Erlotinib
N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)
quinazolin-4-amine
![]()
| Chemical Name: Erlotinib Hydrochloride (Tarceva) |
| Synonyms: N-(3-Ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine Hydrochloride; 6,7-Bis(2-methoxyethoxy)-4-(3-ethynylanilino)quinazoline Hydrochloride; [6,7-Bis(2-methoxyethoxy)quinazolin-4-yl]-(3-ethynylphenyl)amine Hydrochloride; CP 358774; OSI 774; Tarceva; |
| CAS Number: 183319-69-9 |
|
| Mol. Formula: C22H24ClN3O4 |
| Appearance: Off-White Solid |
| Melting Point: 223-225°C |
| Mol. Weight: 429.9 |
Erlotinib hydrochloride (trade name Tarceva) is a drug used to treat non-small celllung cancer, pancreatic cancer and several other types of cancer. It is a reversibletyrosine kinase inhibitor, which acts on the epidermal growth factor receptor (EGFR). It is marketed in the United States by Genentech and OSI Pharmaceuticals and elsewhere by Roche
Erlotinib is an EGFR inhibitor. The drug follows Iressa (gefitinib), which was the first drug of this type. Erlotinib specifically targets the epidermal growth factor receptor (EGFR)tyrosine kinase, which is highly expressed and occasionally mutated in various forms of cancer. It binds in a reversible fashion to the adenosine triphosphate (ATP) binding site of the receptor.[1] For the signal to be transmitted, two EGFR molecules need to come together to form a homodimer. These then use the molecule of ATP to trans-phosphorylate each other on tyrosine residues, which generates phosphotyrosine residues, recruiting the phosphotyrosine-binding proteins to EGFR to assemble protein complexes that transduce signal cascades to the nucleus or activate other cellular biochemical processes. By inhibiting the ATP, formation of phosphotyrosine residues in EGFR is not possible and the signal cascades are not initiated.
Erlotinib hydrochloride (1), chemically named as N-(3-ethynylphenyl)-6,7-bis-(2-meth- oxyethoxy)-4-qumazolimmine monohydro chloride, is an inhibitor of oncogenic and proto- oncogenic protein tyrosine kinases, e.g. epidermal growth factor receptor (EGFR). Erlotinib is therefore useful in the treatment of proliferative disorders and is currently marketed for the treatment of lung cancer and pancreatic cancer.
![]()
(Erlotinib Hydrochloride)
(1)
![]()
It has been reported that erlotinib hydrochloride can exist in different polymorphic forms. The manufacturing process for many pharmaceuticals is hindered by the fact that the organic compound which is the active ingredient can exist in more than one polymorphic form. It is essential in pharmaceutical development to ensure that the manufacturing process for the preparation of the active ingredient affords a single polymorph with a consistent level of polymorphic purity. If the manufacturing process produces a product with varying degrees of polymorphic purity and/ or or where the process does not control polymorphic inter-conversion, it could lead to serious problems in dissolution and/ or bioavailability in the finished pharmaceutical composition comprising the active ingredient, Erlotinibhydrochloride is disclosed in patent US 5,747,498 and details of the disclosed method for the preparation of erlotinib hydrochloride are described in Scheme 1.
Scheme 1
4-Chloro-6,7-bis-(2-methoxyed oxy)qiunazoline (2) was reacted with 3-emynylaniline (3) or its hydrochloride salt using various solvents and pyridine as a base to yield erlotinib hydrochloride (1) which was treated widi a biphasic mixture consisting of saturated aqueous NaHC03, chloroform and methanol, to formerlotinib base (4). The base (4) obtained in the organic phase was purified by flash chromatography to afford purified erlotinib base. The purified base was further treated with hydrochloric acid in the presence of diethyl ether and chloroform to yield erlotinib hydrochloride.
This isolation of purified erlotinib base required the use of a lengthy workup process including column chromatography and required the chlorinated solvent, chloroform, which is not particularly suitable £01 commercial production of pharmaceuticals. Furthermore, the p irification by column chromatography is neither economical nor feasible at industrial scale. In addition, substantially pure erlotinib could not be obtained.
Two crystalline forms of erlotinib hydrochloride (polymorph A and polymorph B), were characterized by XRPD in patent application, WO 01/34574. Erlotinib hydrochloride can be obtained in form A or in a mixture of polymorph A and B, by refluxing 3-ethynylaniline and 4-chloro-6,7-bis-(2-methoxyemoxy)-qitiiiazoline in a mixture of toluene and acetonitrile. This afforded polymorph A or a mixture of polymorph A and B. It was also disclosed that the formation of polymorph A was favoixred by reducing the amounts of acetonitrile with respect to toluene.
Furthermore, erlotinibhydrochloride polymorph A can be converted into polymorph B by refluxing the polymorph A with alcohol/water. Consequently, in the disclosed methods, there was always contamination of form A with form B and vice-versa. In addition, the products of the reaction are not chemically pure and difficult to purify thereafter. Consequently, these methods are not suitable for preparation of commercial quantities of pure polymorph A.
A process for the preparation of erlotinib hydrochloride, polymorph E by condensation reaction of 3-emynylaiiiline and 4-chloro-6,7-bis-(2-memoxyethoxy)quii azoline in ( , , )- trifiuorotoluene and HC1 was disclosed in U.S. Patent application 2004/0162300. Polymorph E was characterized by XRPD, IR and melting point. However, (α,α,α)- trifluorotoluene is a highly flammable and dangerous solvent for the environment and is not suitable for commercial production. A process for the preparation of erlotinib hydrochloride, polymorph A by reaction of erlotinib base widi aqueous or gaseous HC1 was disclosed in US 2009/0131665. In this method, toluene, a mixture of toluene and methanol, TBME, ethyl acetate, 1-butanol or MIBK were used as a solvent.
However, when DCM, diethyl ether, isopropyl acetate, was used as a solvent, polymorph B was formed. In practice, it has been found that the disclosed methods are inconsistent and afford polymorphic mixtures. In particular, example 1 of US 2009/131665 was repeated and erlotinib hydrochloride was obtained with only 97% purity. In addition, XRPD analysis showed d at the example afforded form B or mixtures of forms A and B. Furthermore, several crystallizations of erlotinib hydrochloride, obtained from repetition of the example, using various solvents and their combinations would not yield a product pure enough to comply with ICH guidelines.
A process for the preparation of a hydrate of erlotinib hydrochloride comprising crystallization of erlotinib hydrochloride using water as solvent, preferably in the absence of organic solvent was disclosed in US 20080167327. This patent also disclosed the process to prepare hemihydrate polymorph form I as well as form II.
A process for the preparation of erlotinib hydrochloride, polymorph M, N and P by reaction of erlotinib base and aqueous or gaseous HC1 dissolved in organic solvents was disclosed in WO 2008/102369.
A process for the preparation of erlotinib hydrochloride by condensation reaction of 4- chloro~6,7-bis-(2-me oxyemoxy)-quinazoline and 3-ethynylaniline in isopropyl alcohol as a solvent and pyridine as a base was disclosed in Molecules Journal (Vol, 11, 286, 2006) but no details on the polymorph were disclosed.
