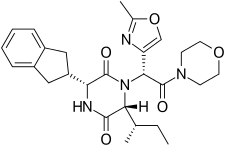
 A
A B
B C
C D
DGSK 2793660
DATA FOR A
HCL SALT CAS 1613458-78-8
BASE CAS 1613458-70-0
C20 H27 N3 O3 . Cl H
MW OF BASE…..357.45
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl]tetrahydr -2H-pyran-4-carboxamide hydrochloride
2H-Pyran-4-carboxamide, 4-amino-N-[(1S,2E)-4-(2,3-dihydro-1H-indol-1-yl)-1-ethyl-4-oxo-2-buten-1-yl]tetrahydro-, hydrochloride (1:1)
DATA FOR B
1613458-79-9 HCL SALT
1613458-71-1 BASE
C22 H31 N3 O3 . Cl H
MW 385.50 OF BASE
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten- l-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
4-Amino-N-[(2E,4S)-1-(2,3-dihydro-1H-indol-1-yl)-6-methyl-1-oxohept-2-en-4-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
DATA FOR C
1-Amino-N-[(3S)-1-(3-cyano-4′-fluorobiphenyl-4-yl)pyrrolidin-3-yl]cyclohexanecarboxamide hydrochloride
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride
C24 H27 F N4 O . Cl H, MW 442.957
CAS OF BASE 1394001-73-0
CAS OF HCL 1394001-71-8
DATA FOR D
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride
CAS OF BASE 1394001-74-1
CAS OF HCL 1394001-72-9
Cathepsin C inhibitors for treating cystic fibrosis, non-cystic fibrosis bronchiectasis, and ANCA-associated vasculitis
Bronchiectasis
Dipeptidyl peptidase I inhibitor
http://www.gsk.com/media/280387/product-pipeline-2014.pdf
This study is the first administration of GSK2793660 to humans and will evaluate the safety, tolerability, PK and PD of single oral ascending doses of GSK2793660, and of repeat oral doses of GSK2793660 in healthy subjects. The study will comprise two parts (Part A and Part B). Part A will consist of two cohorts of subjects, each taking part in a three-way cross over study, with ascending doses of GSK2793660 and placebo. Available safety, PK and PD data will be reviewed before each dose escalation. This will be followed by a food-effect arm in the cohort that received what is deemed to be the target clinical dose. Part B is planned to consist of up to two cohorts of subjects, each taking part in one 14 day repeat dose study period. Subjects will be dosed on Day 1 and then on Days 3-15. It is planned that two doses will be evaluated. The dose(s) to be tested will be selected based on safety, PK, and PD from Part A. The study is intended to provide sufficient confidence in the safety profile of the molecule and information on target engagement to allow progression to further studies………..https://clinicaltrials.gov/ct2/show/NCT02058407
Cathepsin C inhibitors for treating cystic fibrosis, non-cystic fibrosis bronchiectasis, and ANCA-associated vasculitis
Cathepsins are a family of enzymes included in the papain superfamily of cysteine proteases. Cathepsins B, C, F, H, K, L, S, V, and X have been described in the scientific literature. Cathepsin C is also known in the literature as Dipeptidyl Peptidase I or “DPPI.”
A number of recently published studies have begun to describe the role cathepsin C plays in certain inflammatory processes. See e.g. Adkison et al., The Journal of Clinical Investigation 109:363-371 (2002); Tran et al., Archives of Biochemistry and Biophysics 403 : 160-170 (2002); Thiele et al., The Journal of Immunology 158: 5200-5210 (1997);
Bidere et al., The Journal of Biological Chemistry 277: 32339-32347 (2002); Mabee et al., The Journal of Immunology 160: 5880-5885 (1998); McGuire et al., The Journal of
Biological Chemistry, 268: 2458-2467 (1993); and Paris et al., FEBS Letters 369: 326-330 (1995). From these studies, it appears that cathepsin C is co-expressed in granules of neutrophils and other leukocytes with certain serine proteases and cathepsin C functions to process the pro-forms of the serine proteases to active forms. Serine proteases are released from the granules of leukocytes recruited to sites of inflammation. Once activated, these proteases have a number of functions including degradation of various extracellular matrix components, which together can propagate tissue damage and chronic inflammation.
Studies in both cathepsin C deficient mice, and the human cathepsin C deficiency
Papillon-Lefevre syndrome clearly demonstrate that cathepsin C is required for the
activation of the neutrophil serine proteases in azurophilic granules such as neutrophil elastase (NE), cathepsin G, and proteinase 3. See Pham, C. T. et al., J. Immunol. 173 :
7277-7281 (2004).
A number of respiratory diseases are associated with an overabundant
acculumation of neutrophils and the presence of increased levels of at least some
