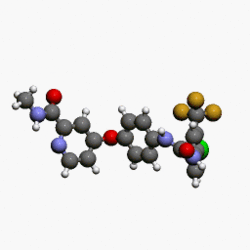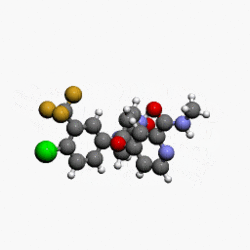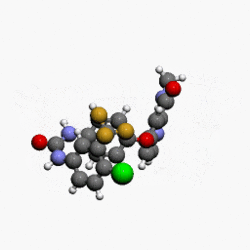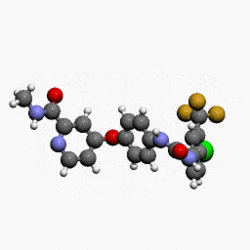
(4-(4-(3-(4-chloro-3-(trifluoromethyl)phenyl)ureido)phenoxy)-N-methylpicolinamide)
Bayer HealthCare has obtained approval from the Japanese Ministry of Health, Labour and Welfare (MHLW) for its Nexavar (sorafenib) for treatment of patients with unresectable differentiated thyroid carcinoma.
Bayer HealthCare has obtained approval from the Japanese Ministry of Health, Labour and Welfare (MHLW) for its Nexavar (sorafenib) for treatment of patients with unresectable differentiated thyroid carcinoma.
Nexavar’s approval in Japan is supported by data from the multicentre, placebo-controlled Phase III DECISION (‘stuDy of sorafEnib in loCally advanced or metastatIc patientS with radioactive Iodine refractory thyrOid caNcer’) study.
The international Phase III DECISION study, which randomised a total of 417 patients, met its primary endpoint of extended progression-free survival. Safety and tolerability profile of sorafenib was generally consistent with the known profile of sorafenib.
The most common treatment-emergent adverse events in the sorafenib arm were hand-foot skin reaction, diarrhea, alopecia, weight loss, fatigue, hypertension and rash.
Nexavar was awarded orphan drug status by the MHLW for thyroid carcinoma in September 2013.
A phase 3 clinical trial has started recruiting (November 2009) to use sorafenib for non-responsive thyroid cancer.[12] The results were presented at the ASCO 13th Annual Meeting and are the base for FDA approval. The Sorafenib in locally advanced or metastatic patients with radioactive iodine-refractory differentiated thyroid cancer: The Phase 3 DECISION trial showed significant improvement in progression-free survival but not in overall survival. However, as is known, the side effects were very frequent, specially hand and foot skin reaction.[13]
The European Commission granted marketing authorization to the drug for the treatment of patients with hepatocellular carcinoma(HCC), the most common form of liver cancer, in October 2007,[22] and FDA approval for this indication followed in November 2007.[23]
As of November 22, 2013, sorafenib has been approved by the FDA for the treatment of locally recurrent or metastatic, progressive differentiated thyroid carcinoma (DTC) refractory to radioactive iodine treatment.[28]
In some kinds of lung cancer (with squamous-cell histology) sorafenib administered in addition to paclitaxel and carboplatin may be detrimental to patients.[29]
In January 2014, Bayer’s CEO stated that Nexavar was developed for “western patients who [could] afford it”. At the prevailing prices, a kidney cancer patient would pay $96,000 (£58,000) for a year’s course of the Bayer-made drug. However, the cost of the Indian version of the generic drug would be around $2,800 (£1,700).[32]

4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-Λ/2-methylpyridine-2- carboxamide is commonly known as sorafenib (I). Sorafenib is prepared as its tosylate salt. Sorafenib blocks the enzyme RAF kinase, a critical component of the RAF/MEK/ERK signaling pathway that controls cell division and proliferation; in addition, sorafenib inhibits the VEGFR-2/PDGFR-beta signaling cascade, thereby blocking tumor angiogenesis.
Sorafenib, marketed as Nexavar by Bayer, is a drug approved for the treatment of advanced renal cell carcinoma (primary kidney cancer). It has also received “Fast Track” designation by the FDA for the treatment of advanced hepatocellular carcinoma (primary liver cancer). It is a small molecular inhibitor of Raf kinase, PDGF (platelet-derived growth factor), VEGF receptor 2 & 3 kinases and c Kit the receptor for Stem cell factor.
Sorafenib and pharmaceutically acceptable salts thereof is disclosed in WO0042012. Sorafenib is also disclosed in WO0041698. Both these patents disclose processes for the preparation of sorafenib.