A method for the preparation of erlotinib hydrochloride polymorph A comprising passing hydrochloride gas onto solid erlotinib base containing residual amounts of isopropanol was disclosed in WO 2010/040212. However, in practice it was found that the process did not afford chemically or polymorphically pure product. Repetition of example 1 (page 8) of WO 2010/040212 to prepare erlotinibhydrochloride, by reaction of erlotinib base and gaseous HQ in IPA as a solvent, afforded a mixture of polymorph A and polymorph B (as checked by XRPD).
A process for the preparation of acid salts of erlotinib by reaction of 4-chloro-6,7-bis-(2- memoxyemoxy)-quinazoline and 3-emynykniline or an acid salt of 3-emynylaniline under acidic conditions to form the corresponding erlotinib salt was disclosed in US 2010/0094004.
In order to complete the reaction, several hours (6 hours) of reflux was required and hence it is not a cost effective process. In addition, in practice it was found that the process did not afford chemically or polymorplxLcally pure product. A process £oi the preparation of erlotinib base, polymorph Gl, G2 and G3 was disclosed in WO 2009/002538 and WO 2010/05924.
Scheme 2
A method for the preparation of eiiotinib hydrochloride was disclosed in US 2009/0306377. The method, illustrated in Scheme 2, involves treating 6,7-dimethoxy- 4(3H)-quinazolone (5) with hydrobiOmic acid or pyridine-hydrochloric acid to afford 6,7- dihydroxy-4(3H)-quinazolone (6), which was diacetylated with acetic anhydride to afford diester (7), which was treated with oxalyl chloride/DMF to afford 4-chloro-6,7- ctiacetoxyquinazoline (8). Compound (8) was condensed with 3-e ynylaniline to afford JV- (3-ethynylphenyl)-6,7-dihydfoxy-4-quinazolinamine hydrochloride (9), which was converted into the diol N-(3-emynylphenyl)-6,7-dmyckOxy-4-quinazolinamine (10) by treatment with aqueous ammonia/methanol.
The diol (10) was treated with 2-iodo-ethylmethyl ether to yield compound (4) which on treatment with HC1 afforded erlotinib hydrochloride (1). However, this preparation of erlotinib hydrochloride is a long synthetic route and gives low yields and requires very toxic reagents like pyridine, HBi and controlled reagents like acetic anhydride. Hence, it is not suitable for large scale production. Object of the invention
The priot art processes described above for the preparation of erlotinib and its salts have major disadvantages with respect to the formation and removal of process related chemical and polymorphic impurities; poor commercial viability due to die use of hazardous reactants; expensive, time consuming separation methods such as column chromatography and/ or low yields and purity of final and intermediate products.
As the commercial production of erlotinib hydrochloride is of great importance, for the treatment of cancer, and in view of the above disadvantages associated with the prior art there is a real need for alternative and improved processes for the preparation of erlotinib hydrochloride which do not involve multiple steps and further eliminates the need for cumbersome purification techniques, particularly for the removal of the chemical and polymorphic impurities. The alternative processes must be economical and high yielding and provide erlotinib and its salts with a high degree of chemical and polymorphic purity.
U.S. Patent No. 5,747,498 disclosed 4-(substituted phenylamino) quinazoline derivatives, processes for their preparation, pharmaceutical compositions in which they are present and method of use thereof. These compounds are Tyrosine Kinase Inhibitors and are useful in the treatment of hyperproliferative diseases, such as cancers, in mammals. Among them, erlotinib hydrochloride, chemically N-(3-ethynylphenyl)-6,7-bis(2-methoxy ethoxy)-4-quinazolinamine hydrochloride is a selective inhibitor of the erbB family of oncogenic and protooncogenic protein tyrosine kinases, such as epidermal growth factor receptor (EGFR), and is useful for the treatment of proliferative disorders, such as cancers, particularly non small cell lung cancer, pancreatic cancer, ovarian cancer, breast cancer, glioma, head cancer or neck cancer.
Polymorphism is defined as “the ability of a substance to exist as two or more crystalline phases that have different arrangement and /or conformations of the molecules in the crystal Lattice. Thus, in the strict sense, polymorphs are different crystalline forms of the same pure substance in which the molecules have different arrangements and / or different configurations of the molecules”. Different polymorphs may differ in their physical properties such as melting point, solubility, X-ray diffraction patterns, etc. Polymorphic forms of a compound can be distinguished in the laboratory by analytical methods such as X-ray diffraction (XRD), Differential Scanning Calorimetry (DSC) and Infrared spectrometry (IR).
Solvent medium and mode of crystallization play very important role in obtaining a crystalline form over the other.
Erlotinib hydrochloride can exist in different polymorphic forms, which differ from each other in terms of stability, physical properties, spectral data and methods of preparation.
The U.S. Patent No. 5,747,498 (herein after referred to as the ‘498 patent) makes no reference to the existence of specific polymorphic forms of erlotinibhydrochloride. In this patent, it is disclosed that the compound is isolated according to conventional techniques; more precisely, according to the embodiments exemplified, crude erlotinib hydrochloride residue (obtained by reaction of 4-chloro-6,7-bis-(2-methoxyethoxy)-quinazoline with 3-ethynylaniline or its hydrochloride salt in a solvent such as a d-Cβ-alcohol, dimethylformamide, N-methylpyrrolidin-2-one, chloroform, acetonitrile, tetrahydrofuran, 1,4-dioxane, pyridine or other aprotic solvents, preferably isopropanol) is basified with saturated aqueous NaHCO3 in the presence of methanol and chloroform followed by flash chromatography on silica using 30% acetone in hexane to afford erlotinib free base, which is further treated with hydrochloric acid in the presence of diethyl ether and chloroform to give erlotinib hydrochloride (melting point: 228° – 2300C).
PCT Patent Publication No. WO 99/55683 disclosed erlotinib mesylate anhydrate and hydrate polymorphic forms, their method of preparation and pharmaceutical compositions containing thereof.
PCT Patent Publication No. WO 01/34574 A1 (herein after referred to as the ‘574 patent publication) described two crystalline forms of erlotinib hydrochloride (polymorph A and polymorph B), characterized by powder X-ray diffraction (p-XRD) pattern. The publication further taught that the synthetic procedure described and exemplified in the ‘498 patent produces the erlotinib hydrochloride as a mixture of the polymorphs A and B.
TARCEVA (erlotinib), a kinase inhibitor, is a quinazolinamine with the chemical name N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine. TARCEVA contains erlotinib as the hydrochloride salt that has the following structural formula:
Erlotinib hydrochloride has the molecular formula C22H23N3O4•HCl and a molecular weight of 429.90. The molecule has a pKa of 5.42 at 25oC. Erlotinib hydrochloride is very slightly soluble in water, slightly soluble in methanol and practically insoluble in acetonitrile, acetone, ethyl acetate and hexane.
Aqueous solubility of erlotinib hydrochloride is dependent on pH with increased solubility at a pH of less than 5 due to protonation of the secondary amine. Over the pH range of 1.4 to 9.6, maximal solubility of approximately 0.4 mg/mL occurs at a pH of approximately 2.
![]()
![]()
WO2012028861
![]()
wo2007060691
………………….
PATENT
http://www.google.com/patents/WO2008122776A2?cl=en
Erlotinib is a Human Epidermal Growth Factor Receptor Type 1 /Epidermal Growth Factor Receptor (HER1/EGFR) tyrosine kinase inhibitor.
Erlotinib is described chemically as N-(3-ethynylpheny!)-6,7-bis(2- methoxyethoxy)quinazolin-4-amine, and its hydrochloride salt is represented by the compound of Formula I.
Erlotinib is disclosed in EP0817775 which also a discloses process for its preparation, which involves adding 3-ethynylaniline and 4-chloro-6,7-bis(2-methoxyethoxy)quinazoline in isopropanol containing pyridine and then refluxing the mixture for 4 hours under the atmosphere of dry nitrogen. The solvent is removed and residue is extracted in 10% methanol in CHCI3 and saturated aqueous NaHCO3. N-(3-ethynylphenyl)-6,7-bis(2- methoxyethoxy)quinazolin-4-amine base is separated chromatographically and converted to the hydrochloride salt in a solvent such as CHCI3 using hydrochloric acid.
EP1044969 claims a method for preparing intermediates and compounds covering erlotinib. This patent discloses a process for preparing N-(3-ethynylphenyl)-6,7-bis(2- methoxyethoxy)quinazolin-4-amine which involves stirring 4-[3-[[6,7-bis(2-methoxyethoxy)- 4-quinazolinyl]amino]phenyl]-2-methyl-3-butyn-2-ol with anhydrous sodium hydroxide and 2-methoxyethanol and heating at reflux for 47 hours. The reaction mixture is cooled to 20- 25°C and concentrated HCI is added to it. The resulting mixture is granulated at 20-25°C to crystallize the product.
Indian patent application 902/CHE/2006 discloses a process for preparation of N-(3- ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine hydrochloride. The process involves reacting 3,4-dihydroxy benzaldehyde with substituted ethylmethyl ether in the presence of an inert solvent and base to obtain 3,4-bis(2-methoxyethoxy) benzaldehyde. This 3,4-bis(2-methoxyethoxy) benzaldehyde is converted to 3,4-bis(2-methoxyethoxy) benzaldoxime in the presence of a base and organic solvent and is further dehydrated to 3,4-bis(2-methoxyethoxy) benzonitrile. The benzonitrile so obtained is nitrated to obtain 4,5-bis(2-methoxyethoxy)-2-nitrobenzonitrile which is further reduced to obtain 2-amino- 4,5-bis(2-methoxyethoxy) benzonitrile. N’-(3-ethynylphenyl)-N,N-dimethyl formamidine obtained on formylation of 3-ethynylaniline with N,N-dimethyl formamidine is coupled with 2-amino-4,5-bis(2-methoxyethoxy) benzonitrile to obtain erlotinib free base which on treatment with a polar solvent containing hydrochloric acid gives erlotinib hydrochloride.
Indian patent application 904/CHE/2006 also discloses a process for preparation of N-(3- ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine hydrochloride. The process involves reacting 3,4-dihydroxy benzaldehyde with substituted ethylmethyl ether in the presence of an inert solvent and base to obtain 3,4-bis(2-methoxyethoxy) benzaldehyde. This 3,4-bis(2-methoxyethoxy) benzaldehyde is converted to 3,4-bis(2-methoxyethoxy) benzaldoxime in the presence of a base and organic solvent and is further dehydrated to 3,4-bis(2-methoxyethoxy) benzonitrile. The benzonitrile so obtained is nitrated to obtain 4,5-bis(2-methoxyethoxy)-2~nitrobenzonitrile which is further reduced to get 2-amino-4,5- bis(2-methoxyethoxy) benzonitrile. 2-amino-4,5-bis(2-methoxyethoxy) benzonitrile is formylated with a formylating agent in the presence of formic acid derivative to obtain N’- [2-cyano-4,5-bis(2-methoxyethoxy)phenyl]-N,N-dimethylformamidine which is coupled with an aniline derivative to obtain erlotinib free base which on treatment with a polar solvent containing hydrochloric acid gives erlotinib hydrochloride.
![]()
EXAMPLES:
Example – 1a:
Preparation of Erlotinib Hydrochloride : 5.O g of 4-chloro-6,7-bis (2-methoxyethoxy) quinazoline was suspended in 75 ml water and 2.55 g of 3-aminophenyl acetylene was charged at 25 – 300C. Further 1.0 ml 50 % hydrochloric acid was added. The reaction mass was stirred at 25 – 300C for 2 hours. The solid obtained was filtered and washed with water. The product was dried at 40 – 45°C to obtain 6.1 g of erlotinib hydrochloride. In a similar manner, different solvents were used for preparing erlotinib hydrochloride under acidic conditions as given in table 1 below :
Table 1
Example – 2a:
Preparation of Erlotinib Hydrochloride :
5.0 g of 4-chloro-6,7-bis(2-methoxyethoxy) quinazoline was suspended in 75 ml of water and 2.55 g of 3-aminophenyl acetylene was added at 25 – 300C followed by 1.0 ml of 50 % hydrochloric acid. The reaction mass was heated at 35 – 400C for 1 hour. The solid obtained was filtered and washed with water. The product was dried at 40 – 45°C to obtain 5.8 g of erlotinib hydrochloride.
In a similar manner, different solvents were used for preparing erlotinib hydrochloride under acidic conditions as given in table 2 below :
Table 2
Example – 3:
Preparation of Erlotinib Hydrochloride :
5 g of 4-chloro-6,7-bis(2-methoxyethoxy) quinazoline was suspended in 150 ml denatured spirit (SPDS) and 4.6 g of 3-aminophenyl acetylene was charged at 25 – 300C. Further 1.0 ml of methane sulphonic acid was added. The reaction mass was stirred at 25 – 300C for 3 hours. Solid obtained was filtered, washed with SPDS and dried under vacuum. This solid was suspended in water, basified with ammonia and stirred for 10 minutes. The resulting erlotinib base was isolated, washed with water and dried under vacuum. The base was suspended in water and acidified to pH 1.0 – 2.0 using hydrochloric acid. The reaction mixture was stirred for 2 hours, filtered, washed with water and dried at 40 – 450C to obtain 5.8 g of erlotinib hydrochloride.
Example – 4: Preparation of Erlotinib Hydrochloride :
10.0 g of 4-chloro-6,7-bis(2-methoxyethoxy) quinazoline was suspended in 300 ml methanol and 9.2 g of 3-aminophenyl acetylene was charged at 25 – 300C. Further 2.0 ml of benzoic acid was added. The reaction mass was stirred at 25 – 300C for 4 hours. Solid obtained was filtered, washed with methanol and dried under vacuum. This solid was suspended in water and then basified with sodium hydroxide and stirred for 10 minutes. The resulting erlotinib base was isolated, washed with water and dried under vacuum. The base was suspended in water and acidified to pH 1.0 – 2.0 using hydrochloric acid. The reaction mixture was stirred for 2 hours, filtered, washed with water and dried to obtain 11.2 g of erlotinib hydrochloride. Example – 5:
Preparation of Erlotinib Hydrochloride :
15.0 g of 4-chloro-6,7-bis(2-methoxyethoxy) quinazoline was suspended in 450 ml ethanol and 13.8 g of 3-aminophenyl acetylene was added at 25 – 30°C. Further 3.0 g tartaric acid was added. The reaction mass was stirred at 25 – 300C for 6 hours. Solid obtained was filtered, washed with water and dried under vacuum. This solid was suspended in water, basified with potassium hydroxide and stirred for 10 minutes. The resulting erlotinib base was isolated by filtration, washed with ethanol and dried under vacuum. The solid obtained was then suspended in water and acidified to pH 1.0 – 2.0 using hydrochloric acid. The reaction mixture was stirred for 2 hours, filtered, washed with water and dried at 40 – 45°C to obtain 18.3 g of erlotinib hydrochloride.
Example – 6: Preparation of Erlotinib Hydrochloride :
50 g of 4-chloro-6,7-bis(2-methoxyethoxy) quinazoline was suspended in 1500 ml acetonitrile and 46 g of 3-aminophenyl acetylene was added at 25 – 300C, followed by 10 ml acetic acid. The reaction mass was stirred at 25 – 30°C for 30 minutes. Solid obtained was filtered, washed with water and dried under vacuum. This solid was suspended in water, basified with potassium hydroxide and stirred for 10 minutes. The resulting erlotinib base was isolated, washed with acetonitrile and dried under vacuum. The solid obtained was then suspended in water and acidified to pH 1.0 – 2.0 using hydrochloric acid. The reaction mixture was stirred for 2 hours, filtered, washed with water and dried at 40 – 45°C to obtain 63 g of erlotinib hydrochloride.
……………………
Org. Process Res. Dev., 2007, 11 (5), pp 813–816
DOI: 10.1021/op700054p
An efficient, economical and large-scale convergent synthesis of epidermal growth factor receptor- tyrosine kinase inhibitors gefitinib (1, Iressa) and erlotinib (2, Tarceva) approved by U.S. FDA for the treatment of non-small-cell lung cancer is described. The formation of 4-anilinoquinazolines are achieved in a simple one-pot reaction of suitable formamidine intermediates and substituted anilines involving Dimroth rearrangement, thereby avoiding the need to make quinazolin-4(3H)-one intermediates, which require a large experimental inputs. Using this process, we have produced drug candidates 1 with overall yield of 66% from 4-methoxy-5-[3-(4-morpholinyl) propoxy]-2-nitrobenzonitrile (3) and 2 with 63% from 4,5-bis(2-methoxyethoxy)-2-nitrobenzonitrile (6) on a multigram scale.
2 as crude material, which was further recrystallized from ethyl acetate (1 L) and then with methanol (500 mL) to give off-white crystalline compound 2 (350 g, 66% yield). FREE BASE ERLOTINIB
HPLC >99%.
Mp 149–153 °C.
1H NMR (CDCl3): δ 3.08 (s, 1H), 3.43 (s, 6H), 3.80 (m, 4H), 4.22 (m, 4H), 7.17 (s, 1H), 7.24–7.37 (m, 3H), 7.61 (brs, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.85 (s, 1H), 8.63 (s, 1H).
MS (m/z): 393 (M+), 334, 276, 230, 59.
[6,7-Bis(2-methoxyethoxy)-quinazolin-4-yl]-(3-ethynylphenyl)Amine Hydrochloride (Erlotinib Hydrochloride, 9)
Through a stirred suspension of erlotinib free base 2 (200 g) in methanol (2 L) was passed dry hydrochloric acid gas for ~0.5 h, keeping the temperature of the reaction mass at 15–20 °C. The solid precipitate was filtered and dried at 50 °C to give off-white crystalline material of erlotinib hydrochloride (9) (200 g, 92% yield).
Mp 228–230 °C.
HPLC >99%.
1H NMR (DMSO-d6, 200 MHz): δ 3.36 (s, 6H), 3.77 (m, 4H), 4.29 (s, 1H), 4.32–4.38 (m, 4H), 7.38–7.55 (m, 3H), 7.78 (d,J = 8.0 Hz, 1H), 7.88 (s, 1H), 8.38 (s, 1H), 8.86 (s, 1H), 11.42 (s, 1H).
Elemental Anal. Calcd for C22H24N3O4Cl: C, 61.32; H, 5.85; N, 9.75. Found: C, 61.45; H, 5.62; N, 9.60. Chloride assay by potentiometric method 98.82%.
SYNTHESIS
![]()
APOTEX PHARMACHEM INC.; KOTHAKONDA, Kiran Kumar; REY, Allan W.; GUNTOORI, Bhaskar Redd Patent: WO2010/40212 A1, 2010 ; Location in patent: Page/Page column 8 ;
Esteve Química, S.A. Patent: EP2348020 A1, 2011 ; Location in patent: Page/Page column 9 ;
Ube Industries, Ltd. Patent: EP1481971 A1, 2004 ; Location in patent: Page 9 ;
F.I.S. Fabbrica Italiana Sintetici S.p.A. Patent: EP2433931 A1, 2012 ; Location in patent: Page/Page column 12 ;
Norris, Timothy; Santafianos, Dinos Journal of the Chemical Society. Perkin Transactions 2, 2000 , # 12 p. 2498 – 2502
Bulletin of the Korean Chemical Society, , vol. 32, # 3 p. 909 – 914
Synthetic Communications, , vol. 37, # 19 p. 3409 – 3415
Heterocycles, , vol. 71, # 1 p. 39 – 48
Molecules, , vol. 11, # 4 p. 286 – 297
WO2011/76813 A1, ;
EP2433931 A1, ;
Journal of the Chemical Society. Perkin Transactions 2, , # 12 p. 2498 – 2502
NMR
![]()
MASS
![]()
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO1996030347A1 * |
Jun 6, 1995 |
Oct 3, 1996 |
Lee D Arnold |
Quinazoline derivatives |
| WO2004072049A1 * |
Feb 11, 2004 |
Aug 26, 2004 |
Andre Gerard Bubendorf |
Polymorph of {6,7-bis(2-methoxy-ethoxy)-quinazolin-4-yl}-(3e) |
| Reference |
| 1 |
* |
PETR KNESL ET AL: “Improved synthesis of substituted 6,7-dihydroxy-4-quinazolineamines: tandutinib, erlotinib and gefitinib” MOLECULES, vol. 11, no. 4, 2006, pages 286-297, XP002456342 ISSN: 1420-3049 |
| 2 |
* |
TIMOTHY NORRIS AND DINOS SANTAFIANOS: “Discovery of a new stable polymorph of 4-(3-ethynylphenylamino)-6,7-bis(2-methoxy ethoxy)quinazolinium methanesulfonate using near-infrared spectroscopy to monitor form change kinetics” JOURNAL OF THE CHEMICAL SOCIETY, PERKIN TRANSACTIONS 2, vol. 12, 2000, pages 2498-2502, XP002261707 ISSN: 1472-779X |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2009007984A2 * |
Jul 11, 2007 |
Jan 15, 2009 |
Hetero Drugs Ltd |
An improved process for erlotinib hydrochloride |
| WO2010057430A1 * |
Nov 18, 2009 |
May 27, 2010 |
Shanghai Institute Of Pharmaceutical Industry |
Polymorph form l of erlotinib, methods of preparation and uses thereof |
| WO2010109443A1 |
Mar 26, 2010 |
Sep 30, 2010 |
Ranbaxy Laboratories Limited |
Process for the preparation of erlotinib or its pharmaceutically acceptable salts thereof |
| WO2011058525A2 |
Nov 12, 2010 |
May 19, 2011 |
Ranbaxy Laboratories Limited |
Processes for the preparation of erlotinib hydrochloride form a and erlotinib hydrochloride form b |
| WO2011068403A2 * |
Dec 2, 2010 |
Jun 9, 2011 |
Ultimorphix Technologies B.V. |
Novel n-{3-ethynylphenylamino)-6,7-bis(2-methoxyethoxy)-4-quinazolinamjne salts |
| WO2011076813A1 |
Dec 21, 2010 |
Jun 30, 2011 |
Esteve Química, S.A. |
Preparation process of erlotinib |
| WO2011147102A1 * |
May 28, 2010 |
Dec 1, 2011 |
Shyang Jen Biotech Co., Ltd. |
Synthetic method for 6,7-substituents-4-aniline quinazoline |
| WO2013054147A2 |
Oct 10, 2012 |
Apr 18, 2013 |
Egis Gyógyszergyár Nyilvánosan Műkődő |
Erlotinib salts |
| CN101863844B |
Apr 16, 2009 |
Oct 3, 2012 |
欧美嘉股份有限公司 |
Synthesis method of 6,7-substituted-4-aniline quinazoline |
| CN101891691A * |
Jul 30, 2010 |
Nov 24, 2010 |
天津市炜杰科技有限公司 |
Method for preparing erlotinib hydrochloride |
| CN102827087A * |
Jun 18, 2012 |
Dec 19, 2012 |
中国科学院成都生物研究所 |
Synthetic method of erlotinib |
| CN102827087B * |
Jun 18, 2012 |
Mar 25, 2015 |
中国科学院成都生物研究所 |
一种厄洛替尼的合成方法 |
| EP2348020A1 * |
Dec 23, 2009 |
Jul 27, 2011 |
Esteve Química, S.A. |
Preparation process of erlotinib |
| US8389531 |
Jul 11, 2007 |
Mar 5, 2013 |
Hetero Drugs Limited |
Process for erlotinib hydrochloride |
| US8440823 |
Mar 26, 2010 |
May 14, 2013 |
Ranbaxy Laboratories Limited |
Process for the preparation of erlotinib or its pharmaceutically acceptable salts thereof |
| US8642758 |
Apr 3, 2008 |
Feb 4, 2014 |
Cipla Limited |
Process for preparation of erlotinib and its pharmaceutically acceptable salts |
Filed under:
Uncategorized Tagged:
erlotinib,
Erlotinib Hydrochloride ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

 AMITIZA (lubiprostone)
AMITIZA (lubiprostone).jpg)

























 .
. .
.





























































![methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide NMR spectra analysis, Chemical CAS NO. 1048007-93-7 NMR spectral analysis, methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-15/001/603/1603135_1h.png)
![methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide NMR spectra analysis, Chemical CAS NO. 1048007-93-7 NMR spectral analysis, methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-15/001/603/1603135_13c.png)





































 Headquartered in India, Strides Arcolab is a pharmaceutical company with a key focus on development and manufacture of IP-led niche generics and bio-pharmaceuticals. It is also among the world’s largest manufacturers of specialty soft gelatin capsules. With world-class manufacturing facilities, an innovative R&D hub in Bangalore and a strong commercial platform to market branded and commodity generics globally, Strides has earned a reputation for building and scaling profitable businesses in a short span of time.
Headquartered in India, Strides Arcolab is a pharmaceutical company with a key focus on development and manufacture of IP-led niche generics and bio-pharmaceuticals. It is also among the world’s largest manufacturers of specialty soft gelatin capsules. With world-class manufacturing facilities, an innovative R&D hub in Bangalore and a strong commercial platform to market branded and commodity generics globally, Strides has earned a reputation for building and scaling profitable businesses in a short span of time.









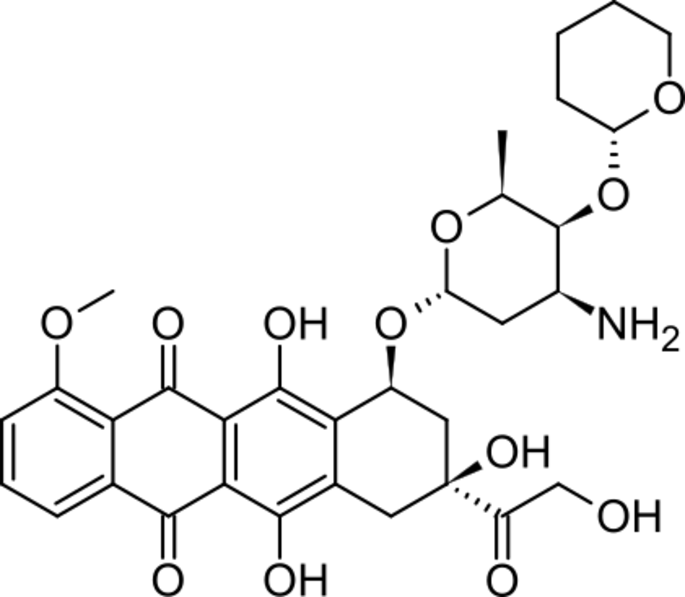
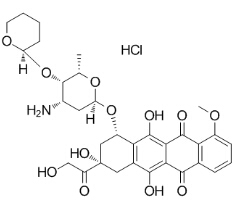 Pirarubicin Hydrochloride
Pirarubicin Hydrochloride 

 .
.")



 .
.






 GOLCONDA
GOLCONDA
















 .
. .
.
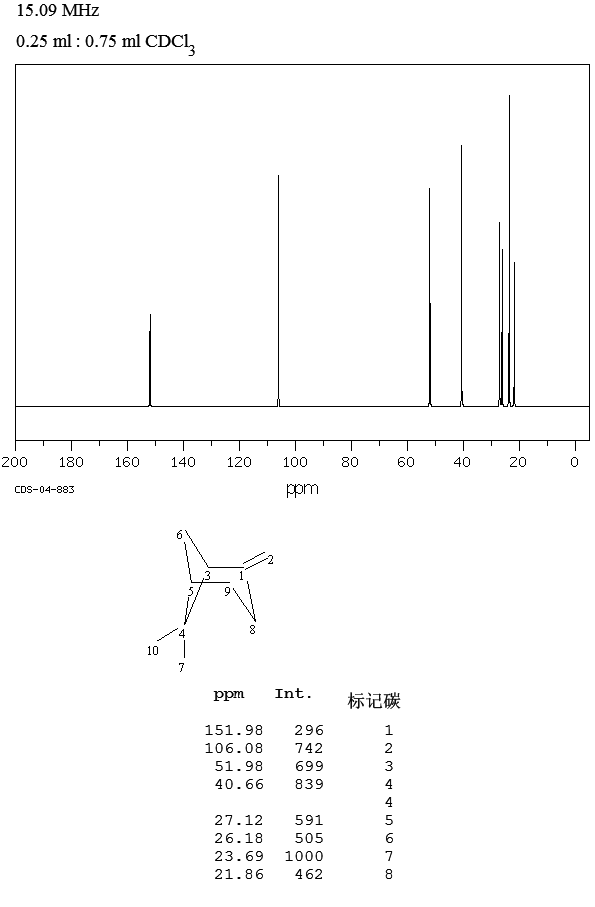 .
. .
. .
. .
. .
.
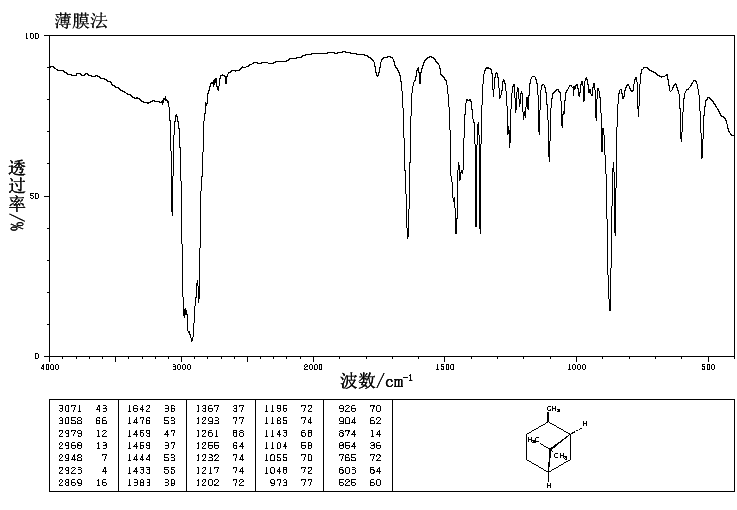 .
.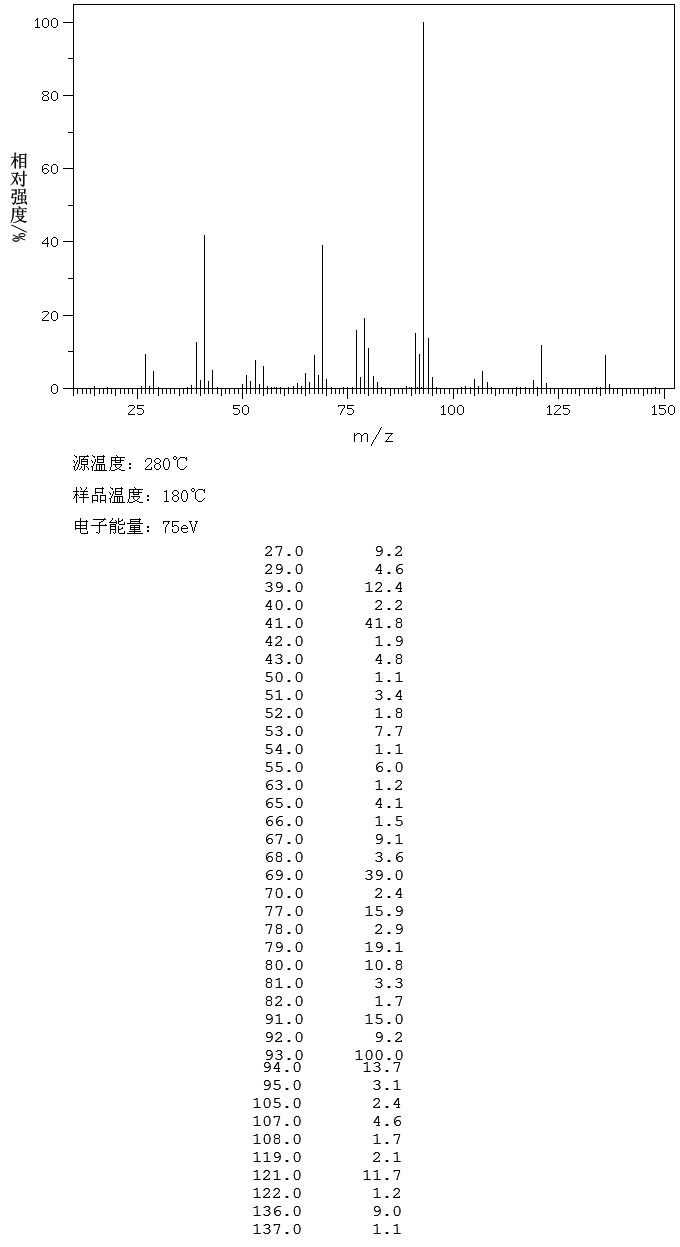 .
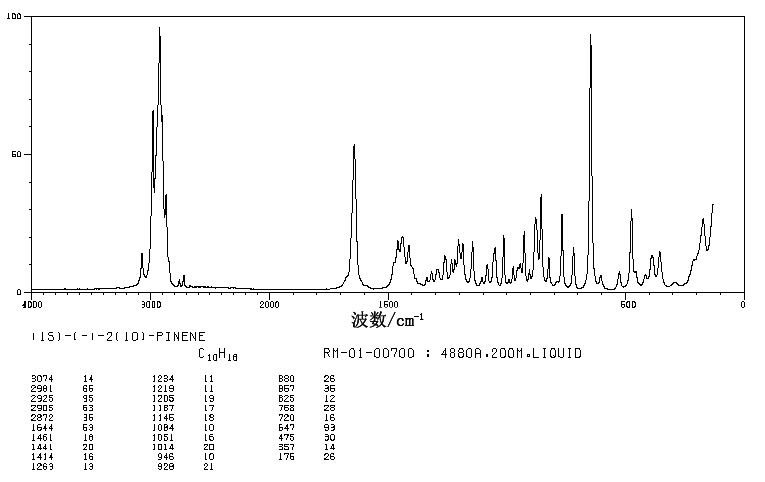
.









































































