Literature References: Prepn: GB858187 (1961 to Hoffmann-La Roche); Villani et al.,J. Med. Pharm. Chem.5, 373 (1962); Winthrop et al.,J. Org. Chem.27, 230 (1962). Pharmacology: C. D. Barnes, W. L. Adams, Neuropharmacology17, 445 (1978); N. N. Share, ibid. 721; and toxicology: J. Metysova et al.,Arch. Int. Pharmacodyn. Ther.144, 481 (1963). Metabolism: G. Belvedere et al.,Biomed. Mass Spectrom.1, 329 (1974); H. B. Hucker et al.,Drug Metab. Dispos.6, 184 (1978). Bioavailability: eidem,J. Clin. Pharmacol.17, 719 (1977). Clinical studies: J. V. Basmajian, Arch. Phys. Med. Rehabil.5, 58 (1978); B. R. Brown, J. Womble, J. Am. Med. Assoc.240, 1151 (1978). Comprehensive description: M. L. Cotton, G. R. B. Down, Anal. Profiles Drug Subs.17, 41-72 (1988).
Properties: bp1 175-180°. uv max: 224, 289 nm (log e 4.57, 4.02), (Villani et al.)
Boiling point: bp1 175-180°
Absorption maximum: uv max: 224, 289 nm (log e 4.57, 4.02), (Villani et al.)
Percent Composition: C 77.03%, H 7.11%, N 4.49%, Cl 11.37%
Literature References: Use as muscle relaxant: N. N. Share, FR2100873 (1972 to Frosst), C.A.78, 47801n (1973).
Properties: Crystals from isopropanol, mp 216-218°. Soly in water: >20 g/100 ml. Freely sol in water, methanol, ethanol; sparingly sol in isopropanol; slightly sol in chloroform, methylene chloride. Practically insol in hydrocarbons. uv max: 226, 295 nm (e 52300, 12000). LD50 in mice (mg/kg): 35 i.v., 250 orally (Metysova).
Melting point: mp 216-218°
Absorption maximum: uv max: 226, 295 nm (e 52300, 12000)
Cyclobenzaprine, a centrally-acting muscle relaxant, was first synthesized in 196111 and has been available for human use since 1977.10 It was initially studied for use as antidepressant given its structural similarity to tricyclic antidepressants – it differs from Amitriptyline by only a single double bond.11,10 Since its approval, it has remained relatively popular as an adjunctive, short-term treatment for acute skeletal muscle spasms secondary to musculoskeletal injury.
Cyclobenzaprine (sold under the brand name Flexeril, among others) is a medication used for muscle spasms from musculoskeletal conditions of sudden onset.[6] It is not useful in cerebral palsy.[6] It is taken by mouth.[6] Use is not recommended for more than a few weeks.[6]
Common side effects include headache, feeling tired, dizziness, and dry mouth.[6] Serious side effects may include an irregular heartbeat.[6] There is no evidence of harm in pregnancy, but it has not been well studied in this population.[6] It should not be used with an MAO inhibitor.[6] How it works is unclear.[6]
Cyclobenzaprine was approved for medical use in the United States in 1977.[6] It is available as a generic medication.[6] In 2019, it was the 45th most commonly prescribed medication in the United States, with more than 15 million prescriptions.[7][8] It was not available in the United Kingdom as of 2012.[9]
Synthesis Reference
Villani, F.J.; US. Patent 3,409,640; November 5,1968; assigned to Schering Corporation.
Paper
By: Gowda, Narendra B.; Rao, Gopal Krishna; Ramakrishna, Ramesha A.
A simple and convenient protocol for deoxygenation of aliphatic and aromatic N-oxides to the corresponding amines in good to excellent yield using sodium borohydride–Raney nickel in water is reported. Other functional moieties such as alkenes, halides, ethers, and amides are unaffected under the present reaction condition.
Graphical abstract
Cyclobenzaprine N-oxide, CAS RN: 6682-26-4
Dissolve (1 mmol) of cyclobenzaprine N-oxide in 2.5 mL of water at 60 °C. 2. Add Raney nickel (0.10 g, W6 grade) to the solution. 3. Stir the reaction mixture for 10 minutes. 4. Add (2 mmol) of sodium borohydride slowly in portions over 15-20 minutes to the reaction mixture. 5. Stir the reaction mixture at the same temperature for 2.5 hours (the completion of the reaction as monitored by TLC). 6. Once the reaction is completed, add chloroform (50 mL) to the reaction mixture. 7. Filter the resulted mixture to remove Raney nickel. 8. Dry the chloroform layer over anhydrous magnesium sulfate. 9. Filter the reaction mixture. 10. Evaporate the solvent under vacuum. 11. Purify the obtained residue through short path flash chromatography with silica gel and chloroform.
Cyclobenzaprine hydrochloride, chemically known as 5-(3-dimethylaminopropylidene)- dibenzo (a,e) cycloheptatriene hydrochloride (Formula I),
Formula I is a commonly prescribed tricyclic amine having muscle relaxant pharmaceutical activity. After sustaining an injury, muscle spasms may occur to stabilize the affected body part and prevent further damage. Cyclobenzaprine hydrochloride is used to treat such muscle spasm associated with acute, painful musculoskeletal conditions.
Few multistep processes for the preparation of this tricyclic amine are already available in the literature which involves isolation and purification of intermediate compounds. The conventional route of synthesis as reported in US3454643, ES8201950 includes preparation of Grignard reagent (GR) of 3-dimethylaminopropyl chloride in a first step, reacting with 5-dibenzosuberenone (Formulall) in a second step. The reaction mass was extracted with benzene, solid obtained was recrystallized from alcohol to produce 5- hydroxy intermediate (Formula III) and further dehydrated in third step using acetyl chloride or acetic anhydride in presence of chloroform as a solvent medium followed by purging HC1 gas to produce hydrochloride salt (Formula I). CH,
CI-(CH2)3 NS
CH,
Dimeth laminopropyl chloide
Di methy lam i nopropy I 5-dibenzosubrenone – y roxy compoun magnesium chloide
(Formula II) (Formula III)
Cyclobenzaprine base Cyclobenzaprine hydrochloride
(Formula IV) (Formula I)
The multistep synthesis is cumbersome and use of hazardous solvents and reagents like chloroform, benzene and acetyl chloride etc are not recommended for the preparation of pharmaceutical substances.
J. Org. Chem. Vol. 27, 230-240 (1961) also portrayed similar procedure for the synthesis of cyclobenzaprine hydrochloride, wherein 5-hydroxy compound of formula III was isolated and recrystallized before dehydration reaction.
Synthetic Comm. 11 (3), 241-246 (1981) described a process which involves isolation and purification of the intermediate at magnesium -complex stage. Hydrolysis of the isolated complex afforded desired tricyclic amine. GB858186 and GB858187 jointly described a process which comprises preparation of 5- hydroxy compound (Formula III) and subsequent conversion of the same to cyclobenzaprine hydrochloride. However the overall yield reported is significantly low.
In a different approach, a high temperature dehydrogenation of amitriptyline base resulting in formation of cyclobenzaprine hydrochloride is reported in Indian patent application 387/CHE/2005.
. EXAMPLE:
In a reaction vessel, THF (1 10ml), magnesium turnings 20gm (0.823mole) were charged and the mixture was warmed to 45-55°C for 20 min. A solution of l OOgm (0.823mole) of 3-dimethylaminopropyl chloride prepared in 1 10ml THF was added dropwise to the reaction mixture by controlling the reflux generated due to reaction initiation and maintained for 2hrs. The formed Grignard reagent was then cooled to 0-5°C and a solution of lOOgm (0.485mole) 5-dibenzosuberenone prepared in 220ml THF was charged to the reaction mass at temperature below 10°C. The reaction mass was stirred for 45 min at temperature 10-15°C. The absence of 5-dibenzosuberenone was checked by TLC and 770ml of 20% aq. HC1 was charged to the reaction mass at a temperature below 10°C. The reaction mass was then heated to 70-80°C for 3 hrs. The acidic mass was neutralized by using aqueous Na2C03 solution and extracted with 900ml methylene dichloride. The solvent was removed completely under reduced pressure and oil thus formed was dissolved in 450ml IPA and acidified with 240 ml of 20% IPA .HC1 solution and stirred for 2 hrs at 0-5°C for complete precipitation. The precipitate is filtered, recrystallized from IPA (800 ml) and dried to obtain 1 18 gm (78%) white crystalline cyclobenzaprine hydrochloride with purity 99.93% by HPLC.
Cyclobenzaprine, N,N-dimethyl-3-(dibenzo[a,d]cyclohepten-5-ylidene) propylamine (15.3.9), is synthesized by reacting 5H-dibenzo[a,d]cyclohepten-5-one with 3-dimethylaminopropylmagnesium chloride and subsequent dehydration of the resulting carbinol (15.3.8) in acidic conditions into cyclobenzaprine (15.3.9) [30–32].
Cyclobenzaprine is structurally similar to tricyclic antidepressants. It acts at the brain stem level. It is used as an adjuvant agent for relieving muscle spasms associated with severe diseased conditions of the muscle. A synonym of this drug is flexeril.