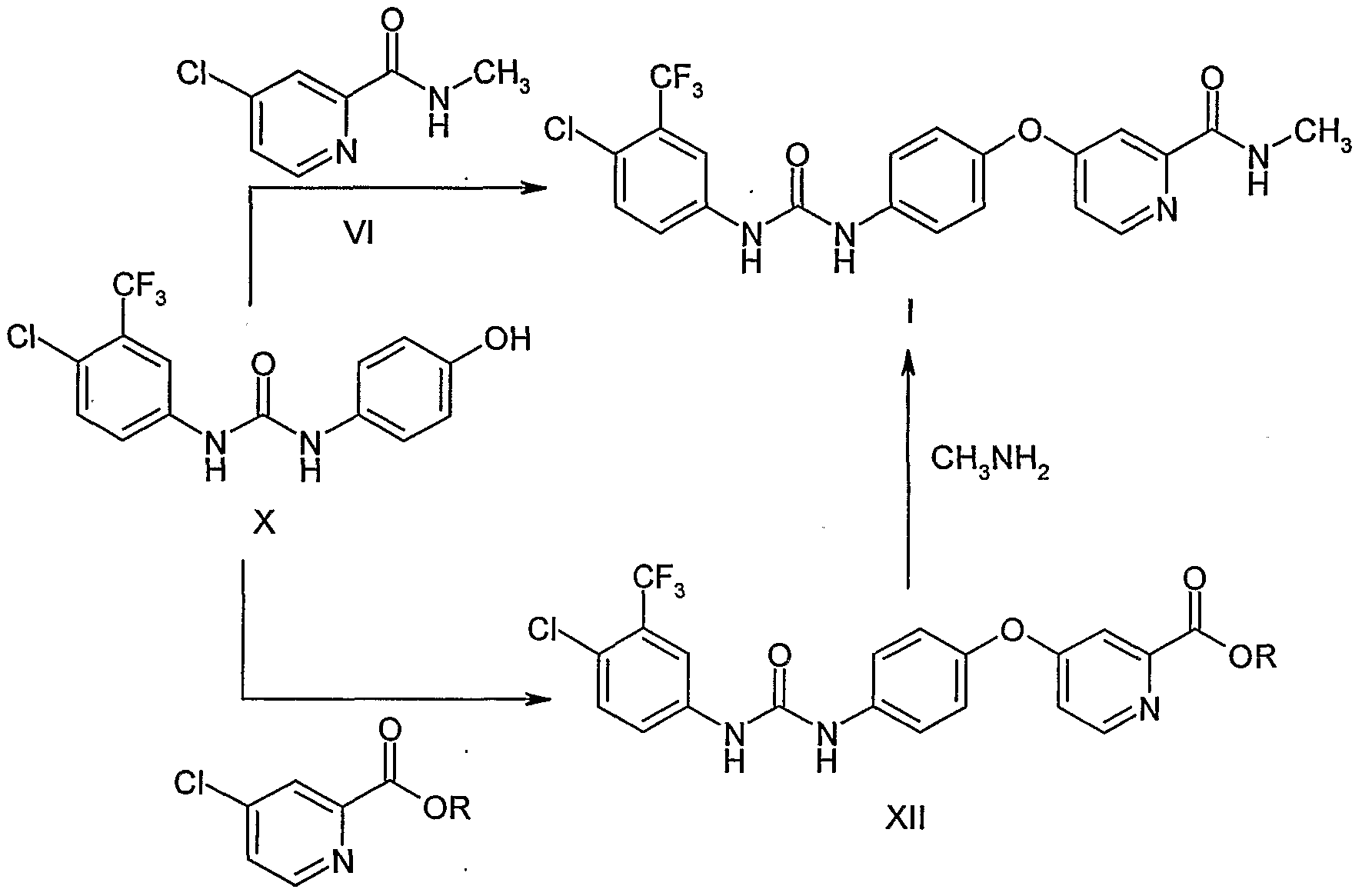
WO0042012 and WO0041698 describe the process as given in scheme I which comprises reacting picolinic acid (II) with thionyl chloride in dimethyl formamide (DMF) to form acid chloride salt (III). This salt is then reacted with methylamine dissolved in tetrahydrofuran (THF) to give carboxamide (IV). This carboxamide when further reacted with 4- aminophenol in anhydrous DMF and potassium tert-butoxide 4-(2-(N-methylcarbamoyl)-4- pyridyloxy)aniline (V) is formed. Subsequent reaction of this aniline with 4-chloro-3- (trifluoromethyl) phenyl isocyanate (Vl) in methylene chloride yields sorafenib (I). The reaction is represented by Scheme I as given below.
Scheme I
Picolini
Sorafenib (I)
WO2006034796 also discloses a process for the preparation of sorafenib and its tosylate salt. The process comprises reacting 2-picolinic acid (II) with thionyl chloride in a solvent inert toward thionyl chloride without using dimethyl formamide to form acid chloride salt (III). This acid salt on further reaction with aqueous solution methylamine or gaseous methylamine gives compound (IV). Compound (IV) is then reacted with 4-aminophenol with addition of a carbonate salt in the presence of a base to yield compound (V).
Compound (V) can also be obtained by reacting compound (IV) with 4-aminophenol in the presence of water with addition of a phase transfer catalyst. Compound (V) when reacted with 4-chloro-3-(trifluoromethyl) phenyl isocyanate (Vl) in a non-chlorinated organic solvent, inert towards isocyanate gives sorafenib (I). Sorafenib by admixing with p- toluenesulfonic acid in a polar solvent gives sorafenib tosylate (VII). The reaction is represented by Scheme Il as given below.
