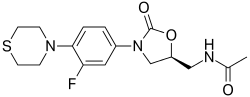
Sutezolid
168828-58-8
N-({(5S)-3-[3-fluoro-4-(thiomorpholin-4-yl)phenyl]-2-oxo-oxazolidin-5-yl}methyl) acetamide
(S)—N-[[3-[3-fluoro-4-(4-thiomorpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide
Sutezolid (PNU-100480, PF-02341272) is an oxazolidinone antibiotic currently in development as a treatment for extensively drug-resistant tuberculosis.
Sutezolid, an antimicrobial oxazolidinone and the thiomorpholine analogue of linezolid, had been in early clinical development for the treatment of tuberculosis. However, development was discontinued.
The compound had been found to be active against Gram-positive bacteria such as multiresistant staphylococci, streptococci and enterococci. It was being developed by Pfizer. In 2011, orphan drug designation was assigned in the U.S. and the E.U. for the treatment of tuberculosis.
In 2013, Sequella acquired an exclusive worldwide license for the development and commercialization of sustezolid.
|
8-5-2011
|
Combination Therapy for Tuberculosis
|
http://www.google.com/patents/US20110190199
Scheme 1 illustrates a general synthetic sequence for preparing compounds of the present invention.

Example 3 Preparation of (5S)-5-{[(4-chlorobenzylidene)amino]methyl}-3-(3-fluoro-4-thiomorpholin-4-ylphenyl)-1,3-oxazolidin-2-one
The title compound in Example 2 (194 g, 0.56 mole), and the title compound of Example 1 (195 g, 0.84 mole), and lithium tert-butoxide (116 g, 1.4 mole) were charged into a 3000 mL three neck round bottom flask under nitrogen. The reactants were slurried with methyl tert-butyl ether (1200 mL) and the mixture was warmed to 56° C. and stirred for 2 h as a yellow solid gradually formed. The reaction was cooled to room temperature, and diluted with 1200 mL water. The mixture was then stirred vigorously over 60 min as the solid changed from dark yellow to a more pale yellow solid. The mixture was cooled to 10° C., filtered, and the filter cake was washed with ice cold methyl tert-butyl ether (450 mL). The resulting light yellow solid was dried in air for 30 min, then placed in a vacuum oven and dried at 40° C. overnight to afford the title compound (243 g, 99% yield). 1H NMR (400 MHz, CDCl3): δ 2.8 (m, 4H), 3.2 (m, 4H), 3.9 (m, 2H), 4.1 (m, 2H), 5.0 (m, 1H), 6.9 (m, 1H), 7.2 (m, 1H), 7.4 (m, 3H), 7.6 (m, 2H), 8.4 (s, 1H).
Example 4 Preparation of N-{[(5S)-3-(3-fluoro-4-thiomorpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl}acetamide
The title compound in Example 3 (243 g, 0.56 mole) was combined with EtOAc (1300 mL) and water (1300 mL) in a 5000 mL three neck round bottom flask equipped with a mechanical stirrer. The mixture was treated drop-wise with 12N HCl (140 mL, 1.68 moles) and the mixture was stirred vigorously for 1 hour at room temperature. The layers were separated and the aqueous layer was washed with EtOAc (1×500 mL). The resulting aqueous solution containing (S)-5-(aminomethyl)-3-(3-fluoro-4-thiomorpholinophenyl)oxazolidin-2-one hydrochloride was combined with a mixture of dichloromethane (1800 mL) and MeOH (120 mL), and the vigorously stirred mixture was charged with acetic anhydride (132 mL, 1.4 mole) in one portion and subsequently treated drop-wise with 10 N NaOH (200 mL, 2.0 mole) over 15 min. An extremely thick reaction mixture resulted from addition of the base, which gradually thinned as the pH rose and the acylation rapidly progressed. The reaction was stirred vigorously for 1 hour after the mixture resolved to two phases. At that time, 10 M NaOH (160 mL, 1.6 mole) was added drop-wise to the mixture until the pH was stable at 7. The layers were separated, the aqueous layer was extracted with dichloromethane (250 mL), and the combined organic layers were dried over anhydrous potassium carbonate. The volatiles were removed in vacuo to give an off-white solid which was titrated with methyl tert-butyl ether (250 mL), collected, and dried in vacuo to give title compound (5) (186.1 g, 94% yield) as a fine white solid with greater than 98% HPLC purity (retention time=3.93 minutes, HPLC conditions reported below).
The crude solid was dissolved in warm 6% methanol in dichloromethane (1250 mL) in a 5000 mL three neck round bottom flask equipped with a mechanical stirrer. The solution was warmed to reflux, diluted by the portion-wise (500 mL) addition of 2500 mL isopropanol (IPA), and, in order to maintain reflux, the temperature was ramped to 50-70° C. On completion of this addition of IPA, the reflux condenser was replaced with a short-path distillation head and distillation was continued into a cooled flask. During distillation, a 500 mL portion of fresh IPA was added after 500 mL of distillate was collected to maintain between 2000 and 2500 mL IPA present at all times. After this addition (internal flask temperature dropped to 60° C.) the mixture became slightly cloudy and remained so for the balance of the distillation, becoming increasingly cloudy as the distillate temperature exceeded 70° C.; particulate matter appeared as the distillate temperature exceeded 75° C. The temperature controller was ramped to 85° C. and held there until the conclusion of the distillation. When the distillate was clearly isopropanol alone (82-83° C.) the volume was reduced to 2500 mL hot IPA, the heating mantle was removed, stirring was discontinued, and the paddle was removed from the flask. The mixture was allowed to continue to crystallize as the flask cooled. The white crystalline solid was then collected by filtration, washed with methyl tert-butyl ether (250 mL), and dried in vacuo at 40° C. to afford 180 g (91% yield) of the title compound in greater than 99% HPLC purity (retention time=3.93 minutes, HPLC conditions reported below). 1H NMR (400 MHz, DMSO-d6): δ 1.8 (s, 3H), 2.7 (m, 4H), 3.2 (m, 4H), 3.4 (m, 2H), 3.7 (m, 1H), 4.7 (m, 1H), 7.1 (m, 1H), 7.15 (m, 1H), 7.2 (m, 1H), 8.2 (m, 1H). Mass Spec. C16H20FN3O3S: m/z 354.1 (M+1).
HPLC conditions for analyses mentioned in the text: HP Series 1100; Column: Symmetry C8 5 uM 4.6×50 mm; Flow rate 1.2 mL/min; Solvent A: water with 0.1% formic acid, Solvent B: acetonitrile with 0.1% formic acid; Injection volume=10 uL of 1 mg/mL (acetonitrile); Gradient: Solvent B 0-100% over 7 minutes then 100% B for 1 minute; wavelength=254 nm.
Identification of a novel oxazolidinone (U-100480) with potent antimycobacterial activity
J Med Chem 1996, 39(3): 680
http://pubs.acs.org/doi/full/10.1021/jm950956y