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Cyclobenzaprine is used, in conjunction with physical therapy, to treat muscle spasms that occur because of acute musculoskeletal conditions.[10] After sustaining an injury, muscle spasms to stabilize the affected body part occur, which may increase pain to prevent further damage. Cyclobenzaprine is used to treat such muscle spasms associated with acute, painful musculoskeletal conditions.[11] It decreases pain in the first two weeks,[12][13] peaking in the first few days, but has no proven benefit after two weeks.[12][14] Since no benefit is proven beyond that, therapy should not be continued long-term.[11] It is the best-studied muscle relaxer.[12] It is not useful for spasticity due to neurologic conditions such as cerebral palsy.[11][15]
A 2004 review found benefit for fibromyalgia symptoms, with a reported number needed to treat of 4.8 (meaning that 1 person out of every 4.8 benefits from treatment) for pain reduction, but no change in fatigue or tender points.[16] A 2009 Cochrane review found insufficient evidence to justify its use in myofascial pain syndrome.[17] It may also be used along with other treatments for tetanus.[18]
Agitation is a common side effect observed, especially in the elderly. Some experts[who?] believe that cyclobenzaprine should be avoided in elderly patients because it can cause confusion, delirium, and cognitive impairment.[20][21] In general, the National Committee for Quality Assurance recommends avoiding the use of cyclobenzaprine in the elderly because of the potential for more severe side effects.[22]
Monoamine oxidase inhibitors taken within two weeks of cyclobenzaprine may result in serious, life-threatening side effects.[11]
Cyclobenzaprine may affect the medications used in surgical sedation and some surgeons request that patients temporarily discontinue its use prior to surgery.[26]
Pharmacology
Cyclobenzaprine is a centrally acting muscle relaxant.[27] Cyclobenzaprine is a 5-HT2 receptor antagonist; it relieves muscle spasm through action on the central nervous system at the brain stem, rather than targeting the peripheral nervous system or muscles themselves.[28]
Values are Ki (nM), unless otherwise noted. The smaller the value, the more strongly the drug binds to the site.
Pharmacokinetics
Cyclobenzaprine has an oral bioavailability of about 55% and approximately 93% is bound to proteins in plasma. The half-life of the drug is 18 hours and it has a plasma clearance of 0.7 litres per minute.[27][30][31]
Comparison to other medications
Cyclobenzaprine has been found to be not inferior to tizanidine, orphenadrine, and carisoprodol in the treatment of acute lower back pain, although none have been proven to be effective for long-term use (beyond two weeks of treatment). No differences in pain or spasm scores were noted among these agents, nor when compared to benzodiazepines.[32] However, nonbenzodiazepine (including cyclobenzaprine) treatment was found to have a lower risk of medication abuse and continuation of use against medical advice.[medical citation needed] Side effects such as sedation and ataxia are also less pronounced with nonbenzodiazepine antispasmodics.[medical citation needed]
In a study on the treatment of musculoskeletal pain treatment with cyclobenzaprine alone or in combination with ibuprofen, no significant differences in pain scores were noted among the three treatment groups. Peak benefit was found to occur on day seven of the treatment for all groups.[33]
Formulations
Cyclobenzaprine 10mg tablets
By mouth, cyclobenzaprine is marketed as Apo-Cyclobenzaprin, Fexmid, Flexeril and Novo-Cycloprine. It is available in generic form. A once-a-day, extended-release formulation, Amrix, is available.[34] Cyclobenzaprine is also used by compounding pharmacies in topical creams.[citation needed]
^ Yang YW, Macdonald JB, Nelson SA, Sekulic A (December 2017). “Treatment of vismodegib-associated muscle cramps with cyclobenzaprine: A retrospective review”. Journal of the American Academy of Dermatology. 77 (6): 1170–1172. doi:10.1016/j.jaad.2016.12.017. PMID29132849. S2CID8265576.
^ “Long-term Use of Cyclobenzaprine for Pain: A Review of the Clinical Effectiveness”. CADTH Rapid Response Reports. Ottawa, Ontario: Canadian Agency for Drugs and Technologies in Health. 23 February 2015. PMID25763449.
^ Keegan MT, Brown DR, Rabinstein AA (December 2006). “Serotonin syndrome from the interaction of cyclobenzaprine with other serotoninergic drugs”. Anesthesia and Analgesia. 103 (6): 1466–8. doi:10.1213/01.ane.0000247699.81580.eb. PMID17122225.
^ Kobayashi H, Hasegawa Y, Ono H (September 1996). “Cyclobenzaprine, a centrally acting muscle relaxant, acts on descending serotonergic systems”. European Journal of Pharmacology. 311 (1): 29–35. doi:10.1016/0014-2999(96)00402-5. PMID8884233.
^ Winchell GA, King JD, Chavez-Eng CM, Constanzer ML, Korn SH (January 2002). “Cyclobenzaprine pharmacokinetics, including the effects of age, gender, and hepatic insufficiency”. Journal of Clinical Pharmacology. 42 (1): 61–9. doi:10.1177/0091270002042001007. PMID11808825. S2CID7749001.
Radotinib dihydrochloride was approved by Korea Food and Drug Administration (KFDA) on January 5, 2012. It was developed and marketed as Supect® by IL-Yang in KR.
Radotinib dihydrochloride is a second-generation tyrosine kinase inhibitor of Bcr-Abl fusion protein and the platelet-derived growth factor receptor (PDGFR). It is indicated for the second-line treatment of patients with Philadelphia chromosome-positive (Ph+) CML that is refractory to Imatinib mesilate.
Supect® is available as capsule for oral use, containing 100 mg or 200 mg of free Radotinib. The recommended dose is 400 mg twice daily.
Radotinib is being developed by Ilyang Pharmaceutical Co., Ltd of South Korea[2] and is co-marketed by Daewoong Pharmaceutical Co. Ltd, in South Korea.[3] Radotinib completed a multi-national Phase II clinical trial study in 2012[4] and in August 2011, Ilyang initiated a Phase III, multinational, multi-center, open-label, randomized study for first-line indication.[5] Its mechanism of action involves inhibition of the Bcr-Abl tyrosine kinase and of platelet-derived growth factor receptor (PDGFR).[6]
In January 2012, radotinib hydrochloride (marketed as Supect ®) obtained its approval from the KFDA (Korea Food and Drug Administration) for the treatment of patients with Philadelphia chromosomepositive chronic myeloid leukemia (CML) who have become resistant to existing drugs such as Gleevec, Tasigna and Sprycel. Originally developed by IL-YANG pharmaceuticals of South Korea as an orally second-generation tyrosine kinase inhibitor, the drug inhibits both Bcr-Abl fusion protein and the platelet-derived growth factor receptor (PDGFR).
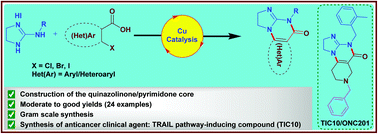
Chemical Synthesis
Because of the structural similarity of radotinib to that of nilotinib (Tasigna ®), the process-scale synthetic route (which is depicted in the scheme) is capable of furnishing both drugs.Claisen condensation of commerical 2-acetylpyrazine (142) with N,N-dimethylformamide dimethylacetal gave rise to the enamino ketone 143 in 81% yield. Under basic conditions, vinylogous amide 143 was coupled with commercial guanidine nitrate 144187 to produce aminopyridine 145. Subsequent condensation with commercial aniline (146) by means of potassium t-butoxide in THF constructed radotinib 147 in 85% yield as the free base, and this material could be converted to the radotinib dihydrochloride (XXII) upon exposure to concentrated hydrochloric acid in chilled acetone.
The compound represented by Formula 1 was disclosed in Korea Patent Registration
No. 10-0674813. A preferred compound according to Formula 1 includes 4-methyl-N- [3-(4-methylimidazole- 1 -yl)-5-trifluoromethyl-phenyl] -3-(4-pyrazine-2-yl -pyrimidine-2-yl amino)benzamide. It has been known that the compound represented by Formula 1 can inhibit at least one kind of tyrosine kinase, for example, c-Abl, Bcr- AbI, and receptor tyrosine kinases (PDGF-R, Flt3, VEGF-R, EGF-R and c-Kit). Accordingly, the compound represented by Formula 1 may be used for treatment of various kinds of cancers in a warm blooded animal, such as lung cancer, stomach cancer, colon cancer, pancreatic cancer, liver cancer, prostate cancer, breast cancer, chronic or acute leukemia, hematological malignancy, brain tumor, bladder cancer, rectal cancer, uterine cervical cancer, lymphoma, etc.
[7] According to a conventional method, the compound represented by Formula 1 is synthesized through hydrolysis of ethyl ester into carboxylic acid and then a reaction with aniline, and herein, diethyl cyano phosphonate is used as a coupling agent (see Reaction Scheme 1).
[8] [Reaction Scheme 1]
NsOIf
( 2 ) { s :
Diethyl cyano phosphate
( 1 )
[10] The above method requires a process of hydrolyzing ethyl ester (2) into carboxylic acid (3). In order to obtain the compound represented by Formula 3 as shown in Reaction Scheme 1, a preparation process and a purifying process require a long time. Also, in the condensation reaction, there have been problems such as high production cost due to a low yield (30 to 40%) of the compound represented by Formula 1. Especially, it is very difficult to treat carboxylic acid (3) after purification and reaction, due to its very low solubility in general organic solvent. Also, diethyl cyano phosphonate used for the condensation reaction is an expensive reagent, and an environmentally harmful and very toxic material, which has LD50 values of 25mg/Kg and 4mg/Kg in mice and rabbits (that is, rodents), respectively. Therefore, there is a requirement for an alternative method of conveniently, consistently, efficiently and rapidly preparing a high-purity compound (represented by Formula 1) with low production cost in high yield, which is not harmful for humans and the environment.
Example 2
[69] Synthesis of 4-methyl-N-[3-(4-methylimidazole-l-yl)-5-trifluoromethyl-phenyl] –
[72] A pale yellow solid final compound (18.7g, yield 85%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4-methyl -3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)-benzoic acid ethyl ester in a similar manner as described in Method A of Example 1, except that 4-methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino) -benzoic acid ethyl ester (15.3g, 45.60mmol) was used, instead of 4-methyl-3-(4-thiazole-2-yl-pyrimidine-2-yl amino)benzoic acid ethyl ester.
[76] A pale yellow solid final compound (18.3g, yield 83%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4-methyl -3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)-benzoic acid methyl ester in a similar manner as described in Method A of Example 1, except that 4-methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino) -benzoic acid methyl ester (14.7g, 45.60mmol) was used, instead of 4-methyl-3-(4-thiazole-2-yl-pyrimidine-2-yl amino)benzoic acid ethyl ester.
[77]
[78] Method C
[79] A pale yellow solid final compound (17.2g, yield 78%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4- methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)benzoic acid methyl ester (14.7g, 45.60mmol) in a similar manner as described in Method A of Example 1, except that sodium tert-butoxide was used, instead of potassium tert-butoxide.
