possible to use a solid. In the method of the present invention, since the ring-opening addition of the amine to epoxy triazole is promoted, 4 – there is no need to use excess methylene piperidine, high yield, and by-products of the compound of Formula 1 under mild conditions It can be produced by reducing compound. Therefore, by the method of the present invention to produce an industrial scale formula 1 compound became possible.
Tschen EH, Bucko AD, Oizumi N, Kawabata H, Olin JT, Pillai R (Feb 2013). “Efinaconazole solution in the treatment of toenail onychomycosis: a phase 2, multicenter, randomized, double-blind study”. J Drugs Dermatol12 (2): 186–192. PMID23377392.
Tatsumi Y, Nagashima M, Shibanushi T, et al. (May 2013). “Mechanism of action of efinaconazole, a novel triazole antifungal agent”. Antimicrob Agents Chemother57 (5): 2405–2509.
“These patients were treated in our Phase 1 clinical trial of Selinexor in … Additional Phase 1 and Phase 2 studies are ongoing or currently planned and … the discovery and development of novel first-in-class drugs directed against …
Achillion Pharmaceuticals Inc. announced the company has begun dosing ACH-3422, a uridine-analog nucleotide polymerase inhibitor, for seven days in patients with genotype 1 chronic hepatitis C viral infection (HCV) in its ongoing Phase 1 clinical trial. Proof-of-concept results from this trial are expected to be reported during the fall of 2014. Furthermore, Achillion announced that the U.S. Food and Drug Administration (FDA) has removed the clinical hold on sovaprevir, an NS3/4A protease inhibitor, to permit the conduct of trials in patients with HCV. Sovaprevir doses of 200 mg once daily, the previously evaluated dose that was well-tolerated with clinical activity in two completed Phase 2 studies, may be used in additional therapeutic clinical trials.
Alan Gray, senior director, oncology commercial strategy group, Kantar Health
As in previous years, ASCO 2014 did not disappoint, revealing exciting data for promising new treatments, especially in the field of immuno-oncology. Other exciting drugs (CDKs) and newer generations of targeted agents for EGFR and ALK mutations continue to advance treatment of
Image may be NSFW. Clik here to view.
Considerations for Value Preparation for ASCO 2014 Drugs
Medical Marketing and Media
Other exciting drugs (CDKs) and newer generations of targeted agents for … that are traditionally completed during Phase III of clinical development.
Will the FDA finally approve Contrave, Orexigen’s hot new diet drug, this week? Expectations are high for this new weight loss medication, which could prove an important anti-obesity aid. The ruling is expected as soon as tomorrow.
Now temporarily renamed NB32, Contrave was eagerly awaited when it got a startling thumbs-down from the FDA in 2011. The agency demanded additional research into cardiovascular risks.
The execs at Dr. Reddy’s Laboratories have taken notice as some of its competitors in India have run afoul of the FDA over loose manufacturing standards. The actions have meant banned plants and plummeting revenues for Ranbaxy Laboratories and Wockhardt. To avoid that fate, Dr. Reddy’s and some other Indian drugmakers have decided it is worth investing hundreds of millions of dollars for new plants and equipment in a country that has traditionally relied on cheap human labor.
 Originally posted on lyranara.me:
Originally posted on lyranara.me:
 Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW.