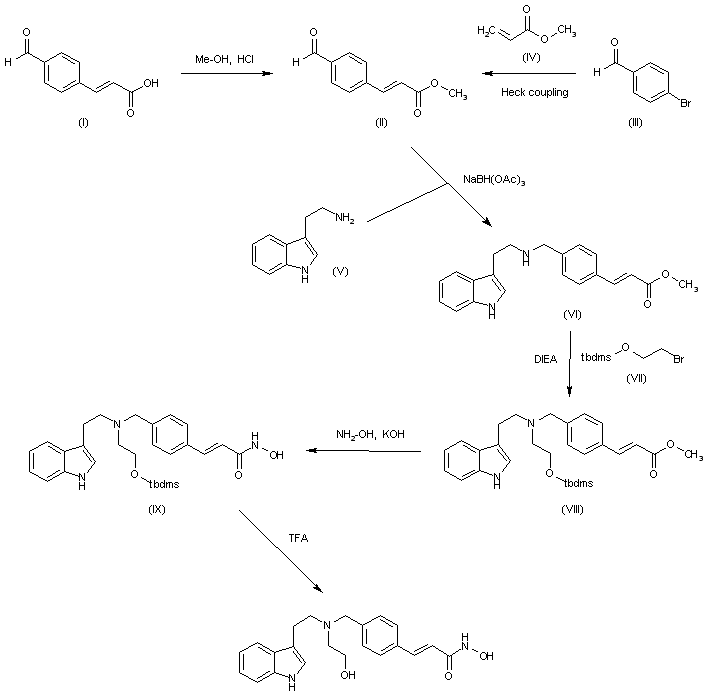
![AMG 925]()
AMG 925
1401033-86-0
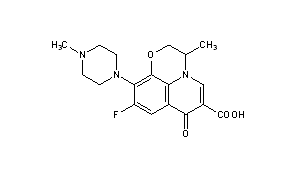
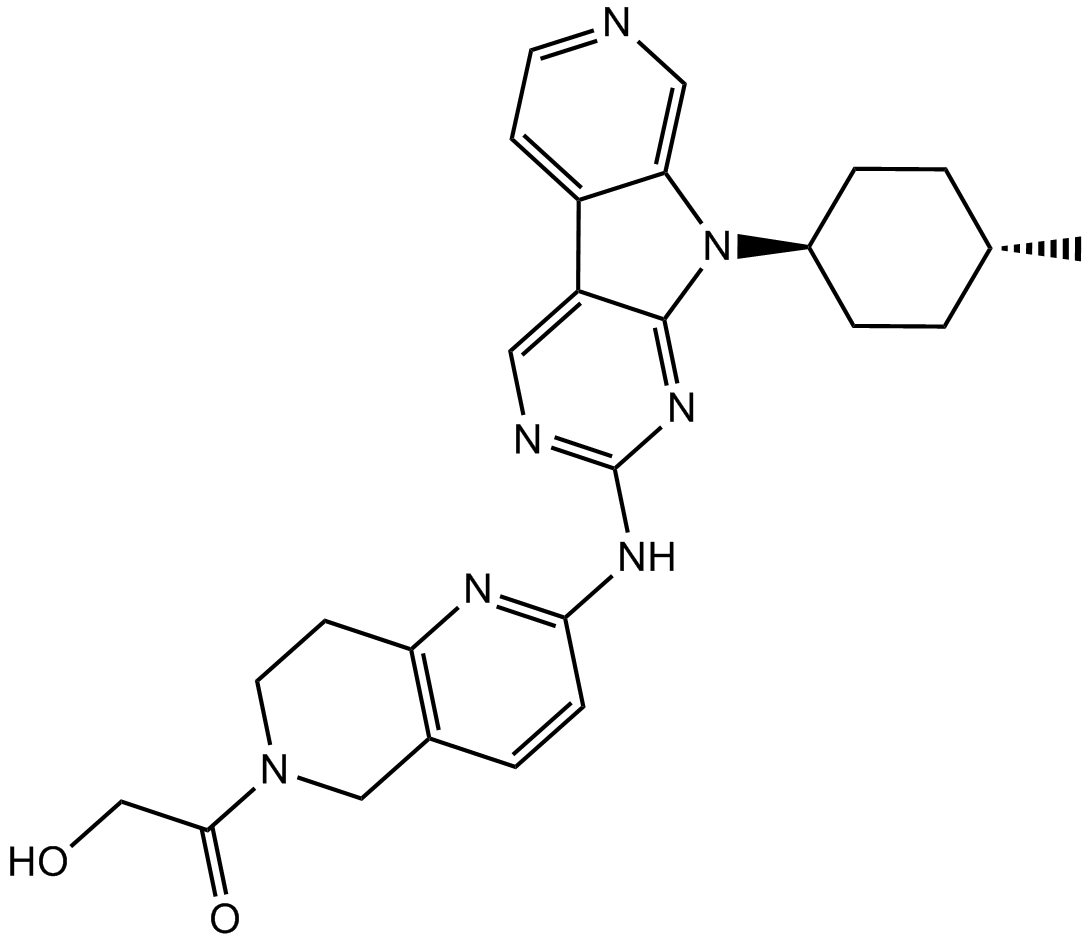
2-Hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone
2-Hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)ethanone
2-Hydroxy-1-(2-((9-((1R,4R)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (AMG 925)
FLT3/CDK4 inhibitor,potent and selective
AMG 925 is a dual kinase inhibitor of FLT3 and CDK4 with IC50 value of 1 nM and 3 nM, respectively
C26H29N7O2., 471.55
Amgen Inc. Innovator
![]()
BY
SECTION 1
STEP A
STEP B
STEP C
STEP D
9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine
tert-butyl 2-chloro-7,8-dihydro-l,6-naphthyridine-6(5H)-carboxylate
STEP E
tert-butyl 2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridine-6(5H)-carboxylate
STEP F
![Figure imgf000129_0001]() COMPD 1
COMPD 1
9-((l r,4r)-4-methylcyclohexyl)-N-(5,6,7,8-tetrahydro- l,6-naphthyridin-2-yl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine (1)
SECTION B
COUPLER2
2,5-dioxopyrrolidin-l-yl 2-acetoxyacetate
STEP G
2-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)-2-oxoethyl acetate
STEP H
![Figure imgf000135_0001]()
AMG 925
STEP I
AMG 925 is a potent, selective, and orally available FLT3/CDK4 dual inhibitor. It also inhibits CDK6 potently in kinase assay. In acute myeloid leukemia (AML) cell lines MOLM13 and Mv4-11, AMG 925 inhibits cell growth (IC50 values of 19nM and 18nM, respectively) through inhibiting P-FLT3 and P-STAT5 and inducing apoptosis. FLT3 mutants cause resistance to the current FLT3 inhibitors. AMG 925 is reported to inhibit cell growth in AML cells with FLT3 mutants FLT3-D835Y and FLT3-D835V. In AML tumor –bearing mice, administration of AMG 925 shows inhibition of P-STAT5 and P-RB as well as cell growth both in subcutaneous MOLM13 xenograft tumor model and systemic MOLM13-Luc xenograft tumor model. AMG 925 is also reported to have antitumor activity in a dose-dependent manner in theRB-positive Colo205 colon adenocarcinoma xenograft model
![]() AMGEN
AMGEN
cute myeloid leukemia (AML) represents a significant unmet medical need. It is a hematological malignancy characterized by a block in differentiation and aberrant proliferation of the myeloid lineage of hematopoietic progenitor cells. There are approximately 13,000 new cases and 9,000 deaths per year in the United States. The survival rate is 25-70% in patients younger than 60 years and 5-15% in older patients, with worse outcomes in patients with poor risk cytogenetics. Current standard of care treatment is daunorubicin and cytarabine chemotherapy with induction and consolidation phases. Bone marrow stem cell transplant is also used for treating AML in younger patients.
Cyclin-dependent kinases (CDKs) are a family of serine/ threonine protein kinases playing important cellular functions. The cyclins are the regulatory subunits that activate the catalytic CDKs. CDKl/Cyclin B 1 , CDK2/Cyclin A, CDK2/Cyclin E, CDK4/Cyclin D, CDK6/Cyclin D are critical regulators of cell cycle progression. CDKs also regulate transcription, DNA repair, differentiation, senescence and apoptosis (Morgan, D. O., Annu. Rev. Cell. Dev. Biol., 13:261-291 (1997)).
Small molecule inhibitors of CDKs have been developed to treat cancer
(de Career, G. et al., Curr. Med. Chem., 14:969-85 (2007)). A large amount of genetic evidence supports that CDKs, their substrates or regulators have been shown to be associated with many human cancers (Malumbres, M. et al, Nature Rev. Cancer, 1 :222- 231 (2001)). Endogenous protein inhibitors of CDKs including p 16, p21 and p27 inhibit CDK activity and their overexpression results in cell cycle arrest and inhibition of tumor growth in preclinical models (Kamb, A., Curr. Top. Microbiolo. Immunol., 227: 139- 148 (1998)).
Small molecule inhibitors of CDKs may also be used to treat variety of other diseases that result from aberrant cell proliferation, including cardiovascular disorders, renal diseases, certain infectious diseases and autoimmune diseases. Cell proliferation pathways including genes involved in the cell cycle Gl and S phase checkpoint (p53, pRb, pi 5, pi 6, and Cyclins A, D, E, CDK 2 and CDK4) have been associated with plaque progression, stenosis and restenosis after angioplasty. Over- expression of the CDK inhibitor protein p21 has been shown to inhibit vascular smooth muscle proliferation and intimal hyperplasia following angioplasty (Chang, M. W. et al., J. Clin. Invest, 96:2260 (1995); Yang, Z-Y. et al., Proc. Natl. Acad. Sci. (USA) 93:9905 (1996)). A small molecule CDK2 inhibitor CVT-313 (Ki = 95 nM) was shown to cause significant inhibition of neointima formation in animal models (Brooks, E. E. et al., J. Biol. Chem., 272:29207-2921 1 (1997)). Disregulation of cell cycle has been associated with polycystic kidney diseases, which are characterized by the growth of fluid-filled cysts in renal tubules. Treatment with small molecule inhibitors of CDKs yielded effective arrest of cystic disease in mouse models (Bukanov, N. O., et al., Nature, 4444:949-952 (2006)).
Infection by a variety of infectious agents, including fungi, protozoan parasites such as Plasmodium falciparum, and DNA and RNA viruses may be treated with CDK inhibitors. CDKs have been shown to be required for replication of herpes simplex virus (HSV) (Schang, L. M. et al., J. Virol., 72:5626 (1998)). Synovial tissue hyperplasia plays important roles in the development of rheumatoid arthritis; inhibition of synovial tissue proliferation may suppress inflammation and prevent joint destruction. It has been shown that over-expression of CDK inhibitor protein pl6 inhibited synovial fibroblast growth (Taniguchi, K. et al., Nat. Med., 5:760-767 (1999)) and joint swelling was substantially inhibited in animal arthritis models.
Selective inhibitors of some CDKs may also be used to protect normal untransformed cells by inhibiting specific phases of cell cycle progression (Chen, et al., J. Natl. Cancer Institute, 92: 1999-2008 (2000)). Pre-treatment with a selective CDK inhibitor prior to the use of a cytotoxic agent that inhibits a different phase of the cell cycle may reduce the side effects associated with the cytotoxic chemotherapy and possibly increase the therapeutic widow. It has been shown that induction of cellular protein inhibitors of CDKs (pi 6, p27 and p21) conferred strong resistance to paclitaxel- or cisplatin-mediated cytotoxicity on the inhibitor-responsive cells but not on the inhibitor-unresponsive cells (Schmidt, M, Oncogene, 2001 20:6164-71).
CDK4 and CDK6 are two functionally indistinguishable cyclin D dependent kinases. They are widely expressed with high levels of expression observed in cells of hematopoeitic lineage (CDK4/6 will be used throughout this document to reference both CDK4 and CDK6). CDK4/6 promotes Gl-S transition of the cell cycle by phosphorylating the retinoblastoma protein (Rb). CDK4 and CDK6 single knockout mice are viable and double knockout mice die around birth with defective
hematopoiesis (Satyanarayana, A. et al., Oncogene, 28:2925-39 (2009); Malumbres, M. et al., Cell, 1 18:493-504 (2004)). Strong evidence supports a significant involvement of the cyclin D-CDK4-pl6INK4A-Rb pathway in cancer development (Malumbres, M. et al., Nature Rev. Cancer, 1 :222-31 (2001)). Rb negatively regulates the cell cycle at Gl by sequestering E2F proteins that are required for initiation of S phase, p 1 is a key member of the ΓΝΚ4 family of CDK4/6 cellular inhibitors. The genes for Rb and pl6INK4A are tumor suppressors that are often deleted or silenced in cancer cells.
Additionally CDK4, CDK6 and cyclin D are reported to be amplified in hematologic malignancies and solid tumors. The importance of this pathway in oncogenesis is further supported by the finding that depletion or inactivation of CDK4 inhibits tumor growth in mouse tumor models (Yu, Q. et al., Cancer Cell, 9:23-32 (2006); Puyol, M. Cancer Cell, 18:63-73 (2010)). Rb and p 16^^ are rarely deleted in AML. However, the plS1^^ gene, another member of the ΓΝΚ4 family, has been reported to be down regulated by hypermethylation in up to 60% of AML (Naofumi, M. et al., Leukemia Res., 29:557-64 (2005); Drexler, H. G. Leukemia, 12:845-59 (1998); Herman, J. G. et al., Cancer Res., 57:837-41 (1997)), suggesting a possible critical role for CDK4/6 in AML cells.
FLT3 (Fms-like tyrosine kinase 3, FLK2) is a class III receptor tyrosine kinase. It is activated by the FLT3 ligand (FL) and signals through the PI3K, RAS, and JAK/STAT pathways (Scholl C. et al., Semin. Oncol., 35:336-45 (2008); Meshinchi S. et al., Clin. Cancer Res., 15:4263-9 (2009)). FLT3 plays a role in early hematopoiesis and FLT3 deficient mice have reduced numbers of progenitors of multiple lymphoid lineages (Mackarehtschian K, et al., Immunity, 3: 147-61 (1995). Activating mutations in FLT3 are found in approximately 30% of AML patients, representing the most frequent genetic alteration in the disease. About 75% of the activating mutations are internal tandem duplications (ITD) and 25% are point mutations in the activation loop of the kinase domain.
The most frequently identified activating point mutation is D835Y (Yamamoto et al., Blood, 97(8): 2434-2439 (2001)). However, mutations have also been found at N841I (Jiang, J. et al., Blood, 104(6): 1855-1858 (2004)) and Y842C (Kindler et al., Blood, 105(1): 335-340 (2005)). Additional point mutations have been identified in the juxtamembrane domain and kinase domain, although these have been shown to result in lower transforming potential (Reindel et al., Blood 107(9): 3700- 3707 (2006)).
Murine bone marrow transplanted with a retrovirus expressing the
FLT3-ITD has been shown to result in the production of a lethal myeloproliferative disease in mice (Kelly et al., Blood 99: 310-318 (2002)) characterized by leukocytosis consisting of mature neutrophils. This disease did not show a block in differentiation as seen in human AML suggesting that FLT3 mutations confer a proliferative or survival advantage to the cells. Additional oncogene mutation producing a block in
differentiation such as AML1/ETO is hypothesized to be required to produce disease that is more similar to human AML.
A number of FLT3 inhibitors have been tested in clinical trials.
Although they have shown initial clinical responses in AML, the responses observed were transient and resistance can develop rapidly (Weisberg, E. et al., Oncogene, 29:5120-34 (2010)). The major resistance mechanism appears to be through the acquisition of secondary mutations in FLT3, which may interfere with the binding of FLT3 inhibitors to the FLT3 receptor (Weisberg, E. et al., Oncogene, 29:5120-34 (2010); Chu, S. H. et al., Drug Resist. Update, 12:8-16 (2009)). One such resistance mutation (N676K) was identified in a patient at the time of clinical relapse while on multi-kinase FLT3 inhibitor midostaurin (PKC412) monotherapy (Heidel, F. et al., Blood, 107:293-300 (2006)). Combinations of FLT3 inhibitors with chemotherapy are being tested in clinical trials despite the recognition that chemotherapy is poorly tolerated. Additional possible mechanisms for lack of durable responses include inadequate target coverage (Pratz, K. W., et al., Blood, 139:3938-46 (2009)) and protection of AML cells in the bone marrow where stromal growth factors may provide proliferative signals in addition to FLT3 activation (Tarn, W. F. et al., Best Pract. Res. Clin. Haematol., 21 : 13-20 (2008)). Inhibitors with combined FLT3 and CDK4/6 inhibitory activities are novel and may prove beneficial in treating various cancers including, but not limited to, AML.
Fused tricyclic pyridine, pyrimidine, and triazine compounds useful for treating diseases mediated by CDK4 are disclosed in WO 2009/085185, published on July 9, 2009, which is hereby incorporated by reference in its entirety and for all purposes as if fully set forth herein. Various gem-disubstituted and spirocyclic compounds useful for treating diseases mediated by CDK4 are disclosed in WO 2009/0126584, published on October 15, 2009, which is hereby incorporated by reference in its entirety and for all purposes as if fully set forth herein.
A continued need exists for new compounds that can be used to modulate CDK4, CDK6, and/or FLT3 and can be used to treat various disease conditions associated with these kinases. The compounds of the present invention provide significant improvements in inhibition in one or more of these kinases and have properties making them excellent therapeutic candidates.
………………………
PATENT WO2012129344
http://www.google.com/patents/WO2012129344A1?cl=en
[01 16] In some embo f Formula I, the compound is
SCHEME 3
R1′-CI
3G …………………………………………………………..3H
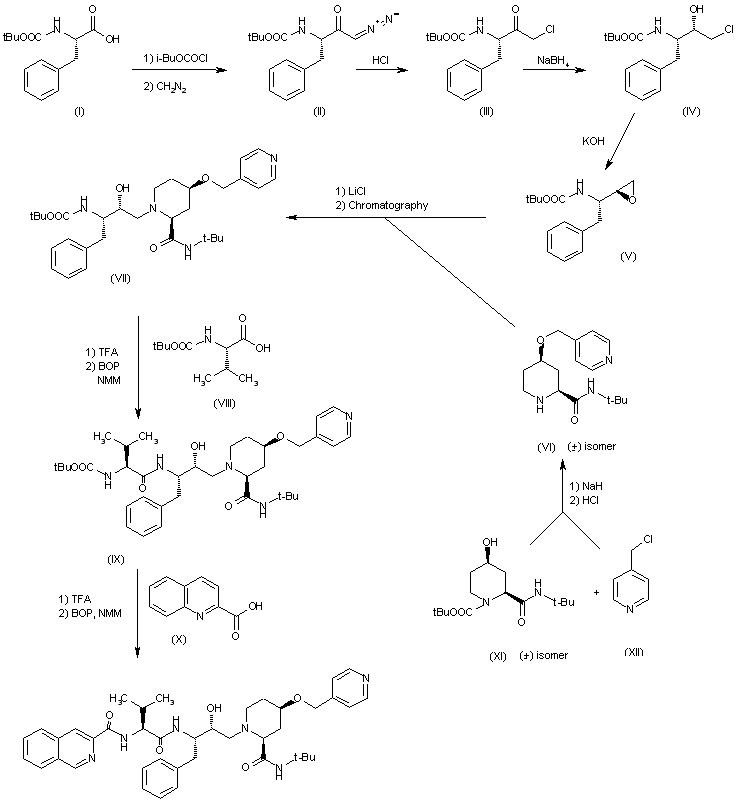
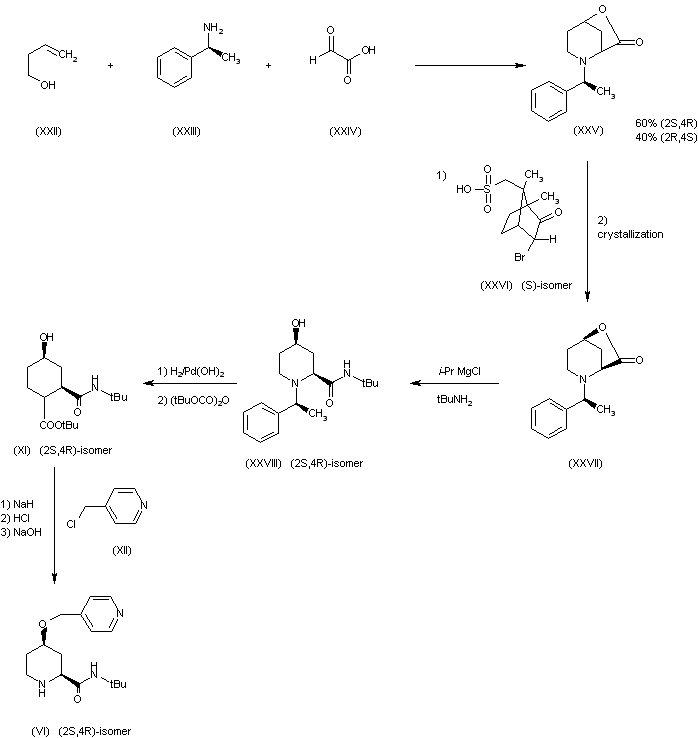
Example 1. 9-((lr,4r)-4-Methylcyclohexyl)-N-(5,6,7,8-tetrahydro-1 ,6-naphthyridin-2-yl)-9H-py 3-d] pyrimidin-2-amine
![Figure imgf000125_0001]() KEY INTERMEDIATE 1
KEY INTERMEDIATE 1
……………………………………..SECTION 1 BELOW
STEP A
4-Chloropyrimidine-2-amine (commercially available from Sigma-Aldrich, St. Louis, MO) (1000 g, 7.72 mol, 1.0 eq), trans- 4-methylcyclohexylamine hydrochloride (commercially available from TCI America, M1780) (1500 g, 10.03 mol, 1.3 eq) and TEA (3.23 L, 23.2 mol, 3.0 eq) were mixed together in n-butanol (8 L). The reaction mixture was heated at reflux for 36 hours and monitored using LCMS. Upon completion, the reaction mixture was cooled to room temperature, diluted with water (8 L) and extracted with EtOAc (2 x 10 L). The organic layers were combined, dried over Na2S04, and concentrated under reduced pressure to give the title compound (1770 g) which was us
STEP B
![Figure imgf000125_0003]()
Synthesis of 5-iodo-A^-((lr,4r)-4-methylcyclohexyl)pyridine-2,4- diamine. N4-((lr,4r)-4-Methylcyclohexyl)pyridine-2,4-diamine (1770 g, 8.58 mol, 1.0 eq) was dissolved in anhydrous DMF (8 L). To this solution under N2 atmosphere at 10 °C was added NIS (1.93 kg, 8.58 mol, 1.0 eq) in portions over 10 minutes. Upon completion of the addition, the reaction mixture was stirred at room temperature for 2 hours. The reaction was monitored using LCMS. Upon completion, the reaction mixture was cooled using an ice bath, quenched with saturated aqueous sodium carbonate (5 L) and extracted with EtOAc (2 x 15 L). The combined organic extracts were washed with saturated aqueous sodium carbonate (2 x 5 L), water (3 x 2 L), dried over Na2S04, and concentrated under reduced pressure. The residue was purified using column chromatography eluting with 25% to 40% EtOAc in hexanes to provide the title compound (1.47 kg, 57% over two steps). ^-NMR (300 MHz, DMSO-d6) δ ppm 0.85 (3H, d, J= 7.2 Hz), 0.98 (1H, dd, J= 12.9, 2.7 Hz), 1.41 – 1.27 (3H, m), 1.66 (2H, d, J = 12.3 Hz), 1.78 (2H, d, J= 12.3 Hz), 3.85 (1H, m), 5.48 (1H, d, J= 8.1 Hz), 6.16 (2H, br s), 7.86
STEPC
![Figure imgf000126_0001]()
Synthesis of 5-(3-fluoropyridin-4-yl)-N -((lr,4r)-4- methylcyclohexyl)pyrimidine-2,4-diamine. To a solution of 2,2,6,6- tetramethylpiperidine (commercially available from Sigma-Aldrich, St. Louis, MO) (997 mL, 5.87 mol, 3 eq) in anhydrous THF (6 L) under N2 atmosphere at 0 °C, was added n-BuLi (2.5 M in hexanes, 2.35 L, 5.87 mol, 3 eq) via an addition funnel over 30 minutes. Upon completion of the addition, the reaction mixture was stirred at 0 °C for 1 hour. The reaction mixture was cooled to -74 °C (acetone/ dry ice bath) and a solution of 3-fluoropyridine (commercially available from Sigma-Aldrich, St. Louis, MO) (561 g, 5.773 mol, 2.95 eq) in anhydrous THF (500 mL) was added over 15 minutes keeping the temperature below -63 °C. Upon completion of the addition, the reaction mixture was stirred at -74 °C for an additional 2 hours. A solution of ZnBr2 (1422 g, 6.32 mol, 3.22 eq) in anhydrous THF (3 L) was then added dropwise over 35 minutes keeping the temperature below -60 °C. Upon completion of the addition, the cold bath was removed and the reaction mixture was allowed to warm to room temperature. Then 5- iodo-N4-((lr,4r)-4-methylcyclohexyl)pyridine-2,4-diamine (650 g, 1.95 mol, 1.0 eq) was added in one portion followed by Pd(PPh3)4 (113 g, 97.8 mmol, 0.05 eq). The reaction mixture was heated at reflux overnight and monitored using LCMS. Upon completion, the reaction mixture was cooled to room temperature, quenched with saturated aqueous NaHC03 (6 L) and extracted with EtOAc (10 L x 2). The organic extracts were washed with saturated NaHC03 (2.5 L x 2) and brine (2.5 L), and were then concentrated under vacuum. The residue was dissolved in 2N HC1 (2.5 L) and washed with DCM (1.25 L x 3). The aqueous phase was adjusted to pH 10-12 by addition of aqueous 4N NaOH and extracted with DCM (1.5 L x 3). The organic extracts were washed with water (1.25 L x 2), dried and concentrated to give the title compound (540 g, 92%). ^-NMR (300 MHz, DMSO-d6) δ ppm 0.85 (3H, d, J= 7.2 Hz), 0.98 (1H, dd, J= 12.9, 2.7 Hz), 1.30 – 1.18 (3H, m), 1.64 (2H, d, J= 12.3 Hz), 1.74 (2H, d, J= 1 1.7 Hz), 3.96 (1H, m), 5.00 (1H, d, J= 8.4 Hz), 6.24 (2H, br s), 7.35 (1H, dd, J= 6.6, 4.4 Hz), 7.58 (1H, s), 8.37 (1H, d, J= 4.8 Hz), 8.50 (1H, d, J= 6.6 Hz) ppm.
STEPD
![Figure imgf000127_0001]()
Synthesis of 9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine. To a solution of 5-(3- fluoropyridin-4-yl)-N4-((lr,4r)-4-methylcyclohexyl)pyrimidine-2,4-diamine (854 g, 2.84 mol, 1.0 eq) in anhydrous 1 -methyl-2-pyrrolidinone (8 L) under N2 atmosphere at room temperature, was added LiHMDS (1.0 M in toluene, 8.5 L, 8.5 mol, 3.0 eq) over 30 minutes. Upon completion of the addition, the reaction mixture was heated at 90 °C overnight and monitored using LCMS. Upon completion, the reaction mixture was cooled to room temperature, quenched with ice cold water (10 L) and extracted with EtOAc (12 L). The organic phase was washed with saturated aqueous NaHC03 (4 L x 2), and water (2 L x 3). The aqueous layers were combined and back extracted with EtOAc (15 L x 2). The organic layers were combined, dried over Na2SO/t, and concentrated under reduced pressure. The solid thus obtained was suspended in DCM (2.5 L) and agitated using a rotary evaporator for 30minutes. The solid was collected by filtration, washed with DCM and dried to afford the title compound (400 g). The mother liquor was purified by column chromatography (eluting with DCM/MeOH = 50: 1) to afford, after triturating with DCM (750 mL), additional title compound (277 g, total: 677 g, yield: 84%). ¾ NMR (300 MHz, CD3OD) δ ppm 1.02 (d, J= 6.3 Hz, 3H), 1.33-1.20 (m, 2H), 1.67-1.60 (m, 2H), 1.95-1.84 (m, 4H), 1.58-1.45 (m, 2H), 4.87-4.77 (m, 1H), 7.94 (d, J= 5.1 Hz, 1H), 8.31 (d, J= 5.1 Hz, 1H), 8.87 (s, 1H), 8.96 (s, 1H) ppm; MS m/z: 28
……………………………………
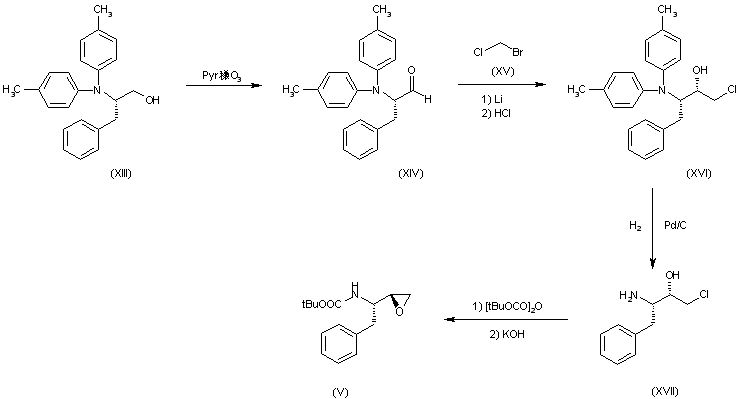
COUPLER 1
Synthesis of tert-butyl 2-chloro-7,8-dihydro-l,6-naphthyridine-6(5H)-carboxylate. To a slurry of 2-chloro-5,6,7,8-tetrahydro-l,6-naphthyridine hydrochloride (106.1 g, 517 mmol, commercially available from D-L Chiral Chemicals, ST-0143) and N,N-diisopropylethylamine (80 g, 108 mL, 621 mmol, 1.2 eq) in DCM (1 L) was added a solution of di-tert-butyl dicarbonate (119 g, 543 mmol, 1.05 eq) in
DCM (100 mL) via an addition funnel within 1 hr. The reaction mixture became a clean solution and the solution thus obtained was stirred at room temperature for an additional hour and monitored using LCMS. Upon completion, the reaction mixture was concentrated. The residue was dissolved in EtOAc (1 L) and washed with water (3 x 300 mL), washed with brine (300 mL) and dried over MgSOzt. The solvent was evaporated under vacuum to give the title compound as an off- white solid (139 g, yield: 100%). lH NMR (400MHz ,CDC13) δ ppm 1.49 (9H, s), 2.97 (2H, t, J= 5.9 Hz), 3.73 (2H, t, J= 6.0 Hz), 4.57 (2H, s), 7.17 (1H, d, J= 8.0 Hz), 7.38 (1H, d, J= 8.0 Hz) ppm;
……………………………
STEP E
![Figure imgf000128_0002]()
Synthesis of tert-butyl 2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridine-6(5H)-carboxylate. To a solution of 9-((lr,4r)-4-methylcyclohexyl)- 9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine (2.81 g, lO mmol) in 1,4-dioxane (45 mL) were added tert-butyl 2-chloro-7,8-dihydro-l,6-naphthyridine-6(5H)- carboxylate (2.57 g, 9.55 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanene (231 mg, 0.40 mmol), and sodium t-butoxide (1.44 g, 15 mmol). Argon was bubbled through the mixture for 10 minutes. Tris(dibenzylideneacetone)dipalladium (0)(183 mg, 0.20 mmol) was added, and argon was again bubbled through the mixture for 5 minutes. The reaction mixture thus obtained was stirred at 100 °C for 3 hours whereupon HPLC-MS analysis indicated that the reaction was complete. The reaction mixture was cooled to 40 °C and diluted with DCM (90 mL) and treated with Si- triamine (functionalized silica gel, from Silicycle, FR31017TR130B) (2.8 g) overnight at room temperature. Celite® brand filter aid 545 (6 g) was added, and the mixture was filtered with a sintered glass funnel and the solid phase was rinsed with DCM (100 mL). The filtrate was concentrated to 25 mL on a rotary evaporator and diluted with a mixture of EtOAc and hexane (20 mL, 4: 1). The resulting slurry was stirred at room temperature for 5 hours. The solid was collected by filtration, washed with a mixture of EtOAc and hexane (20 mL, 1 : 1) and air dried for a few hours to provide the title compound as an off-white solid (4.90 g, 100% yield). lH NMR (500 MHz, CD2C12) δ ppm 1.06 (3H, d, J= 6.4 Hz), 1.34 – 1.22 (2H, m), 1.48 (9H, s), 1.67 (1H, br. s), 2.02 – 1.93 (4H, m), 2.63 (2H, dq, J= 3.1, 12.8 Hz), 2.88 (2H, t, J= 5.7 Hz), 3.74 (2H, t, J= 6.0 Hz), 4.57 (2H, s), 7.51 (1H, d, J= 8.6 Hz), 7.85 (1H, d, J= 5.1 Hz), 8.10 (1H, br. s), 8.42 (1H, d, J= 8.3 Hz), 8.46 (1H, d, J= 4.9 Hz), 8.97 (1H, s), 9.10 (1H, s) ppm;
…………………………
STEP F
1
Synthesis of 9-((l r,4r)-4-methylcyclohexyl)-N-(5,6,7,8-tetrahydro- l,6-naphthyridin-2-yl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine (1).
To a suspension of tert-butyl 2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amm^
6(5H)-carboxylate: 9-((lr,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3- d]pyrimidin-2-amine (4.65 g, 9.05 mmol) in MeOH (30 mL) were added concentrated HC1 (6.74 mL) and water (14 mL). The mixture thus obtained was stirred at room temperature overnight. 50% NaOH in water (4.8 mL) was added at 0 °C to the reaction mixture to adjust the pH value to 9. The precipitated yellow solid was collected by filtration, rinsed with water (25 mL) and air dried for 3 days to give the title compound (3.75 g, 100%). lH NMR (400 MHz, CDC13) δ ppm 1.07 (3H, d, J= 6.5 Hz), 1.29 – 1.25 (3H, m), 2.00 – 1.95 (3H, m), 2.02 (2H, s), 2.69 – 2.53 (2H, m), 2.89 (2H, t, J= 6.0 Hz), 3.26 (2H, t, J= 6.0 Hz), 4.04 (2H, s), 4.71 (1H, m, J= 12.8, 12.8 Hz), 7.41 (1H, d, J= 8.4 Hz), 7.84 (1H, d, J= 6.1 Hz), 7.84 (1H, d, J= 6.1 Hz), 8.03 (1H, s), 8.34 (1H, d, J= 8.4 Hz), 8.50 (1H, d, J= 5.3 Hz), 8.96 (1H, s), 9.08 (1H, s) ppm; LCMS m/z: 414 (M+l).
http://www.google.com/patents/WO2012129344A1?cl=en
SECTION 2 BELOW
SYNTHESIS OF LABEL 5 FROM 1
Example 5. 2-Hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)ethanone
![Figure imgf000134_0001]()
LABEL 5
…………………………
COUPLER 2
Synthesis of 2,5-dioxopyrrolidin-l-yl 2-acetoxyacetate.
A 3-neck round-bottom flask equipped with a mechanical stirrer, thermocouple and addition funnel with nitrogen inlet was charged with N-hydroxysuccinimide (commercially available from Sigma- Aldrich, St. Louis, MO) (21 1 g, 1.83 mol) and DCM (2.25 L) at room temperature, resulting in a suspension. Pyridine (178 mL, 2.2 mol) was added in one portion with no change in the internal temperature. A solution of acetoxyacetyl chloride (commercially available from Sigma-Aldrich, St. Louis, MO) (197 mL, 1.83 mol) in DCM (225 mL) was added dropwise over 60 minutes and the temperature rose to 35 °C. Stirring was continued at room temperature for 2.5 hours. The reaction mixture was washed with water (IxlL), IN HCl (2xlL) and brine (IxlL). The organic layer was concentrated under vacuum and azeotroped with toluene (IxlL) to obtain the product as a white solid (367 g, 93%). lH NMR (400MHz, CDC13) δ 4.96 (2H, s), 2.86 (4H s), 2.19 (3H, s) ppm; LCMS m/z: 238 (M+Na).
…………………………………
STEP G
Synthesis of 2-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)-2-oxoethyl acetate.
To a suspension of 9-((lr,4r)-4- methylcyclohexyl)-N-(5,6,7,8-tetrahydro-l,6-naphthyridin-2-yl)-9H- pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine (1) (827 mg, 2.0 mmol) in chloroform (10 mL) were added diisopropylethylamine (258 mg, 348 uL, 2.0 mmol) and 2,5- dioxopyrrolidin- l-yl 2-acetoxyacetate (560 mg, 2.6 mmol). The reaction mixture thus obtained was stirred at room temperature for 30 minutes whereupon the mixture became a yellow solution. HPLC-MS analysis indicated that the reaction was complete. The reaction mixture was concentrated. MeOH (5 mL) and water (6 mL) were added to form a slurry which was stirred at room temperature for 1 hour. The solid was collected by filtration to give the title compound as a light yellow solid (1.04 g, 98% yield). lH NMR (400 MHz, CDC13, rotamers) δ ppm 1.08 (3H, d, J= 6.5 Hz), 1.37 – 1.20 (2H, m), 2.03 – 1.97 (4H, m), 2.22 (3H, s), 2.69 – 2.52 (2H, m, J= 2.9, 12.8, 12.8, 12.8 Hz), 3.08 – 2.93 (2H, m), 3.75 (1H, t, J= 5.9 Hz), 3.97 (1H, t, J= 5.6 Hz), 4.59 (1H, s), ), 4.80 – 4.65 (2H, m), ), 4.90 – 4.82 (2H, m), 7.57 – 7.45 (1H, m), 7.86 (1H, d, J= 5.7 Hz), 8.21 – 8.10 (1H, m), 8.49 – 8.40 (1H, m), 8.52 (1H, d, J= 5.3 Hz), 8.
…………………………………..
STEPH
LABEL 5
Synthesis of 2-hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)ethanone (5).
To a solution of 2-(2-((9-((lr,4r)-4- methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8- dihydro-l,6-naphthyridin-6(5H)-yl)-2-oxoethyl acetate (514 mg, 1.0 mmol) in DCM (7.5 mL) and MeOH (2.5 mL) was added 0.5 M sodium methoxide solution in MeOH (0.30 mL, 0.15 mmol), and the reaction mixture was stirred at room temperature for 1 hour and monitored using LCMS. Upon completion, the reaction mixture was concentrated. The residue was treated with EtOH (5 mL) and water (10 mL) to provide a solid which was collected by filtration, washed with water, and dried in a vacuum oven at 55 °C overnight to give the title compound (5) as a white solid (468 mg, 99% yield).
lH NMR (500 MHz, acetic acid-d4, 373 K) δ ppm 1.09 (3H, d, J= 6.5 Hz), 1.31-1.43 (2H, m), 1.70-1.80 (1H, m), 1.99-2.03 (2H, m), 2.06-2.13 (2H, m), 2.68 (2H, dq, J= 3.3, 12.7 Hz), 3.10 (2H, t, J= 5.4 Hz), 3.88 (2H, br. s.), 4.46 (2H, br. s.), 4.77 (2H, br. s), 4.90 (1H, tt, J= 3.9, 12.4 Hz), 7.76 (1H, d, J= 8.5 Hz), 8.33 (1H, d, J= 8.5 Hz), 8.40 (1H, d, J= 6.0 Hz), 8.63 (1H, d, J= 6.0 Hz), 9.35 (1H, s), 9.43 (1H, s) ppm; L
………………………………………………
STEP I
5 LABEL HCI Dihydrate
[0222] Synthesis of 2-hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)ethanone monohydrochloride dihydrate. To a suspension of 2-hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3- d]pyrimidin-2-yl)amino)-7,8-dihydro-l,6-naphthyridin-6(5H)-yl)ethanone (472 mg, 1.0 mmol) in water (2 mL) was added 2 N HCI (2 mL). The mixture became a clear solution. The pH value of the solution was adjusted to 4 by addition of 2 N NaOH at 0 °C and the precipitated light yellow solid was collected by filtration. The collected solid was washed with cold water three times. The solid was dried under vacuum to give the title compound as a light yellow solid (469 mg, 92% yield).
¾ NMR (500 MHz, DMSO-d6) δ 1.02 (3H, d, J= 5.0 Hz), 1.20- 1.30 (2H, m), 1.64 (1H, m), 1.88-1.90 (4H, m), 2.59-2.66 (2H, m), 2.85-2.95 (2H, m), 3.71(1H, m), 3.83 (1H, m), 4.19-4.22 (2H, m), 4.60-4.67 (2H, m), 4.85 (1H, m), 7.75 (1H, d, J= 8.5 Hz), 8.19 (1H, d, J= 8.5 Hz), 8.55 (1H, d, J= 5.0 Hz), 8.63 (1H, d, J= 5.0 Hz), 9.47 (1H, s), 9.58 (1H, s), 10.59 (1H, br.s) ppm; LCMS m/z: 472 (M+l). Anal.
Calc: C = 57.40, H = 6.30, N = 18.02; Found: C = 57.06, H = 6.31, N = 17.92. [0223] Alternative Synthesis of Hydrochloride Salt of 2-Hydroxy-l-(2-((9-
((lr,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2- yl)amino)-7,8-dihydro-l,6-naphthyridin-6(5H)-yl)ethanone. To a suspension of 2- hydroxy- 1 -(2-((9-(( 1 r,4r)-4-methylcyclohexyl)-9H-pyrido [4′,3 ‘ :4,5]pyrrolo [2,3 – d]pyrimidin-2-yl)amino)-7,8-dihydro-l,6-naphthyridin-6(5H)-yl)ethanone (2.385 g, 5.0 mmol) in water (10 mL) was added 2N HC1 (10 mL) at 20°C. The mixture became a clear light yellow solution. The pH value of the solution was adjusted to 4 by addition of 2N NaOH through addition funnel at 0° C, and the precipitated yellow solid was collected by filtration. The resulting solid was washed with cold water three times. The solid was dried under vacuum at 50° C for two days to provide 2.49 g of the hydrochloride salt of 2-hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-l,6-naphthyridin- 6(5H)-yl)ethanone as a solid. This salt was also obtained as a hydrate.
………………
US20140163052
http://www.google.com/patents/US20140163052
Example 5
2-Hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone
Synthesis of 2,5-dioxopyrrolidin-1-yl 2-acetoxyacetate
A 3-neck round-bottom flask equipped with a mechanical stirrer, thermocouple and addition funnel with nitrogen inlet was charged with N-hydroxysuccinimide (commercially available from Sigma-Aldrich, St. Louis, Mo.) (211 g, 1.83 mol) and DCM (2.25 L) at room temperature, resulting in a suspension. Pyridine (178 mL, 2.2 mol) was added in one portion with no change in the internal temperature. A solution of acetoxyacetyl chloride (commercially available from Sigma-Aldrich, St. Louis, Mo.) (197 mL, 1.83 mol) in DCM (225 mL) was added dropwise over 60 minutes and the temperature rose to 35° C. Stirring was continued at room temperature for 2.5 hours. The reaction mixture was washed with water (1×1 L), 1N HCl (2×1 L) and brine (1×1 L). The organic layer was concentrated under vacuum and azeotroped with toluene (1×1 L) to obtain the product as a white solid (367 g, 93%). 1H NMR (400 MHz, CDCl3) δ 4.96 (2H, s), 2.86 (4H, s), 2.19 (3H, s) ppm; LCMS m/z: 238 (M+Na).
![Figure US20140163052A1-20140612-C00129]()
Synthesis of 2-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)-2-oxoethyl acetate
To a suspension of 9-((1r,4r)-4-methylcyclohexyl)-N-(5,6,7,8-tetrahydro-1,6-naphthyridin-2-yl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine (1) (827 mg, 2.0 mmol) in chloroform (10 mL) were added diisopropylethylamine (258 mg, 348 uL, 2.0 mmol) and 2,5-dioxopyrrolidin-1-yl 2-acetoxyacetate (560 mg, 2.6 mmol). The reaction mixture thus obtained was stirred at room temperature for 30 minutes whereupon the mixture became a yellow solution. HPLC-MS analysis indicated that the reaction was complete. The reaction mixture was concentrated. MeOH (5 mL) and water (6 mL) were added to form a slurry which was stirred at room temperature for 1 hour. The solid was collected by filtration to give the title compound as a light yellow solid (1.04 g, 98% yield). 1H NMR (400 MHz, CDCl3, rotamers) δ ppm 1.08 (3H, d, J=6.5 Hz), 1.37-1.20 (2H, m), 2.03-1.97 (4H, m), 2.22 (3H, s), 2.69-2.52 (2H, m, J=2.9, 12.8, 12.8, 12.8 Hz), 3.08-2.93 (2H, m), 3.75 (1H, t, J=5.9 Hz), 3.97 (1H, t, J=5.6 Hz), 4.59 (1H, s),), 4.80-4.65 (2H, m),), 4.90-4.82 (2H, m), 7.57-7.45 (1H, m), 7.86 (1H, d, J=5.7 Hz), 8.21-8.10 (1H, m), 8.49-8.40 (1H, m), 8.52 (1H, d, J=5.3 Hz), 8.98 (1H, s), 9.11 (1H, s) ppm; LCMS m/z: 514 (M+1).
![Figure US20140163052A1-20140612-C00130]()
Synthesis of 2-hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (5)
To a solution of 2-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)-2-oxoethyl acetate (514 mg, 1.0 mmol) in DCM (7.5 mL) and MeOH (2.5 mL) was added 0.5 M sodium methoxide solution in MeOH (0.30 mL, 0.15 mmol), and the reaction mixture was stirred at room temperature for 1 hour and monitored using LCMS. Upon completion, the reaction mixture was concentrated. The residue was treated with EtOH (5 mL) and water (10 mL) to provide a solid which was collected by filtration, washed with water, and dried in a vacuum oven at 55° C. overnight to give the title compound (5) as a white solid (468 mg, 99% yield). 1H NMR (500 MHz, acetic acid-d4, 373 K) δ ppm 1.09 (3H, d, J=6.5 Hz), 1.31-1.43 (2H, m), 1.70-1.80 (1H, m), 1.99-2.03 (2H, m), 2.06-2.13 (2H, m), 2.68 (2H, dq, J=3.3, 12.7 Hz), 3.10 (2H, t, J=5.4 Hz), 3.88 (2H, br. s.), 4.46 (2H, br. s.), 4.77 (2H, br. s), 4.90 (1H, tt, J=3.9, 12.4 Hz), 7.76 (1H, d, J=8.5 Hz), 8.33 (1H, d, J=8.5 Hz), 8.40 (1H, d, J=6.0 Hz), 8.63 (1H, d, J=6.0 Hz), 9.35 (1H, s), 9.43 (1H, s) ppm; LCMS m/z: 472 (M+1).
![Figure US20140163052A1-20140612-C00131]()
Synthesis of 2-hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone monohydrochloride dihydrate
To a suspension of 2-hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (472 mg, 1.0 mmol) in water (2 mL) was added 2 N HCl (2 mL). The mixture became a clear solution. The pH value of the solution was adjusted to 4 by addition of 2 N NaOH at 0° C. and the precipitated light yellow solid was collected by filtration. The collected solid was washed with cold water three times. The solid was dried under vacuum to give the title compound as a light yellow solid (469 mg, 92% yield). 1H NMR (500 MHz, DMSO-d6) δ 1.02 (3H, d, J=5.0 Hz), 1.20-1.30 (2H, m), 1.64 (1H, m), 1.88-1.90 (4H, m), 2.59-2.66 (2H, m), 2.85-2.95 (2H, m), 3.71 (1H, m), 3.83 (1H, m), 4.19-4.22 (2H, m), 4.60-4.67 (2H, m), 4.85 (1H, m), 7.75 (1H, d, J=8.5 Hz), 8.19 (1H, d, J=8.5 Hz), 8.55 (1H, d, J=5.0 Hz), 8.63 (1H, d, J=5.0 Hz), 9.47 (1H, s), 9.58 (1H, s), 10.59 (1H, br.s) ppm; LCMS m/z: 472 (M+1). Anal. (C26H29N7O2—HCl.2H2O) Calc: C=57.40, H=6.30, N=18.02. Found: C=57.06, H=6.31, N=17.92.
Alternative Synthesis of Hydrochloride Salt of 2-Hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl) amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl) ethanone
To a suspension of 2-hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (2.385 g, 5.0 mmol) in water (10 mL) was added 2N HCl (10 mL) at 20° C. The mixture became a clear light yellow solution. The pH value of the solution was adjusted to 4 by addition of 2N NaOH through addition funnel at 0° C., and the precipitated yellow solid was collected by filtration. The resulting solid was washed with cold water three times. The solid was dried under vacuum at 50° C. for two days to provide 2.49 g of the hydrochloride salt of 2-hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone as a solid. This salt was also obtained as a hydrate.
………………………
J. Med. Chem., 2014, 57 (8), pp 3430–3449
DOI: 10.1021/jm500118j
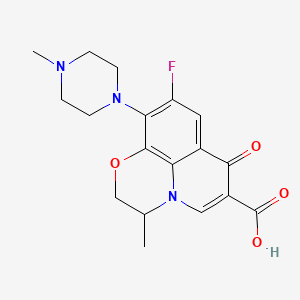
2-Hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (28)
compound 28 as a white solid (468 mg, 99% yield).
1H NMR (500 MHz, acetic acid-d4, 373 K) δ ppm 9.43 (1 H, s), 9.35 (1 H, s), 8.63 (1H, d, J = 6.0 Hz), 8.40 (1 H, d, J = 6.0 Hz), 8.33 (1 H, d, J = 8.5 Hz), 7.76 (1 H, d, J = 8.5 Hz), 4.90 (1 H, m), 4.77 (2 H, br s), 4.46 (2 H, br s), 3.88 (2 H, br s), 3.10 (2 H, t, J = 5.4 Hz), 2.68 (2 H, dq, J = 12.7, 3.3 Hz), 2.06–2.13 (2 H, m), 1.99–2.03 (2 H, m), 1.70–1.80 (1 H, m), 1.31–1.43 (2 H, m), 1.09 (3H, d, J = 6.5 Hz).
HRMS (ESI) m/z: calculated for [M + H]+ 472.2455, found 472.2461.
…………………………
PAPER
OPRD
† Chemical Process R&D, Amgen Inc., One Amgen Center Drive, Thousand Oaks, California 91320
‡ Norchim S.A.S., 33 Quai d’Amont, Saint Leu d’Esserent, France 60340
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/op500367p
The development of a synthetic route to manufacture the drug candidate AMG 925 on kilogram scale is reported herein. The hydrochloride salt of AMG 925 was prepared in 23% overall yield over eight steps from commercially available raw materials, and more than 8 kg of the target molecule were delivered. The synthetic route features a Buchwald–Hartwig amination using BrettPhos as ligand and conducted to afford 12 kg of product in a single batch. In addition, this work highlights the challenges associated with the use of poorly soluble process intermediates in the manufacture of active pharmaceutical ingredients. Creative solutions had to be devised to conduct seemingly routine activities such as salt removal, pH adjustment, and heavy metal scavenging due to the low solubility of the process intermediates. Finally, a slurry-to-slurry amidation protocol was optimized to allow for successful scale-up.
Manufacture of 2-Hydroxy-1-(2-((9-((1R,4R)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (AMG 925)
AMG 925 was isolated in 91.5% yield (8.31 kg), 95.5% overall mass balance, 99.9 wt %, and 99.7 LCAP.
Mp 213–215 °C;
1H NMR (400 MHz, acetic acid-d4, mixture of two rotamers at 20 °C) 9.47–9.59 (m, 2H), 8.76 (d, 1H, J = 6 Hz), 8.55 (d, 1H, J = 6 Hz), 8.48 (d, 1H,J = 9 Hz), 7.79–7.92 (m, 1H), 4.95 (t, 1H, J = 12 Hz), 4.87 and 4.68 (2 singlets, 2H), 4.47–4.59 (m, 2H), 4.04 and 3.80 (2 triplets, 2H, J = 6 Hz), 3.03–3.17 (m, 2H), 2.65–2.82 (m, 2H), 1.96–2.15 (m, 4H), 1.77 (br s, 1H), 1.39 (q, 2H, J = 12 Hz), 1.09 (d, 3H, J = 7 Hz);
13C NMR (100 MHz, acetic acid-d4, mixture of two rotamers at 20 °C) 171.9, 171.8, 158.4, 157.8, 154.7, 149.0, 148.9, 141.6, 135.2, 132.9, 126.3, 124.1, 123.6, 117.7, 113.7, 113.6, 107.5, 107.4, 60.1, 59.9, 56.3, 43.7, 42.6, 40.5, 38.7, 34.0, 31.5, 29.8, 28.8, 28.1, 21.5.
Manufacture of 2-Hydroxy-1-(2-((9-((1R,4R)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone Hydrochloride (AMG 925 HCl)
AMG 925 HCl was isolated in 92.5% yield (7.96 kg), 99.1% overall mass balance, 83.8 wt % AMG 925, 99.75 LCAP AMG 925, 6.2 wt % Cl, 9.6 wt % water, 3800 ppm AcOH, d10 4.0 μm, d50 15.2 μm, d90 38.8 μ, Vm 18.7 μm, BET surface area 1.5 m2/g.
1H NMR (400 MHz, acetic acid-d4, mixture of two rotamers at 20 °C) 9.63 (s, 1H), 9.56 (s, 1H), 8.71–8.76 (m, 1H), 8.60–8.66 (m, 1H), 8.20–8.29 (m, 1H), 7.90–7.98 (m, 1H), 4.90–5.01 (m, 1H), 4.86 and 4.70 (2 singlets, 2H), 4.53 and 4.51 (2 singlets, 2H), 4.05 and 3.82 (2 triplets, 2H, J = 6 Hz), 3.11–3.26 (m, 2H), 2.68 (q, 2H, J = 12 Hz), 1.95–2.13 (m, 4H), 1.74 (br s, 1H), 1.36 (q, 2H, J = 12 Hz), 1.06 (d, 3H, J = 8 Hz);
13C NMR (100 MHz, acetic acid-d4, mixture of two rotamers at 20 °C) 174.9, 174.8, 161.3, 161.2, 160.5, 157.5, 151.6, 151.5, 149.3, 148.9, 145.5, 138.1, 136.0, 129.3, 129.2, 127.1, 126.6, 120.9, 116.7, 116.6, 110.8, 110.7, 63.0, 62.9, 59.3, 46.4, 45.3, 43.2, 41.3, 36.9, 34.3, 32.6, 31.3, 30.6, 24.4; exact mass [C26H29N7O2 + H]+: calculated = 472.2461, measured = 472.2451.
References:
1. K. Keegan et al, Preclinical evaluation of AMG 925, a FLT3/CDK4 dual kinase inhibitor for treating acute myeloid leukemia. Mol Cancer Ther. 2014 Apr;13(4):880-9.
2. ZH Li, et al, Discovery of AMG 925, a FLT3 and CDK4 Dual Kinase Inhibitor with Preferential Affinity for the Activated State of FLT3, J. Med Chem, March 18, 2014
Filed under:
Uncategorized Tagged:
AMG 925,
FLT3,
kinase inhibitor ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()































![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-07-23/000/115/669/161814-49-9-1h.png)
![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-07-23/000/115/669/161814-49-9-13c.png)















































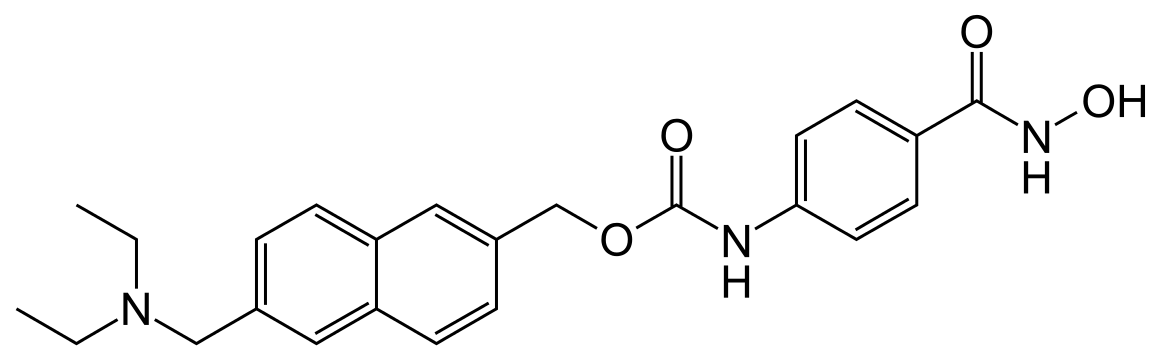
 GIVINOSTAT
GIVINOSTAT







![[6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate NMR spectra analysis, Chemical CAS NO. 497833-27-9 NMR spectral analysis, [6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-20/001/566/1566912_1h.png)
![[6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate NMR spectra analysis, Chemical CAS NO. 497833-27-9 NMR spectral analysis, [6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-20/001/566/1566912_13c.png)



















































 Originally posted on
Originally posted on 






























































































































































 AMG 925 HCL MONOHYDRATE
AMG 925 HCL MONOHYDRATE
























 OLMESARTAN
OLMESARTAN








 Originally posted on
Originally posted on 

















 NCT02216656
NCT02216656