 .
. .
.
 .
.
 .
.



 .
.









 .
.





 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..



 amcrasto@gmail.com
amcrasto@gmail.com







 Originally posted on
Originally posted on 












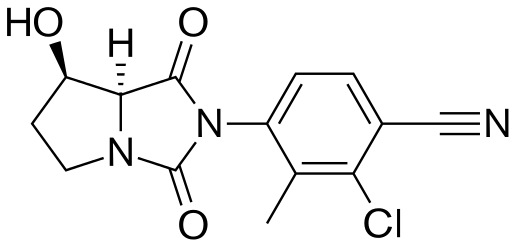
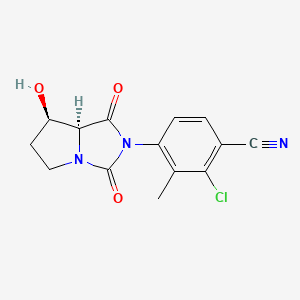
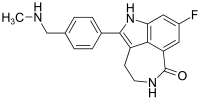
 Novel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.
Novel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.



























 AIRPORT
AIRPORT





 FLAG
FLAG





















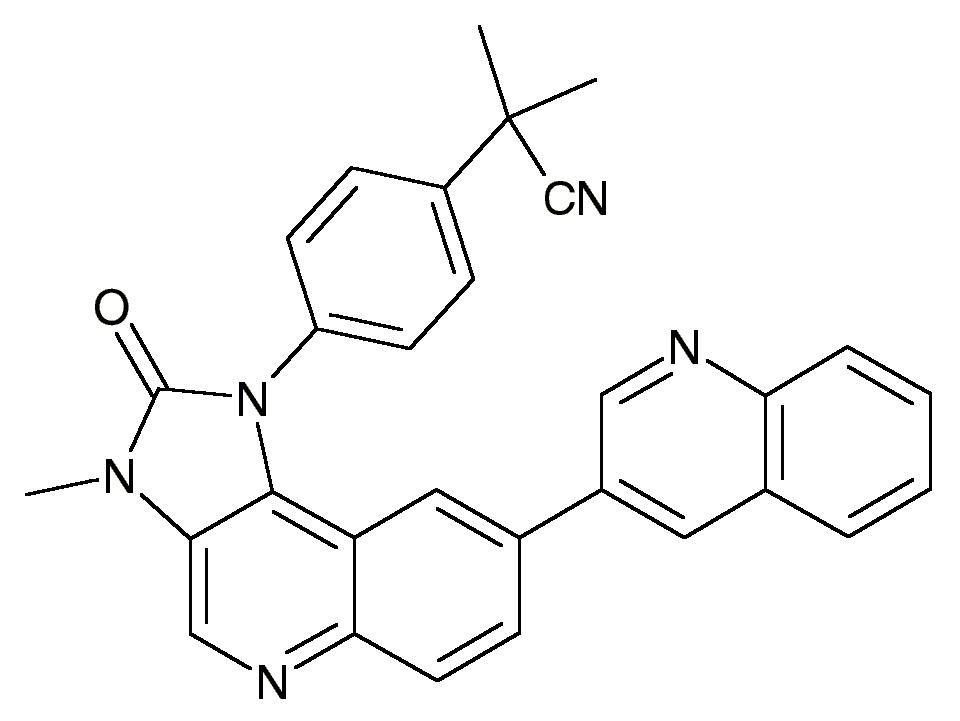




















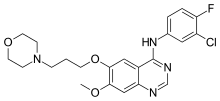














 GEFITINIB
GEFITINIB
























































 .
.





 Tandoori chicken at Surjit Food Plaza. amritsar
Tandoori chicken at Surjit Food Plaza. amritsar







 Golden Temple – Harmandir Sahib: Free food for everyone
Golden Temple – Harmandir Sahib: Free food for everyone


 tandoori chicken
tandoori chicken





















Note: Compound name must be entered under “Substance Identification” and then “Names and Synonyms” selected to view synonyms.