Scheme Il
P
A key step in the synthesis of sorafenib is the formation of the urea bond. The processes disclosed in the prior art involve reactions of an isocyanate with an amine. These isocyanate compounds though commercially available are very expensive. Further synthesis of isocyanate is very difficult which requires careful and skillful handling of reagents.
Isocyanate is prepared by reaction of an amine with phosgene or a phosgene equivalent, such as bis(trichloromethyl) carbonate (triphosgene) or trichloromethyl chloroformate (diphosgene). Isocyanate can also be prepared by using a hazardous reagent such as an azide. Also, the process for preparation of an isocyanate requires harsh reaction conditions such as strong acid, higher temperature etc. Further, this isocyanate is reacted with an amine to give urea.
Reactions of isocyanates suffer from one or more disadvantages. For example phosgene or phosgene equivalents are hazardous and dangerous to use and handle on a large scale. These reagents are also not environment friendly. Isocyanates themselves are thermally unstable compounds and undergo decomposition on storage and they are incompatible with a number of organic compounds. Thus, the use of isocyanate is not well suited for industrial scale application.
Sorafenib and its pharmaceutically acceptable salts and solvates are reported for the first time in WO0041698 (corresponding US 03139605) by Bayer. In the literature only one route is disclosed for the preparation of sorafenib. According to this route (Scheme-I), picolinic acid of formula III is reacted with thionyl chloride to give the 4-chloro derivative which on treatment
VII
Scheme-I with methanol gave the methyl ester of formula V. Compound of formula V is reacted with methylamine to get the corresponding amide of formula VL Compound of formula VI is reacted with 4-aminophenol to get the ether derivative of formula VII. Compound of formula VII is reacted with 4-chloro-3-trifluoromethylphenylisocyante to get sorafenib base of formula I. Overall yield of sorafenib in this process is 10% from commercially available 2-picolinic acid of formula II. Main drawback in this process is chromatographic purification of the intermediates involved in the process and low yield at every step.
Donald Bankston’s (Org. Proc. Res. Dev., 2002, 6, 777-781) development of an improved synthesis of the above basic route afforded sorafenib in an overall yield of 63% without involving any chromatographic purification. Process improvements like reduction of time in thionyl chloride reaction; avoid the isolation of intermediates of formulae IV and V5 reduction of base quantity in p-aminophenol reaction, etc lead to the simplification of process and improvement in yield of final compound of formula I.
Above mentioned improvements could not reduce the number of steps in making sorafenib of formula-I. In the first step all the raw materials are charged and heated to target temperature (72°C). Such a process on commercial scale will lead to sudden evolution of gas emissions such as sulfur dioxide and hydrogen chloride. Also, in the aminophenol reaction two bases (potassium carbonate and potassium t-butoxide) were used in large excess to accomplish the required transformation.
A scalable process for the preparation of sorafenib is disclosed in WO2006034796. In this process also above mentioned chemistry is used in making sorafenib of formula I. In the first step, catalytic quantity. of DMF used in the prior art process is replaced with reagents like hydrogen bromide, thionyl bromide and sodium bromide. Yield of required product remained same without any advantages from newly introduced corrosive reagents. Process improvements like change of solvents, reagents, etc were applied in subsequent steps making the process scalable. Overall yield of sorafenib is increased to 74% from the prior art 63% yield. Purity of sorafenib is only 95% and was obtained as light brown colored solid.
Main drawbacks in this process are production of low quality sorafenib and requirement of corrosive and difficult to handle reagents such as thionyl bromide and hydrogen bromide. Also, there is no major improvement in the yield of sorafenib.
Sorafenib tosylate ( Brand name: Nexavar ®, BAY 43-9006 other name, Chinese name: Nexavar, sorafenib, Leisha Wa) was Approved by U.S. FDA for the treatment of advanced kidney cancer in 2005 and liver cancer in 2007 .
Sorafenib, co-Developed and co-marketed by Germany-based Bayer AG and South San Francisco-based Onyx Pharmaceuticals , is an Oral Multi-kinase inhibitor for VEGFR1, VEGFR2, VEGFR3, PDGFRbeta, Kit, RET and Raf-1.
In March 2012 Indian drugmaker Natco Pharma received the first compulsory license ever from Indian Patent Office to make a generic Version of Bayer’s Nexavar despite the FACT that Nexavar is still on Patent. This Decision was based on the Bayer Drug being too expensive to most patients. The Nexavar price is expected to drop from $ 5,500 per person each month to $ 175, a 97 percent decline. The drug generated $ 934 million in global sales in 2010, according to India’s Patent Office.

Sorafenib tosylate
Chemical Name: 4-Methyl-3-((4 – (3-pyridinyl)-2-pyrimidinyl) amino)-N-(5 – (4-methyl-1H-imidazol-1-yl) -3 – (trifluoromethyl) phenyl) benzamide monomethanesulfonate, Sorafenib tosylate
CAS Number 475207-59-1 (Sorafenib tosylate ) , 284461-73-0 (Sorafenib)
References for the Preparation of Sorafenib References
1) Bernd Riedl, Jacques Dumas, Uday Khire, Timothy B. Lowinger, William J. Scott, Roger A. Smith, Jill E. Wood, Mary-Katherine Monahan, Reina Natero, Joel Renick, Robert N. Sibley; Omega-carboxyaryl Substituted diphenyl Ureas as RAF kinase inhibitors ; U.S. Patent numberUS7235576
2) Rossetto, Pierluigi; Macdonald, Peter, Lindsay; Canavesi, Augusto; Process for preparation of sorafenib and Intermediates thereof , PCT Int. Appl., WO2009111061
3) Lögers, Michael; gehring, Reinhold; Kuhn, Oliver; Matthäus, Mike; Mohrs, Klaus; müller-gliemann, Matthias; Stiehl, jürgen; berwe, Mathias; Lenz, Jana; Heilmann, Werner; Process for the preparation of 4 – {4 – [( {[4-chloro-3-(TRIFLUOROMETHYL) phenyl] amino} carbonyl) amino] phenoxy}-N-methylpyridine-2-carboxamide , PCT Int. Appl., WO2006034796
4) Shikai Xiang, Liu Qingwei, Xieyou Rong, sorafenib preparation methods, invention patent application Publication No. CN102311384 , Application No. CN201010212039
5) Zhao multiply there, Chenlin Jie, Xu Xu, MASS MEDIA Ji Yafei; sorafenib tosylate synthesis ,Chinese Journal of Pharmaceuticals , 2007 (9): 614 -616
Preparation of Sorafenib Tosylate (Nexavar) Nexavar, sorafenib Preparation of methyl sulfonate

Sorafenib (Sorafenib) chemical name 4 – {4 – [({[4 - chloro -3 - (trifluoromethyl) phenyl] amino} carbonyl) amino] phenoxy}-N-methyl-pyridine -2 – formamide by Bayer (Bayer) research and development, in 2005 the U.S. Food and Drug Administration (FDA) approval. Trade name Nexavar (Nexavar). This product is an oral multi-kinase inhibitor, for the treatment of liver cancer and kidney cancer.
Indian Patent Office in March this year for Bayer’s Nexavar in liver and kidney cancer drugs (Nexavar) has released a landmark “compulsory licensing” (compulsory license). Indian Patent Office that due to the high price Nexavar in India, the vast majority of patients can not afford the drug locally, thus requiring local Indian pharmaceutical company Natco cheap Nexavar sales. Nexavar in 2017 before patent expiry, Natco pay only Bayer’s pharmaceutical sales to 6% royalties. The move to make Nexavar patent drug prices, the supply price from $ 5,500 per month dropped to $ 175, the price reduction of 97%. Compulsory licensing in India for other life-saving drugs and patent medicines overpriced open a road, the Indian Patent Office through this decision made it clear that the patent monopoly does not guarantee that the price is too high. Nexavar is a fight against advanced renal cell carcinoma, liver cancer cure. In China, a box of 60 capsules of Nexavar price of more than 25,000 yuan. In accordance with the recommended dose, which barely enough to eat half of patients with advanced cancer. In September this year India a patent court rejected Bayer Group in India cheap drugmaker emergency appeal. Indian government to refuse patent medicine overpriced undo “compulsory licensing rules,” allowing the production of generic drugs Nexavar.
Sorafenat by Natco – Sorafenib – Nexavar – India natco Nexavar

Chemical Synthesis of Sorafenib Tosylate (Nexavar)
Sorafenib tosylate (brand name :Nexavar®, other name BAY 43-9006, was approved by US FDA for the treatment of kidney cancer in 2005 and advanced liver cancer in 2007.

US Patent US7235576, WO2006034796, WO2009111061 and Faming Zhuanli Shenqing(CN102311384) disclosed processes for preparation of sorafenib base and its salt sorafenib tosylate.
References
1)Bernd Riedl, Jacques Dumas, Uday Khire, Timothy B. Lowinger, William J. Scott, Roger A. Smith, Jill E. Wood, Mary-Katherine Monahan, Reina Natero, Joel Renick, Robert N. Sibley; Omega-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors; US patent numberUS7235576
2)Rossetto, pierluigi; Macdonald, peter, lindsay; Canavesi, augusto; Process for preparation of sorafenib and intermediates thereof, PCT Int. Appl., WO2009111061
3)Lögers, michael; gehring, reinhold; kuhn, oliver; matthäus, mike; mohrs, klaus; müller-gliemann, matthias; stiehl, jürgen; berwe, mathias; lenz, jana; heilmann, werner; Process for the preparation of 4-{4-[({[4-chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-n-methylpyridine-2-carboxamide, PCT Int. Appl., WO2006034796CN102311384, CN201010212039
Full Experimental Details for the preparation of Sorafenib Tosylate (Nexavar)
Synthesis of 4-(2-(N-methylcarbamoyl)-4-pyridyloxy)aniline.
A solution of 4-aminophenol (9.60 g, 88.0 mmol) in anh. DMF (150 mL) was treated with potassium tert-butoxide (10.29 g, 91.7 mmol), and the reddish-brown mixture was stirred at room temp. for 2 h. The contents were treated with 4-chloro- N -methyl-2-pyridinecarboxamide (15.0 g, 87.9mmol) and K2CO3 (6.50 g, 47.0 mmol) and then heated at 80°C. for 8 h. The mixture was cooled to room temp. and separated between EtOAc (500 mL) and a saturated NaCl solution (500 mL). The aqueous phase was back-extracted with EtOAc (300 mL). The combined organic layers were washed with a saturated NaCl solution (4×1000 mL), dried (Na2SO4) and concentrated under reduced pressure. The resulting solids were dried under reduced pressure at 35°C. for 3 h to afford 4-(2-(N-methylcarbamoyl)-4-pyridyloxy)aniline as a light-brown solid 17.9 g, 84%):. 1H-NMR (DMSO-d6) δ 2.77 (d, J = 4.8 Hz, 3H), 5.17 (br s, 2H), 6.64, 6.86 (AA’BB’ quartet, J = 8.4 Hz, 4H), 7.06 (dd, J = 5.5, 2.5 Hz, 1H), 7.33 (d, J = 2.5 Hz, 1H), 8.44 (d, J = 5.5 Hz; 1H), 8.73 (br d, 1H); HPLC ES-MS m/z 244 ((M+H)+).
Synthesis of 4-{4-[({[4-Chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-N-methylpyridine-2-carboxamide (sorafenib)
4-(4-Aminophenoxy)-N-methyl-2-pyridinecarboxamide (52.3 kg, 215 mol) is suspended in ethyl acetate (146 kg) and the suspension is heated to approx. 40° C. 4-Chloro-3-trifluoromethylphenyl isocyanate (50 kg, 226 mol), dissolved in ethyl acetate (58 kg), is then added to such a degree that the temperature is kept below 60° C. After cooling to 20° C. within 1 h, the mixture is stirred for a further 30 min and the product is filtered off. After washing with ethyl acetate (30 kg), the product is dried under reduced pressure (50° C., 80 mbar). 93 kg (93% of theory) of the title compound are obtained as colorless to slightly brownish crystals. m.p. 206-208° C. 1H-NMR (DMSO-d6, 500 MHz): δ =2.79 (d, J=4.4 Hz, 3H, NCH3); 7.16 (dd, J=2.5, 5.6 Hz, 1H, 5-H); 7.18 (d, J=8.8 Hz, 2H, 3′-H, 5′-H); 7.38 (d, J=2.4 Hz, 1H, 3-H); 7.60-7.68 (m, 4H, 2′-H, 6′-H, 5″-H, 6″-H); 8.13 (d, J=1.9 Hz, 1H, 2″-H); 8.51 (d, J=5.6 Hz, 1H, 6-H); 8.81 (d, J=4.5 Hz, 1H, NHCH3); 9.05 (br. s, 1H, NHCO); 9.25 (br. s, 1H, NHCO) MS (ESI, CH3CN/H2O): m/e=465 [M+H]+.
Synthesis of Sorafenib Tosylate (Nexavar)
4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-N2-methylpyridine-2-carboxamide (sorafenib) (50g, 0.1076 mol) is suspended in ethyl acetate (500 g) and water (10g). The mixture is heated to 69°C within 0.5 h, and a filtered solution of p-toluenesulfonic acid monohydrate (3.26 g, 0.017 mol) in a mixture of water (0.65 g) and ethyl acetate (7.2 g) is added. After filtration a filtered solution of p-toluenesulfonic acid monohydrate (22g, 0.11 mol) in a mixture of ethyl acetate (48 g) and water (4.34 g) is added. The mixture is cooled to 23°C within 2 h. The product is filtered off, washed twice with ethyl acetate (92.5 g each time) and dried under reduced pressure. The sorafenib tosylate (65.5 g, 96% of theory) is obtained as colorless to slightly brownish crystals.
…………………..
http://www.google.com/patents/EP2195286A2?cl=en
Example 22: Synthesis of Sorafenib
Phenyl 4-chloro-3-(trifluoromethyl)phenylcarbamate (100 g, 0.3174 mol) and 4-(4- aminophenoxy)-N-methylpicolinamide (77.14 g, 0.3174 mol) were dissolved in N1N- dimethyl formamide (300 ml) to obtain a clear reaction mass. The reaction mass was agitated at 40-450C for 2-3 hours, cooled to room temperature and diluted with ethyl acetate (1000 ml). The organic layer was washed with water (250 ml) followed by 1N HCI (250ml) and finally with brine (250 ml). The organic layer was separated, dried over sodium sulfate and degassed to obtain solid. This solid was stripped with ethyl acetate and finally slurried in ethyl acetate (1000 ml) at room temperature. It was then filtered and vacuum dried to give (118 g) of 4-(4-(3-(4-chloro-3- (trifluoromethyl)phenyl)ureido)phenoxy)-N-methylpicolinamide (sorafenib base).
Example 23: Synthesis of 1-(4-chloro-3-(trifluoromethyl)phenyl)urea (Compound 4)
Sodium cyanate (1.7 g, 0.02mol) was dissolved in water (17ml) at room temperature to obtain a clear solution. This solution was then charged drop wise to the clear solution of 3- trifluoromethyl-4-chloroaniline (5 g, 0.025 mol) in acetic acid (25 ml) at 40°C-45°C within 1- 2 hours. The reaction mass was agitated for whole day and cooled gradually to room temperature. The obtained solid was filtered washed with water and vacuum dried at 500C to afford the desired product (5.8 g) i.e. 1-(4-chloro-3-(trifluoromethyl)phenyl)urea.
Example 24: Synthesis of Sorafenib
1-(4-chloro-3-(trifluoromethyl) phenyl)urea (15 g, 0.0628 mol), 1 ,8- diazabicyclo[5.4.0]undec-7-ene (11.75 ml, 0.078 mol) and 4-(4-aminophenoxy)-N- methylpicolinamide (15.27 g, 0.0628 mol) were mixed with dimethyl sulfoxide (45 ml) and the reaction mass was then heated to 110-1200C for 12-18 hours. The reaction mass was cooled to room temperature and quenched in water (250 ml). The quenched mass was extracted repeatedly with ethyl acetate and the combined ethyl acetate layer was then back washed with water. It was dried over sodium sulfate and evaporated under vacuum to obtain solid. The obtained solid was slurried in acetonitrile (150 ml) at ambient temperature and filtered to give 4-(4-(3-(4-chloro-3-(trifluoromethyl) phenyl) ureido) phenoxy)-N-methylpicolinamide (sorafenib base) (17.5 g).
………………………..
http://www.google.com/patents/WO2009054004A2?cl=en
http://worldwide.espacenet.com/publicationDetails/biblio?CC=WO&NR=2009054004A2&KC=A2&FT=D&date=20090430&DB=EPODOC&locale=en_gb
![Figure imgf000006_0002]()
EXAMPLES
Example 1
Preparation of l-(4-chloro-3-(trifluoromethyl)phenyI)-3-(4-hydroxyphenyl)urea Into a 250 ml, four-necked RB flask was charged 1O g of 4-aminophenol and 100 ml of toluene. A solution of 4-chloro-3-(trifluoromethyl)phenyl isocyante (20.4 g) in toluene (50 ml) was added to the reaction mass at 25-300C. The reaction mass was stirred at room temperature for 16 h. The reaction mass was filtered and washed the. solid with 50 ml of toluene. The wet material was dried in the oven at 50-60°C to get 29.8 g of title compound as white solid. M.P. is 218-222°C. IR (KBr): 3306, 1673, 1625, 1590, 1560, 1517, 1482, 1435, 1404, 1328, 1261, 1182, 1160, 1146, 1125, 1095, 1032, 884, 849, 832, 812, 766, 746, 724, 683, 539 and 434 cm“1.
Example 2 Preparation of sorafenib tosylate
Into a 100 ml, three-necked RB flask was charged 2.0 g of l-(4-chloro-3- (trifluoromethyl)-phenyl)-3-(4-hydroxyphenyl)urea and 10 ml of DMF. Potassium tert- butoxide (2.3 g) was added to the reaction mass and stirred for 45 min at RT. 4-Chlro-N- methylpicolinamide (1.14 g) and potassium carbonate (0.42 g) were added to the reaction mass and heated to 80°C. The reaction mass was maintained at 80-85°C for 8 h and cooled to 30°C. The reaction mass was poured into water and extracted with ethyl acetate. Ethyl acetate layer was washed with water, brine and dried over sodium sulphate. Solvent was distilled of under reduced pressure.
The crude compound (4.7 g) was dissolved in 10 ml of IPA and added 1.9 g of p- toluenesulfonic acid. The reaction mass was stirred at RT for 15 h and filtered. The wet solid was washed with 10 ml of IPA and dried at 50-60°C to get 3.4 g of title compound as off-white crystalline solid.
…………………..
A Scaleable Synthesis of BAY 43-9006: A Potent Raf Kinase Inhibitor for the Treatment of Cancer
Bayer Research Center, Pharmaceutical Division, 400 Morgan Lane, West Haven, Connecticut 06516, U.S.A.
Org. Proc. Res. Dev., 2002, 6 (6), pp 777–781
DOI: 10.1021/op020205n
http://pubs.acs.org/doi/abs/10.1021/op020205n
Urea 3 (BAY 43-9006), a potent Raf kinase inhibitor, was prepared in four steps with an overall yield of 63%. Significant process research enabled isolation of each intermediate and target without chromatographic purification, and overall yield increases >50% were observed compared to those from previous methods. This report focuses on improved synthetic strategies for production of scaled quantities of 3 for preclinical, toxicological studies. These improvements may be useful to assemble other urea targets as potential therapeutic agents to combat cancer.
Synthesis of N-[4-Chloro-3-(trifluoromethyl)phenyl]({4-[2-(N-methyl-carbamoyl)(4-pyridyloxy)]phenyl}amino)carboxamide (3, BAY 43-9006).
A suspension of 9 (67.00 g, 275.43 mmol) in methylene chloride ———————-DELETE………………………………The solids were washed with methylene chloride (2 × 50 mL) and dried under vacuum for 4 h at 35 °C to afford 3 (118.19 g, 254.27 mmol, 92%) as an off-white solid.