[80]
[81] Method D
[82] A pale yellow solid final compound (16. Ig, yield 73%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4- methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)benzoic acid phenyl ester in a similar manner as described in Method A of Example 1, except that 4-methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino) -benzoic acid phenyl ester (17.5g, 45.60mmol) was used, instead of 4-methyl-3-(4-thiazole-2-yl-pyrimidine-2-yl amino)benzoic acid ethyl ester.
Radotinib hydrochloride (Supect) In January 2012, radotinib hydrochloride (marketed as Supect) obtained its approval from the KFDA (Korea Food and Drug Administration) for the treatment of patients with Philadelphia chromosome-positive chronic myeloid leukemia (CML) who have become resistant to existing drugs such as Gleevec, Tasigna and Sprycel.181 Originally developed by IL-YANG pharmaceuticals of South Korea as an oral second-generation tyrosine kinase inhibitor, the drug inhibits both Bcr-Abl fusion protein and the platelet-derived growth factor receptor (PDGFR).182 Because of the structural similarity of radotinib to that of nilotinib (Tasigna), the processscale synthetic route (which is depicted in Scheme 27) is capable of furnishing both drugs.183–185 Claisen condensation of commerical 2-acetylpyrazine (142) with N,N-dimethylformamide dimethylacetal gave rise to the enamino ketone 143 in 81% yield.186 Under basic conditions, vinylogous amide 143 was coupled with commercial guanidine nitrate 144187 to produce aminopyridine 145. 184 Subsequent condensation with commercial aniline (146) by means of potassium t-butoxide in THF constructed radotinib 147 in 85% yield as the free base, and this material could be converted to the radotinib dihydrochloride (XXII) upon exposure to concentrated hydrochloric acid in chilled acetone.185
^ Joanne Bronson; Amelia Black; T. G. Murali Dhar; Bruce A. Ellsworth; J. Robert Merritt (2013). “To Market, To Market – 2012”. Radotinib (Anticancer). Annual Reports in Medicinal Chemistry. Vol. 48. pp. 523–524. doi:10.1016/b978-0-12-417150-3.00028-4. ISBN9780124171503.
US9132126B22011-04-192015-09-15Il-Yang Pharm. Co., Ltd.Phenyl-isoxazole derivatives and preparation process thereof
KR20180032784A *2016-09-232018-04-02재단법인 대구경북첨단의료산업진흥재단Novel imidazolyl pyrimidine derivatives, preparation method thereof, and pharmaceutical composition for use in preventing or treating cancer containing the same as an active ingredient
Family To Family Citations
KR101956586B1 *2012-03-272019-03-11일양약품주식회사Pharmaceutical composition and preparation method thereof
S-217622, a therapeutic drug for COVID-19, is a 3CL protease inhibitor created through joint research between Hokkaido University and Shionogi. SARS-CoV-2 has an enzyme called 3CL protease, which is essential for the replication of the virus. S-217622 suppresses the replication of SARS-CoV-2 by selectively inhibiting 3CL protease. Shionogi has already been submitting the non-clinical, manufacturing/CMC data, and clinical trial data obtained so far to the PMDA. Currently the Phase 3 part of a Phase 2/3 clinical trial in patients with mild/moderate symptoms and the Phase 2b/3 part in patients with asymptomatic/only mild symptoms are in progress.
SYN
Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in millions of deaths and threatens public health and safety. Despite the rapid global spread of COVID-19 vaccines, effective oral antiviral drugs are urgently needed. Here, we describe the discovery of S-217622, the first oral non-covalent, non-peptidic SARS-CoV-2 3CL protease inhibitor clinical candidate. S-217622 was discovered via virtual screening followed by biological screening of an in-house compound library, and optimization of the hit compound using a structure-based drug-design strategy. S-217622 exhibited antiviral activity in vitro against current outbreaking SARS-CoV-2 variants and showed favorable pharmacokinetic profiles in vivo for once-daily oral dosing. Furthermore, S-217622 dose-dependently inhibited intrapulmonary replication of SARS-CoV-2 in mice, indicating that this novel non-covalent inhibitor could be a potential oral agent for treating COVID-19.
Chemistry
The synthetic scheme for compound 1 is described in Scheme 1. Starting from the pyrazole derivative 4, cyclization with Ethyl isocyanatoacetate and CDI was conducted, giving 5 in 90% yield. Then, an alkylation with 5-bromomethyl-1,2,3-trifluorobenzene followed by introduction of a 4-difluoromethoxy-2-methylaniline unit, to give 7 (40% in 2 steps). The ester group in 7 was hydrolyzed and then amidated with methylamine, yielding 1 (58% in 2 steps). Compound 2 was synthesized similarly as shown in Scheme 2.
S-217622 (3) was synthesized as described in Scheme 3. Starting from known compound 9,21 an alkylation with 1-(bromomethyl)-2,4,5-trifluorobenzene gave 10 in 93% yield. Then, the 3-tert-Bu group was removed and the triazole unit was introduced, and the substitution of the SEt moiety with the indazole unit finally gave S-217622 (3).
21 Kai, H.; Kameyama, T.; Horiguchi, T.; Asahi, K.; Endoh, T.; Fujii, Y.; Shintani, T.; Nakamura, K.; Matsumoto, S.; Hasegawa, T.; Oohara, M.; Tada, Y.; Maki, T.; Iida, A. Preparation of triazine derivatives and pharmaceutical compound that contains same and exhibits analgesic activity. WO 2012020749 A1, Feb 16, 2012
Scheme 1.
Reagents and Conditions: (a) ethyl isocyanato-acetate, DBU, CDI, DMA, –10 °C to rt, 90%; (b) 5-bromomethyl-1,2,3-trifluorobenzene, N,N-diisopropylethylamine, DMA, 60 °C; (c) 4-difluoromethoxy-2-methylaniline, tert-butanol, 100 °C, 40% in 2 steps; (d) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 58% in 2 steps.
Scheme 2.
Reagents and Conditions: (a) 6-chloro-2-methyl-2H-indazol-5-amine, tert-amyl alcohol, 100 °C, 44% in 2 steps from 5; (b) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 29% in 2 steps.
Scheme 3.
Reagents and Conditions: (a) 1-(bromomethyl)-2,4,5-trifluorobenzene, K2CO3, MeCN, 80 °C, 93%; (b) TFA, rt, 97%; (c) 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride, K2CO3, DMF, 60 °C, 45%; (d) 6-chloro-2-methyl-2H-indazol-5-amine, LHMDS, THF, 0 °C to rt., 25%.
To a solution of 12 (300 mg, 0.727 mmol) and 6-chloro-2-methyl-2H-indazol-5-amine (172 mg, 0.946 mmol) in THF (6 mL) was added LHMDS (1M in THF; 1.46 mL, 1.46 mmol) dropwisely at 0 °C. The reaction mixture was stirred at 0 °C for 2.5 h and then at rt for 40 min. The reaction was quenched with aqueous NH4Cl solution, and the aqueous layer was extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3/MeOH gradient, 0-20% MeOH). The solid was recrystallized from acetone/H2O to afford 3 (S-217622) (95.3 mg, 25%) as a pale brown solid. 1H NMR (400 MHz, DMSO-d6, DCl in D2O) δ 3.90 (3H, s), 4.15 (3H, s), 5.04 (2H, s), 5.26 (2H, s), 7.44 (1H, m), 7.52-7.65 (2H, m), 7.73 (1H, s), 8.40 (1H, s), 9.31 (1H, s). 13C NMR (100 MHz, DMSO-d6, DCl in D2O) δ 37.34, 38.04, 40.06, 40.29, 106.16 (dd, J = 28.2, 21.6 Hz), 116.46-116.70, 116.70, 120.54-120.76, 120.76, 125.93, 129.10, 132.35, 143.84, 145.98, 146.38 (ddd, J = 241.4, 12.5, 3.7 Hz), 146.60, 148.52 (td, J = 247.7, 13.6 Hz), 150.43, 150.50, 155.22 (ddd, J = 244.3, 10.3, 2.2 Hz), 155.58. HRMS-ESI (m/z): [M + H]+ calcd for [C22H18 F3ClN9O2]+ 532.1219; found 532.1221.
Preparation of Compound 3 (S-217622) fumaric acid co-crystal
A mixture of 3 (S-217622) (1.17 g, 2.2 mmol) and fumaric acid (278 mg, 2.4 mmol) in EtOAc (5.9 mL) was stirred at room temperature for 45 min. The suspension was filtrated to afford 3 (S-217622) fumaric acid co-crystal (1.37 g, 95 %) as a white solid. 1H NMR (400 MHz, pyridine-d5) δ 3.64 (s, 3H), 3.99 (s, 3H), 5.56 (s, 2H), 5.61 (s, 2H), 7.16-7.25 (m, 2H), 7.44 (s, 2H), 7.81 (s, 1H), 7.89 (s, 1H), 7.89-7.97 (m, 1H), 8.32 (s, 1H).
Notes
SHIONOGI has applied for a patent covering 1, 2, and 3 (S-217622). Y.U., S.U., K.N., H.N., Y.Y., S.Y., Y.M., Y.T., K.K., T.S., K.K., A.N., S.K., T.S., S.T., K.U., T.M., S.A., A.S., T.S., T.K., and Y.T. are employees of SHIONOGI & Co., Ltd. S.U., K.N., H.N., Y.M., Y.T., K.K., T.S., K.K., S.K., TS, S.T., K.U., T.S., and T.K. are shareholders in SHIONOGI & Co., Ltd. M.S., Y.O., and H.S. are financially supported by the joint research fund from SHIONOGI & Co., Ltd.
Oral antiviral medications, in addition to vaccines, are expected to play an important role in treating coronavirus disease 2019 (COVID-19), which is caused by infection with the severe acute respiratory disease coronavirus-2 (SARS-CoV-2).
These drugs must have significant antiviral activity, as well as target specificity, oral bioavailability, and metabolic stability. Although several antiviral compounds have been reported as possible SARS-CoV-2 inhibitors in vitro, only a few of these drugs have been shown to be effective in vivo.