neutrophil serine proteases. These enzymes are believed to play a role in the pathology of several respiratory diseases, such as Chronic Obstructive Pulmonary Disease (“COPD”), cystic fibrosis (CF), and non-cystic fibrosis (non-CF) bronchiectasis. Each of these diseases is associated with increased levels of E in particular, and E at least is considered to play a role in the progression of disease. See Ranes, J. and Stoller, J. K., Semin. Respir. Crit. Care Med 26: 154-166 (2005); Saget, S. D. et al., Am. J. Resp. Crit. Care Med. 186: 857-865 (2012); Tsang, K. W. et al., Chest 117: 420-426 (2000).
Additional roles of the other proteases is emerging. See Hartl, D. et al., Nature Med. 13 : 1423-1430 (2007); Korkmaz, B. et al., Pharm. Rev. 62: 726-759 (2010).
Cigarette smoking is a significant risk factor for developing COPD. Exposure to cigarette smoke and other noxious particles and gases may result in chronic inflammation of the lung. In response to such exposure, inflammatory cells such as CD8+ T cells, macrophages, and neutrophils are recruited to the area. These recruited inflammatory cells release proteases, which are believed to play a major role in the disease etiology by a number of mechanisms. Proteases released from recruited cells include the serine proteases NE as above; granzymes A and B, released from cytotoxic T cells or natural killer cells; and chymases, released from mast cells. Cathepsin C appears to be involved in activating all of these enzymes to some extent.
A number of studies with cathepsin C deficient mice have suggested roles for cathepsin C in disease models. Cathepsin C knockout mice are resistant to lung airspace enlargement and inflammatory cell infiltration in both cigarette smoke and ozone exposure models of COPD. See Guay et al., Current Topics in Medicinal Chemistry, 2010, 10, 708- 716; See also Podolin et al. (2008), Inflammation Research, 57(Suppl 2) S104.
In a model of rheumatoid arthritis (“RA”), another chronic inflammatory disease where cathepsin C may play a role, neutrophils are recruited to the site of joint
inflammation and release cathepsin G, NE, and proteinase 3, which are believed to be responsible in part for cartilage destruction associated with RA (Hu, Y. and Pham, C. T. Arthritis Rheum. 52: 2553-2558 (2005); Zen, K. et al, Blood 117:4885-4894 (2011)). Other models where cathepsin C may play a role include osteoarthritis, asthma, Multiple Sclerosis, and Anti-Neutrophil Cytoplasmic Autoantibody (ANCA)-related diseases (e.g. ANCA-associated vasculitis). See e.g. Matsui, K., Yuyama, N., Akaiwa, M., Yoshida, N. L., Maeda, M., Sugita, Y., Izuhara, K., Gene 293(1-2): 1-7 (2002); Wolters, P. J., Laig- Webster, M., Caughey, G. H., American Journal of Respiratory Cell & Molecular Biology 22(2): 183-90 (2000); Schreiber et al., J. Am. Soc. Nephrol. 23 :470-482 (2012). Cathepsin C has been demonstrated to have a role in neutrophil migration in the development of aortic aneurysms by a mechanism which has not been clearly elucidated (Pagano, M. B. et al., PNAS 104: 2855-2860 (2007)).
One approach to treating these conditions is to inhibit the activity of the serine proteases involved in the inflammatory process, especially NE activity. See e.g.,
Ohbayashi, Expert Opin. Investig. Drugs 11(7): 965-980 (2002); Shapiro, Am. J. Respir. Cell Mol. Biol. 26: 266-268 (2002). Indeed, a potent and selective inhibitor of NE was found to improve lung function in patients with bronchiectasis (Stockley, R. et al. Respir. Med. 107, 524-533 (2013)). In light of the role cathepsin C plays in activating certain serine proteases, especially NE, it is desirable to prepare compounds that inhibit its activity, which thereby inhibit serine protease activity. Thus, there is a need to identify compounds that inhibit cathepsin C, which can be used in the treatment of a variety of conditions mediated by cathepsin C.
There are additional activities of cathepsin C that may also be related to disease etiology. Cathepsin C is highly expressed in the lung epithelium where it may play a role in the processing of other enzymes not yet identified. Cathepsin C has also been reported to cleave kallikrein-4, which is believed to play a role in dental enamel maturation (Tye, C. E. et al. J. Dental Res. 88: 323-327 (2009)). Finally, cathepsin C is itself released from cells and may play a direct role in the degradation of matrix proteins.
DATA FOR A
WO 2014091443
http://www.google.com/patents/WO2014091443A1?cl=en

synthesis







Intermediate 1
1,1-dimethylethyl ((l -l-{[methyl(methyloxy)amino]carbonyl}propyl)carbamate
To a solution of (2,S)-2-({[(l,l-dimethylethyl)oxy]carbonyl}amino)butanoic acid (2.50 g, 12.3 mmol) in THF (15.0 mL) was added Ι,Γ-carbonyldiimidazole (2.39 g, 14.8 mmol) portionwise over about 10 min. After stirring 30 min at RT, a solution of Ν,Ο- dimethylhydroxylamine hydrochloride (1.32 g, 13.5 mmol) and DIPEA (2.36 mL, 13.5 mmol) in DMF (4.0 mL) was added. The reaction mixture was stirred for 2 h at RT, followed by concentration in vacuo. The residue was diluted with EtOAc (50 mL) and washed with 1 M aq. HC1 (2 x 20 mL), saturated aq. NaHC03 (2 x 20 mL), and brine (20 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound (2.60 g, 88%) as a clear, colorless oil. LC-MS m/z 247 (M+H)+, 0.94 min (ret time).
Intermediate 2
1,1-dimethylethyl [(lS -l-formylpropyl] carbamate
To a solution of L1AIH4 (0.453 g, 11.9 mmol) in Et20 (20 mL) at 0 °C was added dropwise a solution of 1, 1-dimethylethyl ((l,S)-l-{[methyl(methyloxy)amino]carbonyl}- propyl)carbamate (2.67 g, 10.8 mmol) in Et20 (15 mL). The reaction mixture was stirred for 30 min at 0 °C and quenched with EtOAc (6.5 mL) followed by 5% aq. potassium bisulfate (6.5 mL). The reaction mixture was washed with 1 M aq. HC1 (3 x 10 mL), saturated aq. NaHC03 (3 x 10 mL), and brine (10 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound as a clear, colorless oil.
Intermediate 3
methyl (2E V)-4-({ [(1 , l-dimethylethyl)oxy] car bonyl} amino)-2-hexenoate
To a stirred solution of methyl (triphenylphosphoranylidene) acetate (4.35 g, 13.0 mmol) in Et20 (25 mL) at RT was added a solution of Intermediate 2 in Et20 (15 mL). The reaction mixture was stirred at RT overnight. The solid was removed by filtration and the solution was concentrated in vacuo. Purification via flash column chromatography (0-50% EtOAc/hexanes) afforded the title compound (1.44 g, 55% over two steps) as a clear, colorless oil. LC-MS m/z 244 (M+H)+, 0.98 min (ret time). Intermediate 4
(2E,4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-2-hexenoic acid
Li OH (2.95 g, 123 mmol) was added to a solution of methyl (2£, S 4-({[(1, 1- dimethylethyl)oxy]carbonyl}amino)-2-hexenoate (6 g, 24.66 mmol) in THF (50 mL), MeOH (10.00 mL), and water (50.0 mL). The reaction was stirred overnight at RT. After 18.5 h, the reaction mixture was concentrated under reduced pressure to remove the THF and MeOH. Water (40 mL) was added, and aqueous mixture was adjusted to pH = 3 with 6 M aq. HC1, as measured by pH paper. EtOAc (80 mL) was added, the layers were separated, and the aqueous layer was extracted with EtOAc (2 x 40 mL). The combined organic layers were dried over Na2S04, concentrated under reduced pressure, and dried under high vacuum, giving 6.09 g of the title compound. LC-MS m/z 230 (M+H)+, 0.77 min (ret time).
Intermediate 5
1,1-dimethylethyl [(lS,2E)-4-(2,3-dihydro-li -indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl] carbamate
A solution of 50 wt% *T3P in EtOAc (22.00 mL, 37.0 mmol) was added dropwise via addition funnel to a solution of (2£,,4,S)-4-({[(l, l-dimethylethyl)oxy]carbonyl}- amino)-2-hexenoic acid (5.65 g, 24.64 mmol), 2,3-dihydro-lH-indole (2.76 mL, 24.64 mmol), and Et3N (11 mL, 79 mmol) in CH2C12 (90 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 30 min, the reaction was quenched by dropwise addition of saturated aq. NaHC03 (50 mL). The layers were separated, and the reaction was washed with 10% citric acid (1 x 50 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 7.21 g (89%) of the title compound. LC-MS m/z 331 (M+H)+, 1.05 (ret time). Intermediate 6
[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]amine
trifluoroacetate
TFA (25 mL, 324 mmol) was added to a solution of 1, 1-dimethylethyl [(1^,2£)-4- (2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]carbamate (7.21 g, 21.82 mmol) in CH2C12 (25 mL). The reaction was stirred at RT. After 3.5 h, CH2C12 (200 mL) was added, and the reaction was concentrated under reduced pressure and dried under high vacuum. LC-MS m/z 231 (M+H)+, 0.69 (ret time).
Intermediate 7
1,1-dimethylethyl [4-({[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten- l-yl]amino carbonyl)tetrahydro-2H-pyran-4-yl]carbamate
A solution of 50 wt% UT3P in EtOAc (1.3 mL, 2.184 mmol) was added dropwise to a solution of [(l,S’,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]amine trifluoroacetate (500 mg, 1.452 mmol), 4-((tert-butoxycarbonyl)amino)tetrahydro-2H- pyran-4-carboxylic acid (356 mg, 1.452 mmol), and Et3N (1 mL, 7.21 mmol) in CH2C12 (5 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 1 h 20 min, the reaction mixture was washed with saturated aq. NaHC03 (1 x 5 mL) and 10% citric acid (1 x 5 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 251 mg (38%) of the title compound. LC-MS m/z 458 (M+H)+, 0.96 (ret time).
Example 1
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl]tetrahydr -2H-pyran-4-carboxamide hydrochloride
A solution of concentrated aq. HCI (0.23 mL, 2.76 mmol) was added to a solution of 1,1-dimethylethyl [4-({[(l^,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten- l-yl]amino}carbonyl)tetrahydro-2H-pyran-4-yl]carbamate (251 mg, 0.549 mmol) in isopropanol (2.5 mL). The reaction flask was fitted with an air condenser, and the reaction mixture was heated to 65 °C (bath temp) for 1 h 45 min. The solvent was evaporated under reduced pressure. Water (5 mL) was added to the residue, and the mixture was concentrated under reduced pressure at 65 °C. Water (2 mL) was added to the residue, and the mixture was lyophilized, giving 193.3 mg (89%) of the title compound. LC-MS m/z 358 (M+H)+, 0.68 (ret time).
1H MR (400 MHz, METHANOL-^) δ ppm 8.14 (br. s., 1 H); 7.25 (d, J=7.03 Hz, 1 H); 7.18 (t, J=7.53 Hz, 1 H); 7.02 – 7.09 (m, 1 H); 6.83 (dd, J=15.18, 6.65 Hz, 1 H); 6.49 (d, 7=14.8 Hz, 1 H); 4.56 (d, 7=7.28 Hz, 1 H); 4.22 (br. s., 2 H); 3.95 (d, 7=7.53 Hz, 1 H); 3.88 – 3.94 (m, 1 H); 3.71 – 3.78 (m, 2 H); 3.23 (br. s., 2 H); 2.39 – 2.46 (m, 2 H); 1.79 – 1.86 (m, 2 H); 1.75 (s, 1 H); 1.72 (d, 7=8.28 Hz, 1 H); 1.00 (t, 7=7.40 Hz, 3 H)
DATA FOR B
4-Amino-N-[(2E,4S)-1-(2,3-dihydro-1H-indol-1-yl)-6-methyl-1-oxohept-2-en-4-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride







http://www.google.com/patents/WO2014091443A1?cl=en
Intermediate 8
N -{[(l,l-dimethylet leucinamide
To a solution ofN-(tert-butoxycarbonyl)-L-leucine (3.00 g, 13.0 mmol) in THF (25.0 mL) was added Ι,Γ-carbonyldiimidazole (2.52 g, 15.6 mmol) portionwise over about 10 min. After stirring 1 h at RT, a solution of N,O-dimethylhydroxylamine hydrochloride (1.39 g, 14.3 mmol) and DIPEA (2.49 mL, 14.3 mmol) in DMF (6.0 mL) was added. The reaction mixture was stirred for 2.5 h at RT, followed by concentration in vacuo. The residue was diluted with EtOAc (50 mL) and washed with 1 M aq. HCl (2 x 20 mL), saturated aq. NaHC03 (2 x 20 mL), and brine (20 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound (2.34 g, 66%) as a clear, colorless oil. LC-MS m/z 275 (M+H)+, 1.17 min (ret time).
Intermediate 9
1,1-dimethylethyl [(lS -l-formyl-3-methylbutyl]carbamate
To a solution of L1AIH4 (0.356 g, 9.38 mmol) in Et20 (20 mL) at 0 °C was added dropwise a solution ofN2-{[(l, l-dimethylethyl)oxy]carbonyl}-N1-methyl-N1-(methyloxy)-L- leucinamide (2.34 g, 8.53 mmol) in Et20 (15 mL). The reaction mixture was stirred for 30 min at 0 °C and quenched with EtOAc (6 mL) followed by 5% aq. potassium bisulfate (6 mL). The reaction mixture was washed with 1 M aq. HCl (2 x 10 mL), saturated aq. NaHC03 (2 x 10 mL), and brine (10 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound as a clear, colorless oil. Intermediate 10
methyl (2E 4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2-heptenoate
To a stirred solution of methyl (triphenylphosphoranylidene) acetate (3.42 g, 10.2 mmol) in Et20 (25 mL) at RT was added a solution of Intermediate 9 in Et20 (15 mL). The reaction mixture was stirred for 15 h at RT. The solid was removed by filtration and the solution was concentrated in vacuo. Purification via flash column chromatography (0-50% EtOAc/hexanes) afforded the title compound (1.74 g, 75% over two steps) as a clear, colorless oil. LC-MS m/z 272 (M+H)+, 1.22 min (ret time).
Intermediate 11
(2E,4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2-heptenoic acid
To a solution of methyl (2£,,4,S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6- methyl-2-heptenoate (5.00 g, 18.43 mmol) in THF (15 mL), MeOH (15.0 mL), and water (15 mL) was added Li OH (2.206 g, 92.00 mmol). After stirring for 2 h at RT, the reaction mixture was concentrated in vacuo. The reaction mixture was acidified with 6 M aq. HC1 to pH = 5 and then extracted with EtOAc. The organic layer was washed with water, dried over Na2SC”4, filtered, and concentrated in vacuo to afford the title compound (4.7 g, 99%) as a white semi-solid. LC-MS m/z 158 (M+H-Boc)+, 0.94 min (ret time).
Intermediate 12
1,1-dimethylethyl [(lS,2E)-4-(2,3-dihydro-li -indol-l-yl)-l-(2-methylpropyl)-4-oxo-2- buten-l-yl]carbamate
To a solution of (2£,,4,S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2- heptenoic acid (4.70 g, 18.26 mmol) in DMF (30.0 mL) were added BOP reagent (8.08 g, 18.26 mmol) and DIPEA (6.38 mL, 36.5 mmol). After stirring at RT for 5 min, 2,3-dihydro- lH-indole (2.053 mL, 18.26 mmol) was added and stirring continued overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was washed with brine, dried over Na2S04, filtered, concentrated in vacuo and purified by flash column chromatography (0-20% EtOAc/hexanes) to afford the title compound (4.83 g, 74%) as a white solid. LC-MS m/z 359 (M+H)+, 1.18 min (ret time).
Intermediate 13
[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten-l-yl]amine trifluoroacetate
To a solution of 1, 1-dimethylethyl [(l^,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2- methylpropyl)-4-oxo-2-buten-l-yl]carbamate (3.21 g, 8.95 mmol) in CH2C12 (10.0 mL) was added TFA (10 mL, 130 mmol). The reaction mixture was stirred for 17.5 h at RT and then concentrated under reduced pressure and dried under high vacuum to afford the title compound. LC-MS m/z 259 (M+H)+, 0.76 min (ret time).
Intermediate 14
1,1-dimethylethyl [4-({[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4- oxo-2-buten-l- l]amino}carbonyl)tetrahydro-2H- ran-4-yl]carbamate
A solution of 50 wt% ¾P in EtOAc (1.2 mL, 2.016 mmol) was added dropwise to a solution of [(15′,2JE)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2- buten-l-yl]amine trifluoroacetate (500 mg, 1.343 mmol), 4-((tert- butoxycarbonyl)amino)tetrahydro-2H-pyran-4-carboxylic acid (329 mg, 1.343 mmol), and Et3N (0.93 mL, 6.71 mmol) in CH2C12 (5 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 1 h 20 min, the reaction was washed with saturated aq. NaHC03 (1 x 5 mL) and 10% citric acid (1 x 5 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 204 mg (31%) of the title compound. LC-MS m/z 486 (M+H)+, 1.07 min (ret time).
Example 2
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten- l-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
A solution of concentrated aq. HCI (0.22 mL, 2.64 mmol) was added to a solution of 1,1-dimethylethyl [4-({[(1^2JE)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4- oxo-2-buten-l-yl]amino}carbonyl)tetrahydro-2H-pyran-4-yl]carbamate (251 mg, 0.517 mmol) in isopropanol (2.5 mL). The reaction flask was fitted with an air condenser, and the reaction mixture was heated to 65 °C (bath temp). After 1 h 45 min, the solvent was evaporated under reduced pressure at 60 °C. Water (5 mL) was added to the residue, and the mixture was concentrated under reduced pressure at 65 °C. Water (2 mL) was added to the residue, and the mixture was lyophilized, giving 130.6 mg (60%) of the title compound. LC-MS m/z 386 (M+H)+, 0.79 (ret time). 1H MR (400 MHz, METHANOL- d4) δ ppm 8.15 (d, J=7.03 Hz, 1 H); 7.25 (d, J=7.03 Hz, 1 H); 7.18 (t, J=7.65 Hz, 1 H); 7.06 (t, J=7.91 Hz, 1 H); 6.81 (dd, J=15.18, 6.40 Hz, 1 H); 6.49 (br. s., 1 H); 4.73 – 4.85 (m, 2 H); 4.21 (t, J=8.28 Hz, 2 H); 3.91 – 3.97 (m, 2 H); 3.70 – 3.77 (m, 2 H); 3.25 – 3.21 (m, 2 H); 2.35 – 2.48 (m, 2 H); 1.82 (d, J=14.31 Hz, 2 H); 1.63 – 1.71 (m, 2 H); 1.50 – 1.57 (m, 1 H); 0.98 (dd, J=11.92, 6.40 Hz, 6 H).
DATA FOR C
1-Amino-N-[(3S)-1-(3-cyano-4′-fluorobiphenyl-4-yl)pyrrolidin-3-yl]cyclohexanecarboxamide hydrochloride
http://www.google.im/patents/WO2012112733A1?cl=en
Example 1
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride
HCI salt
A solution of 1,1-dimethylethyl [l-({[(35)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidinyl]amino}carbonyl)cyclohexyl]carbamate (44 mg, 0.087 mmol) in HCI (4 M solution in 1,4-dioxane, 1.0 mL, 4.00 mmol) was stirred at RT for 1 h. The reaction mixture was diluted with Et20 (5 mL), and the mixture was filtered and washed with Et20 (2 x 2 mL). Residual solid was dissolved in MeOH and concentrated under a stream of nitrogen at 50 °C and dried under high vacuum. Water (2 mL) was added to the residue, and the mixture was lyophilized with a Genevac® HT-4X to afford the title compound (33.5 mg, 87%). LC-MS m/z 407 (M+H)+, 0.94 min (ret time). 1H NMR (400 MHz, METHANOL-^) δ ppm 7.65 – 7.72 (m, 2 H), 7.52 – 7.59 (m, 2 H), 7.10 – 7.17 (m, 2 H), 6.89 (d, J=8.53 Hz, 1 H), 4.50 – 4.58 (m, 1 H), 3.94 (dd, J=10.29, 6.53 Hz, 1 H), 3.80 (dt, J=9.41, 7.09 Hz, 1 H), 3.67-3.71 (m, 1 H), 3.64 (dd, J=10.29, 4.52 Hz, 1 H), 2.29 – 2.37 (m, 1 H), 2.04 – 2.16 (m, 3 H), 1.78 – 1.88 (m, 5 H), 1.45 – 1.62 (m, 3 H).
DATA FOR D
http://www.google.im/patents/WO2012112733A1?cl=en
Example 2
4-amino- V-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3-pyrrolidinyl]tetrahydro-2H- pyr -4-carboxamide hydrochloride
HCI salt
A solution of 1,1-dimethylethyl [4-({[(35)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidinyl] amino }carbonyl)tetrahydro-2H-pyran-4-yl] carbamate (183 mg, 0.360 mmol) in HC1 (4 M solution in 1,4-dioxane, 2.0 mL, 8.00 mmol) was stirred at RT for 0.5 h. The reaction mixture was diluted with Et20 (10 mL), and the mixture was filtered and washed with Et20 (2 x 5 mL). Residual solid was dissolved in MeOH and concentrated under a stream of nitrogen at 50 °C and dried under high vacuum. Water (2 mL) was added to the residue, and the mixture was lyophilized with a Genevac® HT-4X to afford the title compound (122.8 mg, 77%). LC-MS m/z 409 (M+H)+, 0.87 min (ret time). 1H NMR (400 MHz, METHANOL-^) δ ppm 7.66 – 7.72 (m, 2 H), 7.53 – 7.60 (m, 2 H), 7.11 – 7.18 (m, 2 H), 6.89 (d, J=8.78 Hz, 1 H), 4.53 – 4.60 (m, 1 H), 3.87 – 3.97 (m, 3 H), 3.78 – 3.84 (m, 1 H), 3.64 – 3.76 (m, 4 H), 2.30 – 2.44 (m, 3 H), 2.11 – 2.19 (m, 1 H), 1.77 – 1.84 (m, 2 H).
| WO2004002491A1 * | 25 Jun 2003 | 8 Jan 2004 | David J Aldous | Morpholine and tetrahydropyran drivatives and their use as cathepsin inhibitors |
| WO2008121065A1 * | 28 Mar 2008 | 9 Oct 2008 | Astrazeneca Ab | Novel pyrrolidine derivatives as antagonists of the chemokine receptor |
| US20070032484 * | 25 Jul 2006 | 8 Feb 2007 | Roche Palo Alto Llc | Cathepsin K inhibitors |
| US20020107266 * | Dec 11, 2001 | Aug 8, 2002 | Marguerita Lim-Wilby | Amides used particularly in the treatment, prevention or amelioration of one or more symptoms of malaria or Chagas’ disease; inhibiting the activity of falcipain or cruzain |
| US20100286118 * | May 6, 2010 | Nov 11, 2010 | Rhonan Ford | Substituted 1-cyanoethylheterocyclylcarboxamide compounds 750 |
| WO2012109415A1 | Feb 9, 2012 | Aug 16, 2012 | Glaxosmithkline Llc | Cathepsin c inhibitors |
Filed under: PHASE1 Tagged: GLAXO, GSK 2793660, PHASE 1


















 COMPD A
COMPD A





























































 CARMEN
CARMEN



















 It’s a pleasure and a privilege to be interviewed, Bora.
It’s a pleasure and a privilege to be interviewed, Bora. By the time I discovered science blogs I knew my career goals were changing. I’d already been lucky enough to audit a science writing course at Princeton taught by Mike Lemonick from TIME, and thought that maybe science writing was a good choice for me. After reading chemistry blogs for a while I realized “Hey, I can do this!” and started my own blog,
By the time I discovered science blogs I knew my career goals were changing. I’d already been lucky enough to audit a science writing course at Princeton taught by Mike Lemonick from TIME, and thought that maybe science writing was a good choice for me. After reading chemistry blogs for a while I realized “Hey, I can do this!” and started my own blog, 











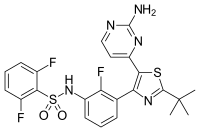

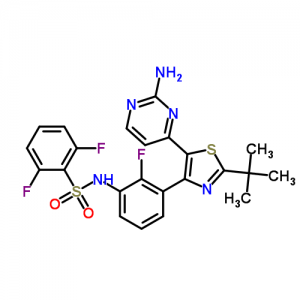
















![N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide NMR spectra analysis, Chemical CAS NO. 1195765-45-7 NMR spectral analysis, N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-11/001/566/1566739_1h.png)
![N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide NMR spectra analysis, Chemical CAS NO. 1195765-45-7 NMR spectral analysis, N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-11/001/566/1566739_13c.png)

 methyl 3-bromo-2-fluorobenzylate; PC3663; fluorobromobenzoic acid methyl ester;
methyl 3-bromo-2-fluorobenzylate; PC3663; fluorobromobenzoic acid methyl ester;




 WO2011/47238 A1, ;
WO2011/47238 A1, ;



































 Originally posted on
Originally posted on 








 MY BLOGS………
MY BLOGS……… 






















 .
.









 DR ANTHONY CRASTO
DR ANTHONY CRASTO




































































































