Mp = 210−212 °C.
1H NMR (DMSO-d6, 300 MHz):
δ 2.77 (d, J = 4.8 Hz, 3H, −NHCH3);
7.16 (m, 3H, aromatic);
7.37 (d, J = 2.5 Hz, 1H, aromatic);
7.62 (m, 4H, aromatic);
8.11 (d, J = 2.5 Hz, 1H, aromatic);
8.49 (d, J = 5.5 Hz, 1H, aromatic);
8.77 (br d, 1H, −NHCH3);
8.99 (s, 1H, −NHCO−); 9.21 (s, 1H, −NHCO−).
Mass spectrum (HPLC/ES): m/e = 465 (M + 1).
Anal. Calcd for C21H16N4ClF3O3: C, 54.26; H, 3.47; N, 12.05. Found: C, 54.11; H, 3.49; N, 12.03.
HPLC (ELS) purity >98%: tR = 3.5 min.
Synthesis of N-[4-Chloro-3-(trifluoromethyl)phenyl]({4-[2-(N-methyl-carbamoyl)(4-pyridyloxy)]phenyl}amino)carboxamide (3, BAY 43-9006): Use of CDI.
A solution of 11 (1.25 g, 6.39 mmol) in methylene chloride———————-DELETED……………………. high vacuum at 35 °C for 2 h to afford 3 (2.55 g, 5.49 mmol, 91%) as a white solid. Proton NMR and mass-spectral data were consistent with structure.
Anal. Calcd for C21H16N4ClF3O3: C, 54.26; H, 3.47; N, 12.05; Cl, 7.63. Found: C, 54.24; H, 3.31; N, 12.30; Cl, 7.84.
Mp (differential scanning calorimetry, 10 °C/min): 205.6 °C;
no polymorphs observed.
References
- “FDA Approves Nexavar for Patients with Inoperable Liver Cancer” (Press release). FDA. November 19, 2007. Retrieved November 10, 2012.
- “Nexavar (sorafenib) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 26 December 2013.
- “NEXAVAR (sorafenib) tablet, film coated [Bayer HealthCare Pharmaceuticals Inc.]“. DailyMed. Bayer HealthCare Pharmaceuticals Inc. November 2013. Retrieved 26 December 2013.
- “Nexavar 200mg film-coated tablets – Summary of Product Characteristics (SPC) – (eMC)”. electronic Medicines Compendium. Bayer plc. 27 March 2013. Retrieved 26 December 2013.
- “PRODUCT INFORMATION NEXAVAR® (sorafenib tosylate)” (PDF). TGA eBusiness Services. Bayer Australia Ltd. 12 December 2012. Retrieved 26 December 2013.
- Escudier, B; Eisen, T; Stadler, WM; Szczylik, C; Oudard, S; Siebels, M; Negrier, S; Chevreau, C; Solska, E; Desai, AA; Rolland, F; Demkow, T; Hutson, TE; Gore, M; Freeman, S; Schwartz, B; Shan, M; Simantov, R; Bukowski, RM (January 2007). “Sorafenib in advanced clear-cell renal-cell carcinoma”. New England Journal of Medicine 356 (2): 125–34. doi:10.1056/NEJMoa060655. PMID 17215530.
- Walid, MS; Johnston, KW (October 2009). “Successful treatment of a brain-metastasized renal cell carcinoma”. German Medical Science 7: Doc28. doi:10.3205/000087. PMC 2775194. PMID 19911072.
- “Pharmaceutical Benefits Scheme (PBS) -SORAFENIB”. Pharmaceutical Benefits Scheme. Australian Government Department of Health. Retrieved 27 December 2013.
- Llovet, et al. (2008). “Sorafenib in Advanced Hepatocellular Carcinoma” (PDF). New England Journal of Medicine 359 (4): 378–90.
- Keating GM, Santoro A (2009). “Sorafenib: a review of its use in advanced hepatocellular carcinoma”. Drugs 69 (2): 223–40. doi:10.2165/00003495-200969020-00006. PMID 19228077.
- Pawlik TM, Reyes DK, Cosgrove D, Kamel IR, Bhagat N, Geschwind JF (October 2011). “Phase II trial of sorafenib combined with concurrent transarterial chemoembolization with drug-eluting beads for hepatocellular carcinoma”. J. Clin. Oncol. 29 (30): 3960–7. doi:10.1200/JCO.2011.37.1021. PMID 21911714.
- “Phase 3 Trial of Nexavar in Patients With Non-Responsive Thyroid Cancer”[dead link]
- [1]
- “Chemotherapy-Induced Nausea and Vomiting Treatment & Management”. Medscape Reference. WebMD. 3 July 2012. Retrieved 26 December 2013.
- Hagopian, Benjamin (August 2010). “Unusually Severe Bullous Skin Reaction to Sorafenib: A Case Report”. Journal of Medical Cases 1 (1): 1–3. doi:10.4021/jmc112e. Retrieved 11 February 2014.
- Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, Van Belle P, Elder DE, Wang Y, Nathanson KL, Herlyn M (January 2009). “CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations”. Oncogene 28 (1): 85–94. doi:10.1038/onc.2008.362. PMC 2898184. PMID 18794803.
- Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M (October 2008). “Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling”. Mol. Cancer Ther. 7 (10): 3129–40. doi:10.1158/1535-7163.MCT-08-0013. PMID 18852116.
- Zhang Y (Jan 2014). “Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways.”. J Mol Med Rep 9 (1): 83–90. PMID 24213221.
- Gauthier A (Feb 2013). “Role of sorafenib in the treatment of advanced hepatocellular carcinoma: An update..”. Hepatol Res 43 (2): 147–154. doi:10.1111/j.1872-034x.2012.01113.x. PMID 23145926.
- FDA Approval letter for use of sorafenib in advanced renal cancer
- European Commission – Enterprise and industry. Nexavar. Retrieved April 24, 2007.
- “Nexavar® (Sorafenib) Approved for Hepatocellular Carcinoma in Europe” (Press release). Bayer HealthCare Pharmaceuticals and Onyx Pharmaceuticals. October 30, 2007. Retrieved November 10, 2012.
- FDA Approval letter for use of sorafenib in inoperable hepatocellular carcinoma
- “Liver drug ‘too expensive‘“. BBC News. November 19, 2009. Retrieved November 10, 2012.
- http://www.ipindia.nic.in/ipoNew/compulsory_License_12032012.pdf
- “Seven days: 9–15 March 2012″. Nature 483 (7389): 250–1. 2012. doi:10.1038/483250a.
- “India Patents (Amendment) Act, 2005″. WIPO. Retrieved 16 January 2013.
- [2]
- “Addition of Sorafenib May Be Detrimental in Some Lung Cancer Patients”
- ClinicalTrials.gov NCT00329719 Sorafenib and Temsirolimus in Treating Patients With Recurrent Glioblastoma
- “Activity of sorafenib against desmoid tumor/deep fibromatosis”
- “‘We didn’t make this medicine for Indians… we made it for western patients who can afford it‘“. Daily Mail Reporter. 24 Jan 2014.
External links
| Reference |
| 1 |
* |
D. BANKSTON ET AL.: “A Scalable Synthesis of BAY 43-9006: A Potent Raf Kinase Inhibitor for the Treatment of Cancer” ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 6, no. 6, 2002, pages 777-781, XP002523918 cited in the application |
| 2 |
* |
PAN W ET AL: “Pyrimido-oxazepine as a versatile template for the development of inhibitors of specific kinases” BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, ELSEVIER SCIENCE, GB, vol. 15, no. 24, 15 December 2005 (2005-12-15), pages 5474-5477, XP025314229 ISSN: 0960-894X [retrieved on 2005-12-15] |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2011036647A1 |
Sep 24, 2010 |
Mar 31, 2011 |
Ranbaxy Laboratories Limited |
Process for the preparation of sorafenib tosylate |
| WO2011036648A1 |
Sep 24, 2010 |
Mar 31, 2011 |
Ranbaxy Laboratories Limited |
Polymorphs of sorafenib acid addition salts |
| WO2011058522A1 |
Nov 12, 2010 |
May 19, 2011 |
Ranbaxy Laboratories Limited |
Sorafenib ethylsulfonate salt, process for preparation and use |
| WO2011092663A2 |
Jan 28, 2011 |
Aug 4, 2011 |
Ranbaxy Laboratories Limited |
4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-n2-methylpyridine-2-carboxamide dimethyl sulphoxide solvate |
| WO2011113367A1 * |
Mar 17, 2011 |
Sep 22, 2011 |
Suzhou Zelgen Biopharmaceutical Co., Ltd. |
Method and process for preparation and production of deuterated ω-diphenylurea |
| US8552197 |
Nov 12, 2010 |
Oct 8, 2013 |
Ranbaxy Laboratories Limited |
Sorafenib ethylsulfonate salt, process for preparation and use |
| US8604208 |
Sep 24, 2010 |
Dec 10, 2013 |
Ranbaxy Laboratories Limited |
Polymorphs of sorafenib acid addition salts |
| US8609854 |
Sep 24, 2010 |
Dec 17, 2013 |
Ranbaxy Laboratories Limited |
Process for the preparation of sorafenib tosylate |
| US8618305 |
Jan 28, 2011 |
Dec 31, 2013 |
Ranbaxy Laboratories Limited |
Sorafenib dimethyl sulphoxide solvate |
| US8669369 |
Mar 17, 2011 |
Mar 11, 2014 |
Suzhou Zelgen Biopharmaceutical Co., Ltd. |
Method and process for preparation and production of deuterated Ω-diphenylurea |


































































 Originally posted on
Originally posted on 



































 Originally posted on
Originally posted on 



































 Originally posted on
Originally posted on