Ensitrelvir, a novel SARS-CoV-2 antiviral
Ensitrelvir (code name S-217622, brand name Xocova), is a new inhibitor of the SARS-CoV-2 major protease (Mpro), also known as 3C-like protease, has been shown to reduce the viral load and help alleviate the severity of SARS-CoV-2 in infected hamsters. In cells, low nanomolar to sub-micromolar doses of S-217622 suppress viral growth. In hamsters, oral treatment of S-217622 showed excellent pharmacokinetic qualities and hastened recovery from acute SARS-CoV-2 infection.
S-217622 also demonstrated antiviral effectiveness against SARS-CoV-2 variants of concern (VOCs), such as the highly pathogenic Delta variant and the newly discovered Omicron variant. Overall, these findings show that S-217622, which is an antiviral drug that is currently being tested in Phase II/III clinical trials, has impressive antiviral efficiency and effectiveness against SARS-CoV-2 and could be a viable oral treatment option for COVID-19.
History
It has reached Phase III clinical trials.[3] The Japanese government is reportedly considering allowing Shionogi permission to apply for approval for medical use before the final steps of trials are completed, potentially speeding up the release for sale. This conditional early approval system has previously been used in Japan to accelerate the progression to market of other antiviral drugs targeting COVID-19, including remdesivir and molnupiravir.[6] In a study of 428 patients, viral load was reduced, but symptoms were not significantly reduced. [7]
It became the first Japanese domestic pill to treat COVID-19, third to be regulatorally approved in Japan; in February 2022.[8]
Percent Composition: C 63.95%, H 6.90%, F 4.82%, O 24.34%
Literature References: Prepn: Bernstein et al.,J. Am. Chem. Soc.78, 5693 (1956); 81, 1689 (1959); Thoma et al.,ibid.79, 4818 (1957); Bernstein et al., Allen et al.,US2789118; US3021347 (1957, 1962, both to Am. Cyanamid). Comprehensive description: K. Florey, Anal. Profiles Drug Subs.1, 367-396, 423-442 (1972); D. H. Sieh, ibid.11, 593-614, 651-661 (1982).
Properties: Crystals, mp 269-271°. mp also reported as 260-262.5°. [a]D25 +75° (acetone). uv max: 238 nm (e 15800).
Melting point: mp 269-271°; mp also reported as 260-262.5°
Optical Rotation: [a]D25 +75° (acetone)
Absorption maximum: uv max: 238 nm (e 15800)
………………………………
Derivative Type: 16,21-Diacetate
CAS Registry Number: 67-78-7
CAS Name: (11b,16a)-16,21-Bis(acetyloxy)-9-fluoro-11,17-dihydroxypregna-1,4-diene-3,20-dione
Percent Composition: C 66.34%, H 7.19%, F 4.37%, O 22.09%
Literature References: Prepd by stirring a suspension of triamcinolone in acetone in the presence of a trace of perchloric acid: Fried et al.,J. Am. Chem. Soc.80, 2338 (1958); Bernstein et al.,ibid.81, 1689 (1959); Bernstein, Allen, US2990401 (1961 to Am. Cyanamid). Alternate synthesis using 2,3-dibromo-5,6-dicyanoquinone: Hydorn, US3035050 (1962 to Olin Mathieson). Clinical trial in chronic asthma: I. L. Bernstein et al.,Chest81, 20 (1982). Comprehensive description: K. Florey, Anal. Profiles Drug Subs.1, 397-421 (1972); D. H. Sieh, ibid.11, 615-649 (1982).
Properties: Crystals, mp 292-294°. [a]D23 +109° (c = 0.75 in chloroform). uv max (abs alc.): 238 nm (e 14600). Sparingly sol in methanol, acetone, ethyl acetate.
Melting point: mp 292-294°
Optical Rotation: [a]D23 +109° (c = 0.75 in chloroform)
Absorption maximum: uv max (abs alc.): 238 nm (e 14600)
Percent Composition: C 67.40%, H 6.79%, F 3.05%, N 2.25%, O 20.52%
Literature References: Prepn: C. Cavazza et al.,DE2047218; eidem,US3749712 (1971, 1973 both to Sigma-Tau). Pharmacology: E. T. Ordonez, Arzneim.-Forsch.21, 248 (1971). Percutaneous absorption by rats and rabbits: W. H. Down et al.,Toxicol. Lett.1, 95 (1977). Clinical study: D. J. Tazelaar, J. Int. Med. Res.5, 338 (1977). HPLC analysis: S. Muck et al.,Boll. Chim. Farm.120, 240 (1981). For structure see Triamcinolone Acetonide.
Properties: Crystalline powder, mp 203-207°. [a]D20 +96 ±3° (c = 1 in ethanol). Sol in methanol, acetone, ethanol, dioxane, pyridine, DMF, chloroform. Insol in water.
Melting point: mp 203-207°
Optical Rotation: [a]D20 +96 ±3° (c = 1 in ethanol)
Percent Composition: C 67.65%, H 7.76%, F 3.57%, O 21.03%
Literature References: The hexacetonide ester of the potent glucocorticoid, triamcinolone, q.v. Prepn of syringeable suspension: Nash, Naeger, US3457348 (1969 to Am. Cyanamid). Anti-inflammatory activity in rabbits: I. M. Hunneyball, Agents Actions11, 490 (1981). Early clinical studies: Bilka, Minn. Med.50, 483 (1967); Layman, Peterson, ibid. 669. Clinical studies of intra-articular therapy in arthritis: R. C. Allen et al.,Arthritis Rheum.29, 997 (1986); M. Talke, Fortschr. Med.104, 742 (1986). Toxicity study: Tonelli, Steroids8, 857 (1966). Comprehensive description: V. Zbinovsky, G. P. Chrekian, Anal. Profiles Drug Subs.6, 579-595 (1977). For structure see Triamcinolone Acetonide.
Properties: Fine, white, needle-like crystals, mp 295-296° (dec), also reported as mp 271-272° (dec). uv max (ethanol): 238 nm (e 15500). [a]D25 +90±2° (c = 1.13% in chloroform). Soly in g/100 ml at 25°: chloroform and dimethylacetamide >5; ethyl acetate 0.77, methanol 0.59, diethyl carbonate 0.50, glycerin 0.42, propylene glycol 0.13; absolute alcohol 0.03; water 0.0004.
Triamcinolone was patented in 1956 and came into medical use in 1958.[8] It is available as a generic medication.[9] In 2019, it was the 107th most commonly prescribed medication in the United States, with more than 6 million prescriptions.[10][11]
Skin is the layer of usually soft, flexible outer tissue covering the body of a vertebrate animal, with three main functions: protection, regulation, and sensation. Skin diseases are the medical condition that affects the skin, hair, nails and related muscle and glands.
Skin disorders vary greatly in symptoms and severity. They can be temporary or permanent, and may be painless or painful. Some have situational causes, while others may be genetic. Some skin conditions are minor, and others can be lifethreatening.
There are many different types of skin disorders which include rashes, dermatoses or skin eruptions. Such rashes, dermatoses or skin eruptions include acute, inflammatory reactions of the skin caused by an allergic or irritant reaction, other forms of eczema, lichen simplex chronicus. Chronic nature includes seborrheic dermatitis, psoriasis, and atopic dermatitis or caused by infection, irritation or aggravation of another condition such as occurs with acne, other rashes, dermatoses or skin eruptions, inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses, contact dermatitis, impetigo, urticarial and scabies.
Typical symptoms of the skin disorders include but not limited to raised bumps that are red or white, a rash, which might be painful or itchy, scaly or rough skin peeling skin, ulcers, open sores or lesions, dry, cracked skin, discolored patches of skin, fleshy bumps, warts, or other skin growths, changes in mole color or size a loss of skin pigment, excessive flushing or the like.
Atopic dermatitis (AD), also known as eczema or atopic eczema, is a type of inflammation of the skin (dermatitis). Atopic dermatitis (AD) is common worldwide. People of all ages from newborns to adults and older live with this condition. Symptoms range from excessively dry, itchy skin to painful, itchy rashes that cause sleepless nights and interfere with everyday life.
Topical corticosteroids have been the mainstay of treatment for atopic dermatitis over the past years, further the cure for atopic dermatitis involves Lifestyle modification, balanced diet intake, self-care measures, phototherapy, wet wrap therapy, use of medications like tacrolimus, pimecrolimus, crisaborole, dupilumab, ciclosporin, methotrexate, interferon gamma- lb, mycophenolate mofetil, and azathioprine or the like.
Triamcinolone Acetonide is a synthetic corticosteroid. Chemically it is [Pregna-1, 4-diene-3, 20-dione, 9-fluoro-l l, 21 -dihydroxy- 16, 17-[(1 methylethylidene) bis-(oxy)]-, (HP, 16a)-] with the empirical formula C24H31FO6 and molecular weight 434.50. Triamcinolone Acetonide is represented by compound of structural formula I
Triamcinolone Acetonide topical cream and ointment with strengths 0.025%, 0.1% and 0.5% (containing 0.25 mg/gm, 1 mg/gm & 5 mg/gm Triamcinolone Acetonide respectively) were approved in USA prior to Jan 1, 1982 under the trade name “Triamcinolone Acetonide” and were indicated for the relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses.
The commercially available products or product known in the prior art produces side effects such as burning, itching, irritation, or dryness of skin at site of application, folliculitis, hypertrichosis, acneiform eruptions, hypopigmentation, perioral dermatitis, allergic contact dermatitis, maceration of the skin, secondary infection, skin atrophy, striae and miliaria.
Pediatric patients may demonstrate greater susceptibility to topical triamcinolone -induced HPA axis suppression and Cushing’s syndrome than mature patients because of a larger skin surface area to body weight ratio. Hypothalamic -pituitary-adrenal (HPA) axis suppression, Cushing’s syndrome and intracranial hypertension have been reported in children receiving topical triamcinolone. Manifestations of adrenal suppression in children include linear growth retardation, delayed weight gain, low plasma cortisol levels, and absence of response to ACTH stimulation. Manifestations of intracranial hypertension include bulging fontanelles, headaches, and bilateral papilledema. Chronic corticosteroid therapy may interfere with the growth and development of children.
Making low dose compositions can present technical and economic challenges that are not present for higher dose formulations.
Examples
The following table 1 shows cream formulation containing lOO.OOmcg per gm, 50.00mcg per gm and 25.00mcg per gm of Triamcinolone Acetonide
Table – 1: cream
Drug Strength IQOmcg/gm 50mcg/gm 25mcg/gm
lOO.OOmcg per gm and for lOOgm, it is lO.OOmg*
50.00mcg per gm and for lOOgm, it is 5.00mg*
25.00mcg per gm and for lOOgm, it is 2.50mg**
Manufacturing process:
a) Dispensing following excipients – isopropyl myristate, glyceryl monostearate and white soft paraffin in vessel I;
b) Dispensing the following excipients – polysorbate 40 and purified water in vessel II;
c) Dispensing the following excipients methyl paraben, propylene glycol in vessel III; wherein methyl paraben is dissolved in propylene glycol to form a clear solution;
d) Dispensing the following active & excipients triamcinolone acetonide or salt thereof, propylene glycol in vessel IV; wherein triamcinolone acetonide or salt thereof is dissolved in propylene glycol to form clear solution;
e) Adding content of step (c) into content of step (b) and stirring to form uniform and homogeneous emulsion;
f) Heating content of step (b) and step (a) at about 75 °C and stirring to form a homogenous uniform emulsion;
g) Cooling the above emulsion gradually to temperature of about 25 °C – 30°C h) Adding the content of step (d) to the primary emulsion of (f) with constant stirring; and
i) Making up the volume of the emulsion with purified water to the required quantity.
Triamcinolone, 9a-fluoro-11b,16a,17,21-tetrahydroxypregna-1, 4-dien-3,20-dione (27.1.61), differs from dexamethsone in terms of chemical structure in that the a methyl group at C16 is replaced with a hydroxyl group. It is synthesized from the 21-O-acetate of hydrocortisone 27.1.17. In the first stage, both carbonyl groups of this compound undergo ketalization by ethylene glycol. Next, the hydroxyl group in the resulting diketal 27.1.53 is replaced with chlorine using thionyl chloride, and the product undergoes dehydrochlorination using an alkaline, during which the 21-O-acetyl group also is hydrolyzed. Acetylating the hydroxyl group once again with acetic anhydride gives a triene 27.1.54. Reacting this with osmium tetroxide gives the vicinal diol 27.1.55. The secondary hydroxyl group at C16 of this product undergoes acetylation by acetic anhydride in pyridine, which forms the diacetate 27.1.56. Treating the product with N-bromoacetamide in chloric acid gives a bromohydrin (27.1.57), which upon reaction with potassium acetate is transformed to an epoxide (27.1.58). Opening of the epoxide ring, using hydrofluoric acid, gives the corresponding 9-fluoro-11-hydroxy derivative 27.1.59. Upon microbiological dehydrogenation, the C1–C2 bond is oxidized to a double bond, forming triamcinolone acetate (27.1.60), the acetyl group of which is hydrolyzed, forming the desired triamcinolone (27.1.61) [30–32].
Triamcinolone is similar to dexamethasone in terms of pharmacological action, and it is better tolerated in some cases. Synonyms of this drug are ledercort, cenocort, delsolon, and others.
The synthesis of triamcinolone (23.2.1) starts from ketalization of cortisol 21-acetate (23.2.8) using ethylene glycol. Dehydration of the obtained compound (23.2.9) for creation of a double bond in position 16,17 of the steroid skeleton through the series of sequential reactions of chlorination, dehydrochlorination, hydrolysis, and acetylation produces 21-acetoxy-4,9(11),16-pregnatriene-3,20-dione (23.2.10), treatment of which with osmium tetroxide in benzene and pyridine produced diol (23.2.11), the secondary hydroxyl group of which, in position 16, was acetylated with acetic anhydride in pyridine to produce the diacetate (23.2.12). The obtained compound in dioxane and water was treated with N-bromoacetamide and 10% perchloric acid to yield bromohydrine (23.2.13). Dehydrobromination of the bromohydrine (23.2.13) with anhydrous potassium acetate in refluxing ethanol produced the epoxy-derivative (23.2.14). Opening of the epoxide ring in (23.2.14) with anhydrous hydrogen fluoride in chloroform produced (23.2.15). Microbiological dehydrogenation of the obtained product with Corynebacterium simplex produced crude diacetate (23.2.16), saponification of which produced triamcinolone (23.2.1) [108-110] (Scheme 23.7.).
Scheme 23.7. Synthesis of triamcinolone.
Triamcinolone is commonly used in the treatment of respiratory inflammation and improves airway reactivity, decreasing respiratory problems. Strangely, there are only few reviews of the pharmacotherapy of triamcinolone [111-113].
Glucocorticoids have a number of diverse effects in different body tissues. Glucocorticoids, in topical, oral and inhaled formulations, are useful for their anti-inflammatory and immunosuppressive properties. Several glucocorticoids such as budesonide and ciclesonide are used for treatment of several disorders.
The synthesis and purification of glucocorticoids have been disclosed at different instances. However, most of these synthetic procedures involve toxic solvents or long reaction times and are ineffective for large scale synthesis. For instance, US 3,92,9768 discloses a process for preparation of budesonide by reacting 16, 17-dihydroxy compound with aldehyde in solvents such as dioxane, methylene chloride or their combinations.
DE 4129535 discloses a process for the synthesis of Ciclesonide which involves the intermediate 16A, 17-[(7?,S)-cyclohexylmethylenedioxy]-l 13, 21-dihydroxy-pregna-l 4- dien-3,20-one which is obtained by an acid catalysed reaction of 11 , 16 , 17, 21-tetra hydroxypregna-l,4-dien-3,20-one with cyclohexane aldehyde.
WO 02/38584 discloses the synthesis of Ciclesonide by reacting corresponding 16, 17-ketals with a cyclohexane aldehyde in the presence of 70% perchloric acid, 1-nitropropane as solvent. However, perchloric acid is a dangerous solvent and can cause serious accidents with fatal consequences.
US Patent No. 6169178 relates to a process for the preparation of budesonide and of 16, 17- acetals of pregnane derivatives structurally co-related thereto comprising treating 16, 17-dios or of 16, 17-ketals or cyclic acetals with aldehydes in the presence of aqueous hydrobromic acid or hydroiodic acid used as reaction catalyst or solvents. However, hydroiodic and other hydrohalic solvents are corrosive, light sensitive and expensive. Further, these acids also post environmental problems. Notwithstanding the use of hydrohalo acids requires use of special equipment since they are extremely corrosive and consequently increase the cost of production.
US 5,55,6964 discloses a process for the preparation of budesonide by reacting 16 – Hydroxy Prednisolone in acetonitrile in the presence of /^-toluene sulfonic acid as a catalyst. There are certain other patents that use alkyl sulfonic acid instead of aryl sulfonic acid for the synthesis of budesonide or similar compounds. However, sulfonic acids are hazardous solvents and FDA has expressed significant concern over the presence or traces of sulfonic acid in pharmaceutical products. Hence, there is a need to have a process for the synthesis 16, 17- acetals of pregnane compounds that is industrially scalable and which does not involve the use of harmful solvents.
Example- 1: Process for preparation of 16-HPN from 3TR
Stage-I
Stage- 1 Stage-I I
Stage-IV
1 6-HPN acetate 1 6-HPN
Scheme 2: Synthesis of 16HPN from 3TR
Stage-I (oxidation)
Charge 750L of acetone (50 volume), 39L of purified water (2.60 volume) and 15 Kg of 3TR (40.93mol) in a SS Reactor at ambient temperature. Cool to -7°C to -5°C than added 6.0L of formic acid (159.03 mol) and 9.0 kg of potassium permanganate (56.95 mol). Maintain at – 5°Cto -3°C for 30 minutes. In-process check by TLC, 3TR should be less than 1.0%. Added 1.5kg sodium metabisulphite (7.89 mol solution in 12L of purified water at -5°C to -3°C then added 3.0 kg of hyflow super cell at 15°C (+2°C) and filter through 10.0 kg of hyflowbed at 27°C(+3°C) and wash with 150L of acetone Added 1.5 kg of activated charcoal, Stir and filter through hyflow bed and wash with 60L of acetone. Total filtrate was distilled under reduced pressure, while maintaining temperature below 45°C. Added 81L of purified water and cool to 5°C+5°C. Filter through centrifuge and wash with 156L of purified water. Wet material is dry at 60°+5°C till moisture less than 0.50%, Yield=15 kg, HPLC purity=98%.
Stage-II (Bromination)
Charge 75L of tetrahydrofuran, 16L of purified water and 15.0 kg of Stage-I (37.46 mol) in a glass reactor. Cool to -6°C (+2°C) and added 7.50 kg of dibromantin (26.23 mol) and 0.60L of perchloric acid (9.38 mol) and maintain at -6°C (+2°C) for one hour. In-process check by TLC, stage-I should be less than 0.50%. Reaction mass is quench in 390L of purified water at ~5°C. Raised the temperature to 25°C and maintained for 01 hour, filter through centrifuge and wash with 828L of purified water or till neutral pH. Wet material is dry at 40°C+5°C till moisture content should be less than 10%, Yield=21.0kg, HPLC purity=97%.
Stage-Ill (Debromination)
Charge 68.0L of N, N-dimethyl formamide(3.238volume) and 21.0kg of stage-II (42.22 mol) in glass reactor, start argon gas purging and cool to -5°C. Charge 13.0L of N,N- dimethylformamide (0.619volume) , 9.70L of dimethylsulfoxide(0.462volume), 1.62kg of chromium chloride hexahydrate (6.51 mol) and 1.94 kg of zinc dust (0.703 mol). Cool to – 10°Cand added 5.50L of thioglycolic acid (79.21 mol). Maintain for one hour while maintaining temperature around -10°C. In-process check by TLC, stage-II should be less than 1.0%. Added 310 L of purified water and cool to 0°C. Filter through centrifuge and wash with 1600L of purified water. Wet material is dry at 60°C+ (5°C) till moisture content less than 6.0%, Yield=15.0kg, HPLC Purity=90%.