(S)-N-[[3-[3-Fluoro-4-(4-thiomorpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide (6, U-100480). A solution of (R)-[3-[3-fluoro-4-(4-thiomorpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl azide (19.662 g, 58.28 mmol) in dry THF (290 mL) was treated with triphenylphosphine (16.815 g, 64.11 mmol) over 10 min. After 2.0 h, TLC analysis (10% MeOH/CHCl3) revealed the conversion to iminophosphorane was complete. H2O (2.10 mL, 116.56 mmol) was added and the reaction mixture heated to 40 °C (internal temperature) for 5 h and then allowed to cool to ambient temperature overnight. At this point, TLC analysis (10% MeOH/CHCl3) indicated incomplete hydrolysis of the iminophosphorane intermediate. More H2O (8.40 mL) was added, and the reaction was heated to 40 °C for 5 h. At this time, TLC indicated complete conversion to the 5-(aminomethyl)oxazolidinone intermediate. The reaction mixture was first concentrated by rotary evaporation (benzene was added several times to azeotrope off the H2O) and then under high vacuum to give the crude amine as an off-white solid. This material was dissolved in CH2Cl2 (250 mL), treated with pyridine (46.099 g, 47.10 mL, 582.79 mmol) and acetic anhydride (29.749 g, 27.49 mL, 291.40 mmol), and then stirred overnight at ambient temperature. TLC analysis (10% MeOH/CHCl3) showed complete conversion to 6. The reaction mixture was diluted with CH2Cl2, transferred to a separatory funnel, and then washed with 1 N HCl until the washings were acidic. The organic layer was then washed with saturated aqueous NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated in vacuo to give crude 6 (U-100480) as a cream-colored solid. The crude product was triturated with hot CHCl3; most but not all of the solids dissolved. After cooling to ambient temperature, the solids were filtered off (cold CHCl3 wash) and dried in vacuo to furnish 13.174 g of analytically pure title compound as a white solid. A second crop of 3.478 g, also analytically pure, afforded a combined yield of 81%:
mp 186.5−187.0 oC; [α]D −8° (c 1.00, CHCl3);
IR (mull) 1749, 1746, 1641, 1656, 1518, 1448, 1419, 1225, 1215, 1158, 1106, 1083, 867 cm-1;
1H NMR (300 MHz, CDCl3) δ 7.42 (dd, 1H, J = 2.6, 14.0 Hz), 7.06 (ddd, 1H, J = 1.0, 2.6, 8.8 Hz), 6.95 (dd, 1H, J = 9.0, 9.0 Hz), 6.61 (br t, 1H, J = 6.0 Hz), 4.81−4.72 (m, 1H), 4.02 (dd, 1H, J = 9.0, 9.0 Hz), 3.75 (dd, 1H, J = 6.7, 9.1 Hz), 3.71−3.55 (m, 2H), 3.32−3.27 (m, 4H), 2.84−2.79 (m, 4H), 2.02 (s, 3H);
MS m/z (rel intensity) 353 (M+, 100), 309 (31), 279 (5), 250 (17), 235 (14), 225 (20), 212 (7), 176 (19), 138 (18), 42 (28);
HRMS calcd for C16H20N3O3FS 353.1209, found 353.1200. Anal. (C16H20N3O3FS) C, H, N.
see aLSO
WO 1995007271
WO 2010026526
Repurposed drugs for tuberculosis treatment.
http://www.nature.com/nrd/journal/v12/n5/fig_tab/nrd4001_F1.html

Filed under: Uncategorized Tagged: PNU-100480, Sutezolid