Charge 150L of methylene chloride (10 volume), 150L of methanol (10 volume.) and 15.0kg (30.16 mol) of stage-Ill in a SS Reactor. Heat to clear solution then added 3.0 kg of activated charcoal (20%) and reflux for 04 hours, Filter through hyflow bed and wash with 75L of methylene chloride (5 volume), and 75L of methanol (5 volume) mixture. Total filtrate is distilled till last drop and added 75L (5 volume) of methylene dichloride, reflux for 04 hours than cool to 40°C+(5°C), Filter through centrifuge and wash with 15L (one volume) of methylene chloride. Wet material is dry at 60°C (+5°C) till moisture contents less than 1.0% (Yield =13.0kg, HPLC Purity=96%). Further charge 65.0L (5volume) of ethyl acetate and 13.0 kg (1.0 mol) of purified material. Heat to reflux and maintain for 04 hours under reflux, then cool to 40°C. Filter through centrifuge and wash with 13.0L (one volume) of ethyl acetate. Wet material is dry at 60°C (+5°C) till moisture contents less than 0.50%, Yield=12.0kg, HPLC Purity=98.6%.
Stage-IV (Deacetylation)
Charge 5.83L of methanol (10 volume) and 5.83L of methylene chloride (10 volume) in a glass flask and added 583 gm of 16-HPN acetate(1.397mol) at RT. Start argon gas purging and cool to 0°C to 5°C under argon purging. Prepare 11.66 gm of sodium hydroxide (0.2915mol) solution in 0.583L of methanol (one volume) under argon purging and cool to 0°Cto 5°C. Sodium hydroxide solution is charge in reaction mass at 0°C to 5°C. Maintained the reaction mass at 0°C to 5°C for one hour, In-process check by TLC against 16-HPN acetate it should be nil. Adjust pH to neutral by 21.40ml of acetic acid (0.3742 mol); distill under reduced pressure while maintaining temperature below 40°C, till dry. Cool to ambient temperature and added 1.166L of purified water (02 volume). Cool to 0°C and maintain for one hour. Filter and wash with 300ml of purified water. Dry at 60°C (+5°C) till moisture content less than 1.0%, Yield=490gm (93.50%), HPLC Purity=98.97%, Single impurity= 0.40%. Example 2: Process of synthesis of Budesonide from 16-HPN
16-HPN Budesonide
Charge 800 ml of aqueous hydrochloric acid (8 volume) in a glass flask, start nitrogen gas purging and Cool to -5°C and maintain for 15 min. then added 100 gm of stage-I (0.27 mol) at -5°C and stir for 15 min., added 30 ml of N-butyraldehyde (0.33 mol) while maintaining temperature -5°C to 0°C in around 30 minutes and maintain at 0°C to 5°C for 150 min. under stirring. In-process check by TLC against stage-I, it should be nil. Reaction mass is quench in 1200 ml of purified water (12 volume) at 5°C to 10°C and stir for 15 min. Added solution of 100 kg of sodium bicarbonate (1.19 mol) and 1 ml of purified water (10 volume) in reaction mass at 5°C to 10°C. Stir at 5°C to 10°C for 15 min. Filter and wash with purified water till neutral pH. Wet material is dry at 50°C (+5°C) till moisture contents less than 1.0 %, Yield =110 gm (96.49%), HPLC purity=96.45%, single impurity=1.29%, Epimer-A=47.76%, Epimer-B=49.69%.
(Purification)
Charge 2.5 L of methanol (25 volume) in a Glass flask and added 100 gm of above mentioned crude product. Dissolved at 25°C+5°C till clear solution, added 10 gm of activated charcoal and stir for 30 min. than filter through hyflow bed and wash with 200 ml of methanol (2 volume). Combined filtrates charged in a Glass flask and cool to 10°C to 15°C and added 5.40 L of purified water (54 volume) at 5°Cto 10°C, stir for 15min., filter and wash with purified water. Wet material is dry at 50°C (+5°C) under vacuum till moisture content less than 0.50%, Output=90.0gm, HPLC purity=99.66%, single impurity=0.1%, Epimer-A=44.47%, Epimer-B=55.01%.
Example 2.1: Scale-up process of manufacturing of Budesonide from 16-HPN
Charge 40 L of aqueous hydrochloric acid (8 volume) in a glass flask, start nitrogen gas purging and Cool to – 10°C and maintain for 15 min. then added 5.0 kg of stage-I (13.315 mol) at – 10°C and stir for 45 min. added 1.5 L of N-butyraldehyde (16.68 mol) while maintaining temperature -7°C to – 11°C in around 30 minutes and maintain at -2°C to -6°C for 60 min. under stirring In-process check by TLC against stage-I, it should be nil. Reaction mass is quench in 60 L of purified water (12 volume) at 5°C to 10°C and stir for 15 min. Added solution of 5.0 kg of sodium bicarbonate (59.525 mol) and 50L of purified water (10 volume) in reaction mass at 5°Cto 10°C. Stir at 5°C to 10°C for 15 min. Filter and wash with purified water till neutral pH. Wet material is dry at 50°C (+5°C) till moisture contents less than 1.0 %, Yield =5.293 kg (94.46%), HPLC purity=95.45%, single impurity=1.45%, Epimer-A=53.51 %, Epimer-B=43.78% Effect of temperature and its variation on epimer ratio (A and B) with respect to batch size (From lab to commercial batch)
Example 3: Process for synthesis ofCiclesonide from 16HPN
Preparation of cyclohexane carboxaldehydemetabisulphite complex
200gm of Cyclohexane carboxaldehyde (1.786 mol) was dissolved in 3.0L of denatured sprit (15 volume) and a solution of 190gm of sodium metabisulphite (1.827 mol) in 300ml of purified water (1.5 volume) was added. The resulting precipitate was filtered and washed with 1.0L of denatured sprit(5.0 volume) and dried under vacuum at 50°C, till moisture content less than 6.00%, Yield=400gm (97 %)
Stage I: Preparation of stage-I from 16-HPN
Cyclohexane carboxaldehyde
sodium metabisulphite complex
170gm of 16-HPN (0.4528 mol) was suspended in 3.40L of dichloromethane (20 volume) and treated with 340ml of 70% perchloric acid. (5.65 mol) and 110.5gm of cyclohexane carboxaldehyde metabisulphite complex (0.512 mol) was added in lots while maintaining the temperature between 0°Cto 5°C. The reaction mass was stirred at 0°C to 5°C for 03 hours. In- process check by TLC 16-HPN should be nil and then neutralized with 10% aqueous sodium bicarbonate solution. The organic layer was separated and concentrated under vacuum to obtain a residue which was stripped with methanol (1.0 volume). The solvent was concentrated and the residue was dissolved by refluxing in methanol (5.0 volume). The clear solution was cooled to 0°C to 5.0°C and the resulting solid was filtered and dried at 50°C till moisture content less than 0.50%, Yield=170.0gm (80.0%), HPLC purity=91.68%.
Stage -II Preparation of Ciclesonide from Stage -I
Stage-I Ciclesonide
158gm of stage-I (0.34mol) was suspended in 1.58L of methylene chloride (10.0 volume) at 25°C to 30°C. The reaction mass was chilled to 0°C to 5°C and 81.0ml of triethylamine(0.581 mol) was added, followed by the addition of 79.0ml of isobutyryl chloride [0.75 mol; diluted with 79.0 ml of methylene chloride (0.50 volume)] slowly at 0° to 5°C and maintained at same temperature for 60min. In-process check by TLC, Stage-I should be nil. The reaction mass was diluted with 2.53L of purified water (16.0 volume) , the organic layer was separated and washed with purified water till neutral pH, than organic layer was separated and concentrated under vacuum to obtained a residue. The residue was dissolved by refluxing in 948ml of methanol (6.0 volume); the clear solution was cooled to 0°C to 5°C under stirring and filtered. The product was dried under vacuum at ~50°C till moisture contents comes less than 0.50%, Yield=158.0 gm (87.0%), HPLC purity=95.74%.
(Purification)
120gm of Ciclesonide crude was dissolved by refluxing in 600ml of methanol. The clear solution was chilled to 20°C under stirring and filtered. The product was dried under vacuum at 90°C till moisture content less than 0.50%. Yield=105 gm (87.50%), HPLC purity=99.7 %.
Example 4: Process for synthesis of Desonide from 16HPN acetate
Stage-I : Preparation of Desonide acetate from 16 HPN acetate
Desonide acetate
16HPN acetate 190.0 ml of acetone (7.0 volume) was charged in a glass flask under nitrogen blanketing than added 27 gm of 16HPN acetate (0.0645mol) at ambient temperature. Temperature raised to 28°C (+2°C) and stir for 20 minutes. 1.35 ml of perchloric acid 70% (0.02 lmol) was added at 28°C (+2°C) and stir for 30 minutes. Temperature further raised to 35°C and stir for 60 minutes. In-process check by TLC against 16HPN acetate, it should be nil. Reaction mass cooled to 10°C, filtered and washed with purified water till neutral pH (~7) and finally washed with acetone. Wet material dried at 50°C+5°C till moisture content less than 0.50% to get stage-I. Yield =23gm (77.76%), HPLC Purity=98.28%
Stage-II: Preparation of Desonide from Desonide acetate
Desonide
Desonide acetate
200 ml of methanol (10 volume) and 200ml of methylene dichloride (10 volume) was charged in a glass flask and start argon gas purging. 20 gm of stage- 1st (0.0436mol) was added at ambient temperature. Cool to 0°C+5°C. 0.40gm of sodium hydroxide (O.Olmol) solution in 20ml of methanol (l.Ovolume) was added at 0°C+5°C. Stir at 0°C+5°C for 120 minutes. In-process check by TLC against stage- 1st it should be nil. Adjust pH to neutral (~7) by 2.0ml of acetic acid at 0°C+5°C. Distilled the solvent from reaction mass under vacuum while maintaining temperature below 40°C till the volume get reduced to 3 to 4 volume of the input. Cool to 0°C and further added 60ml of purified water and stir for 30 minutes. Filtered, washed with purified water till neutral pH (~7). Wet material dried at 50°C+5°C till moisture content less than 0.50% to get crude Desonide. Yield =14.70gm (80.92%), HPLC Purity=88.15%.
(Purification)
140 ml of methanol (10 volume) and 140 ml of methylene chloride (10 volume) was charged in a glass flask and added 14.0 gm of crude material (0.034mol) than stir till clear solution. Added 1.5 gm of activated charcoal and stir for 30 minutes than filtered through hyflow supercel bed and washed with 30ml of methanol and 30ml of methylene chloride mixture. Combined filtrate and distilled the solvent from reaction mass under vacuum while maintaining temperature below 40°C till the volume reduced to 3 to 4 volume of the input. Cool to 0°C. Filtered the reaction mass and washed with 10ml of precooled methanol. Wet material was dried at 50°C+5°C till moisture content less than 0.50% to get Desonide. Yield=8.60gm, HPLC Purity= 99.43%
lOOgm of 3TR (0.27 mol.)was suspended in 1300ml (13 volume) acetone. Cooled it to -5°C to -10°C than added 4.0 ml (0.062 mol.) perchloric acid solution and 50gm of dibromantin. Maintained the reaction at same temperature for 02 hours. In-process check by TLC against 3TR it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol.) in 5 lots and reaction was maintained at 35°C+2°C. In-process check by TLC against step-I reaction mass, it should be nil. Cooled to 0°C (+5°C) and adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol.). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C (+2°C) till moisture content less than 0.50%. Yield =87gm, (83.36%), HPLC Purity=97.883%.
Stage – II:
80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C (+2°C) than added 32ml of formic acid (0.85 mol.) and 48gm of potassium permagnate (0.30 mol.) at -5°C (+2°C). Reaction was maintained at – 5°C+2°Cfor one hour. In-process check by TLC against stage-I it should be nil. Added 8gm of sodium metabisulphite (0.042 mol.) In 80 ml purified water (01 volume) solution at -5°C (+2°C). Temperature raised up to 27°C and filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4volume of stage-I than cool to 0°C to 5°C and added 480ml of purified water stir and filter and washed with purified water to get wet stage-II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0%. Yield =78.30gm, (89.88%), HPLC Purity=99.178%. Stage -III:
Stage-ll Stage-
300ml of hydrofluoric acid (12.60mol) was cooled at -25°C to -30°C than added 75gm of stage-II (0.180mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II, it should be nil. Reaction mass was cooled to -50°C than added 45ml of acetone (0.60volume) at -45°C to -50°C. Reaction was maintained at -45°C to -50°C for 02 hours. In-process check by TLC against before acetone reaction mass. Added 565ml of purified water at 0°C and 340ml of liq. ammonia at ~20°C than reaction mass was quenched in 410ml of liq. ammonia and 735ml of purified water solution at 15°C (+2°C), stir and filter and washed with purified water till neutral pH. Wet material was dried at 45°C to 50°C, Yield =78.50gm, (91.48%), HPLC Purity=91.593%.
(Purification)
76 gm of stage-Ill Crude (0.16 mol.) was dissolved in 760ml of methylene chloride (lOvolume) and 760ml of methanol (lOvolume) mixture at ambient temperature. Stir till clear solution and added 7.6gm of activated charcoal (0. lOvolume) than stir for 30minutes, filter through hyflow bed and washed with methanol (one volume) and methylene chloride (one volume) mixture. Total filtrate was distilled under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°C and stir for 02 hours. Filtered and washed with minimum precooled methanol, Wet material was dried 45°C to 50°C till moisture contents less than 0.50%, Yield=62gm, HPLC Purity=98.633%.
Stage – IV (Process for synthesis of Triamcinolone acetonide from Stage – III):
Stage- Ill Triamcinolone acetonide
60gm of stage-Ill (0.13 mol) was dissolved in 600ml of methanol (lOvolume) and 600ml of methylene chloride (lOvolume) mixture under argon bubbling. Cool to -5°C+2°C and added 1.2gm of sodium hydroxide (0.03mol.) solution in 60ml of methanol (Olvolume) at -5°C (+2°C). Reaction maintained at -5°C (+2°C) for 03 hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral (~7) by adding 1.8ml of acetic acid at -5°C (+2°C). Reaction mass was distilled at below 40°C under vacuum till 3 to 4 volume of input. Cool to 30°C and added 120ml of purified water, stir for one hour than filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°C till moisture content less than 0.50%, Yield =52gm, (95.04%), HPLC Purity=99.21%
(Purification)
50gm of crude material (0.12 mol.) was dissolved in 1100ml of acetone (22volume) and 100ml of purified water (02volume) at 50°C than added 2.5gm of activated charcoal and stir for one hour at same temperature, Filter through hyflow bed and washed with 120ml of acetone (2.40volume). Filtrate was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°Cand maintained for one hour at same temperature. Filter and washed with water. Wet material was dried at 45°C to 50°C till moisture content less than 0.50%, Yield=43gm, HPLC Purity=99.40%.
Example 6: Process for synthesis of Flunisolide from 16HPN acetate Stage -I (Preparation of Desonide acetate from 16HPN acetate):
1 6 H PN acetate eson e acetate
140ml of acetone (7 volume) was charged in glass flask and start argon blanketing than added 20 gm of 16-HPN acetate (0.048mol) at ambient temperature. Cooled to 28°C (+2°C). 1.0ml of perchloric acid 70% (0.016mol) was added at 28°C (+2°) C and stirred for 30 minutes. Temperature raised up to 35°Cand stirred for 60 minutes. In-process check by TLC against 16-HPN acetate, it should be nil. Reaction mass was cooled to 10°C (+2°C). Reaction mass was filtered and washed with purified water till neutral pH (~7) to get wet material. Wet material was dried at 50°C+5°C till moisture content less than 0.50% to get stage-lst. Yield=17.40gm, (79.40%), HPLC Purity=98.241%.
Stage -II (Preparation of Desonide from Desonide acetate):
170ml of methanol (lOvolume) and 170ml of methylene chloride (lOvolume) was charged in a glass flask and start inert atmosphere. 17gm of stage-lst (0.037mol) was added at ambient temperature. Cooled to -5°C. 0.4gm of sodium hydroxide (O.Olmol) solution in 17ml of methanol was added at 0°C (+5°C). Reaction mass was stirred for 02 hours at 0°C (+5°C). In- process check by TLC against stage- 1st it should be nil. Neutral pH (~7) was adjusted by acetic acid. Reaction mass was distilled under vacuum at below 40°C till ~ 100ml. Concentrated mass was cooled to 0°C (+5°C) and stir for one hour. Reaction mass was filtered and washed with precooled methanol to get wet material. Wet material was dried at 50°C (+5°C) till moisture content less than 0.50% to get stage-2nd. Yield=14.0gm, (90.67%), HPLC Purity=99.426%, Single impurity=0.136%.
Stage -III (Preparation of Flunisolide acetate from Desonide):
Desonide Flunisolide acetate
50ml of isopropenyl acetate (5 volume) was charged in a glass flask and added lOgm of stage-2nd (0.024mol) at ambient temperature than heated to 65°C and added 1.5ml of methane sulphonic acid (0.023mol) and temperature raised up to 80°C and stir for one hour. In-process check by TLC against stage-2, it should be nil. Reaction mass cooled to 25°C and adjust pH neutral (~7) by triethylamine. Reaction mass was distilled under vacuum till last drop and degases with acetonitrile. 90ml of acetonitrile (09 volume) was added and cooled to -5°C and than further added 10ml of purified water. lOgm of selectfluor(0.028mol) was added in two lots at 0°C(+5°C) in 02 volume of acetonitrile. Reaction mass was stirred at 10°C to 15°C for 12 hours. In-process check by TLC against before selectfluor reaction mass it should be nil. Adjust pH neutral (~7) by liq. ammonia solution at 0°C+5°C. Reaction mass was quenched in 500ml of purified water (lOOvolume) at ambient temperature. Reaction mass was filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C+5°C till moisture content less than 0.50% to get stage-3rd. Yield=8.60gm, (75.17%), HPLC Purity= 94.12%.
Stage -IV (Preparation of Flunisolide from Flunisolide acetate):
Flunisolide acetate Flunisolide
80ml of methanol (lOvolume) and 80ml of methylene chloride (lOvolume) was charged in a glass flask under inert atmosphere at ambient temperature than added 8.0gm of stage-3r (0.017mol) at ambient temperature. Cooled to -5°C and added 0.16gm of sodium hydroxide (0.004mol) solution in 8ml of methanol at -5°C(+5°C) and stir for 02 hours at -5°C(+5°C). In-process check by TLC against stage-3 ‘ it should be nil. Adjust pH neutral(~7) by acetic acid and reaction mass was distilled under vacuum at below 40°C(+5°C) till ~40ml of volume. Cool to 0°C to 5°C and stir for one hour. Reaction mass was filtered and washed with precooled methanol to get wet material. Wet material was dried at 45°C (+5°C) till moisture content less than 0.50% to get Flunisolide crude. Yield=6.0gm, (82.30%), HPLC Purity=86.50%.
(Purification)
6.0gm of crude Flunisolide(0.014mol) was dissolved in 65ml of ethyl acetate (10.83volume) and 35ml of n-hexane (5.83volume) mixture and clear solution was passed through 60gm of silica gel column. Column was washed with 975ml of ethyl acetate (162.5volume) and 525ml of ft-hexane (87.5volume) mixture. Eluted fraction was distilled under vacuum till 3 to 4 volume of input than cooled it to 0°C and filter to get wet material. Wet material was dried at 50°C (+5°C) till moisture content less than 0.50% to get Flunisolide. Yield=4.28gm, HPLC Purity=95.60%.
Example 7: Process for synthesis of Triamcinolone from 3TR
S
lOOgm of 3TR (0.27mol) was suspended in 1300ml (13 volume) acetone. Cool to -5°C to- 10°C than added 4.0 ml (0.062mol) perchloric acid solution and 50gm of dibromantin. Reaction maintained at same temperature for 02 hours. In-process check by TLC against 3TR, it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol) in 5 lots and reaction was maintained at 35°C (+2°C). In-process check by TLC against step-I reaction mass, it should be nil. Cool to 0°C+5°Cand adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C (+2°C) till moisture content less than 0.50% to get stage-I. Yield=85.30gm, (81.74%), HPLC Purity=96.54%. Stage -II:
80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C (+2°C) than added 32ml of formic acid (0.85 mol.) and 48gm of potassium per magnate (0.30 mol) at -5°C (+2°C). Reaction was maintained at same temperature for one hour. In-process check by TLC against stage-I, it should be nil. Added sodiummetabisulphite solution (8 gm in 80 ml of water) at -5°C+2°C. Temperature was raised up to 27°C and filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4 volume of stage-I than further cooled to 0°C to 5°C and added 480ml of purified water, stirred, filter and washed with purified water to get wet stage- II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0% to get stage-II. Yield=82gm, (94.13%), HPLC Purity=97.75%.
Stage -III:
Stage-II Triamcinolone acetate
160ml of hydrofluoric acid (70%) (6.72mol) was cooled at -25°C to -30°C than added 40gm of stage-II (0.096mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II, it should be nil. Added 280ml of purified water at 0°C and 650ml of liq. ammonia at 20°C than reaction mass was quenched in 200ml of liq. ammonia and 500ml of purified water solution at 15°C(+2°C), stirred, filtered and washed with purified water till neutral pH(~7). Wet material was dried at 45°C to 50°C to get stage-Ill Yield=40gm, (95.42%), HPLC Purity=88.71%
(Purification)
40gm of stage-Ill crude (0.0916 mol) was refluxed in 160ml of acetone. Cool to 0°C. Filtered and washed with minimum precooled acetone. Wet material was dried at 50°C+5°C till moisture content comes less than 0.50% to get stage-Ill. Yield=24.9gm HPLC Purity=95.17%.
24gm of stage-Ill (0.055mol) was dissolved in 240ml of methanol (lOvolume) and 240ml of methylene chloride (lOvolume) mixture under argon bubbling. Cool to -5°C+2°C and added 0.48gm of sodium hydroxide (0.012mol) solution in 24ml of methanol (Olvolume) at – 5°C+2°C. Reaction was maintaining at -5°C (+2°C) for 03hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral by adding 0.70ml of acetic acid at -5°C (+2°C). Reaction mass distilled at below 40°C under vacuum till 04-05 volume of input. Cooled to 0°C+5°Cand stir for one hour than filtered and washed with minimum precooled methanol. Wet material was dried at 45°C to 50°C till moisture content less than 0.50%. Yield=18.50gm, (85.29%), HPLC Purity=98.60%.
Example 8: Process for synthesis of Triamcinolone Hexacetonide from 3TR
S
lOOgm of 3TR (0.27288 mol) was suspended in 1300ml (13 volume) acetone. Cool to -5°C to -10°C than added 4.0 ml (0.0625 mol) perchloric acid solution and 50gm of dibromantin. Reaction was maintained at same temperature for 02 hours. In-process check by TLC against 3TR, it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol) in 5 lots and reaction was maintained at 35°C (+2°C). In-process check by TLC against step-I reaction mass, it should be nil. Cool to 0°C (+5°C) and adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH. Wet material was dried at45°C(+2°C) till moisture content less than 0.50% to get stage-lst. Yield =87gm, (83.36%), HPLC Purity=97.883%. Stage-II :
80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C than added 32ml of formic acid (0.85 mol.) and 48gm of potassium permanganate (0.30 mol) at -5°C+2°C. Reaction maintained at -5°C (+2°C) for one hour. In-process check by TLC against stage-I, it should be nil. Added sodium metabisulphite solution (8 gm in 80 ml water) at -5°C (+2°C). Temperature raised up to 27°Cand filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4 volume of stage-I than cooled to 0°C to 5°C and added 480ml of purified water, stirred, filtered and washed with purified water to get wet stage-II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0% to get stage-2nd. Yield=78.30gm, (89.88%), HPLC Purity=99.18%.
Stage – III:
300ml of hydrofluoric acid (12.60mol) was cooled at -25°C to -30°C than added 75gm of stage-II (0.180mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II. It should be nil. Reaction mass was cooled to -50°C than added 45ml of acetone (0.60volume) at -45°C to -50°C. Reaction maintained at -45°Cto – 50°C for 02 hours. In-process check by TLC against reaction input, it should be nil. Added 565ml of purified water at 0°C and 340ml of liq. ammonia at 20°C than reaction mass was quenched in 410ml of liq. ammonia and 735ml of purified water solution at 15°C(+2°C), stirred, filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°Cto get stage-3rd. Yield=78.50gm, (91.48%), HPLC Purity=91.59%.
(Purification)
76 gm of stage-Ill Crude (0.16 mol) was dissolved in 760ml of methylene chloride (01 volume) and 760ml of methanol (lOvolume) mixture at ambient temperature. Stirred till clear solution and added 7.6gm of activated charcoal (0. lOvolume) than further stir for 30 minutes and filtered through hyflow bed and washed with methanol (one volume) and methylene chloride (one volume) mixture. Total filtrate was distilled under vacuum till 3 to 4 volume of input. Cooled to 0°C to 5°Cand stir for 02 hours. Filtered and washed with minimum precooled methanol. Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get purified stage-3rd. Yield=62gm, HPLC Purity=98.633%
Stage -IV : (Preparation of Triamcinolone acetonide from Stage – III)
Stage- Ill Triamcinolone acetonide
60gm of stage-Ill (0.1259 mol) dissolved in 600ml of methanol (lOvolume) and 600ml of methylene chloride (lOvolume) mixture under inert atmosphere. Cool to -5°C and added 1.2gm of sodium hydroxide (0.03mol) solution in 60ml of methanol (Olvolume) at -5°C (+2°C). Reaction maintained at -5°C+2°C for 03 hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral (~7) by adding 1.8ml of acetic acid at -5°C+2°C. Reaction mass was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to
30°C and added 120ml of purified water, stir for one hour than filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get stage-4111 (Triamcinolone acetonide). Yield=52gm, (95.04%), HPLC Purity=99.21%.
(Purification)
50gm of crude material (0.12 mol) dissolved in 1100ml of acetone (22volume) and 100ml of purified water (02volume) at 50°C than added 2.5gm of activated charcoal and stirred for one hour at same temperature. Filter through hyflow bed and washed with 120ml acetone (2.40volume). Filtrate was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°C and maintained for one hour at same temperature. Filtered and washed with water. Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get purified stage-4th. Yield =43gm, HPLC Purity=99.40%
-V: (Preparation of Triamcinolone Hexacetonide from Triamcinolone acetonide):
50ml of pyridine (lOvolume) charged in a glass flask and added lOgm of Triamcinolone acetonide (0.023mol) at ambient temperature. Heated to 80°C to 90°C than added 10ml of 3, 3-dimethyl butyryl chloride (O.l lmol) at 80°C to 90°C. Stirred at 80°C to 90°C for 02 hours. In-process check by TLC against Triamcinolone acetonide, it should be nil. Reaction mass cooled to ambient temperature and reaction mass was quenched in 1000ml of purified water (lOOvolume) at ambient temperature, stir for one hour than filtered and washed with purified water till neutral pH (~7). Wet material was dried at 50°C (+5°C) till moisture content less than 1.0% to get stage-5th (Triamcinolone Hexacetonide). Yield=12gm, (97.90%), HPLC Purity=98.63%.
(Purification)
120ml of methanol and 120ml of methylene chloride charged in a glass flask and added 12gm of crude material, stir till clear solution than added 1.2gm of activated charcoal and stir for 30 minutes. Filtered through hyflow bed and washed with 12ml of methanol and 12ml of methylene chloride mixture. Total filtrate was distilled under vacuum at below 40°C till 5 to 6 volume of crude. Cooled to 0°C+5°C and stir for one hour. Filtered and washed with 12ml of precooled methanol. Wet material was dried at 40°C+5°C till moisture content less than 0.50% to get TrimcinolneHexacetonide. Yield=8.8gm, HPLC Purity=99.625%//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The derivativetriamcinolone acetonide is the active ingredient in various topical skin preparations (cream, lotion, ointment, aerosol spray) designed to treat skin conditions such as rash, inflammation, redness, or intense itching due to eczema[15] and dermatitis.[16]
Side effects of triamcinolone are similar to other corticoids. In short-term treatment up to ten days, it has very few adverse effects; however, sometimes gastrointestinal bleeding is seen, as well as acute infections (mainly viral) and impaired glucose tolerance.[4]
No acute overdosing of triamcinolone has been described.[17]
Interactions
Drug interactions are mainly pharmacodynamic, that is, they result from other drugs either adding to triamcinolone’s corticoid side effects or working against its desired effects. They include:[4][17]
Triamcinolone and other drugs can also influence each other’s concentrations in the body, amounting to pharmacokinetic interactions such as:[4][17]
Rifampicin, phenytoin, carbamazepine and other inducers of the liver enzyme CYP3A4[19] speed up metabolization of triamcinolone and can therefore reduce its effectiveness.
Conversely, CYP3A4 inhibitors such as ketoconazole and itraconazole can increase its concentrations in the body and the risk for adverse effects.
Blood concentrations of ciclosporin can be increased.
Triamcinolone is a glucocorticoid that is about five times as potent as cortisol, but has very little mineralocorticoid effects.[4]
Pharmacokinetics
When taken by mouth, the drug’s bioavailability is over 90%. It reaches highest concentrations in the blood plasma after one to two hours and is bound to plasma proteins to about 80%. The biological half-life from the plasma is 200 to 300 minutes; due to stable complexes of triamcinolone and its receptor in the intracellular fluid, the total half-life is significantly longer at about 36 hours.[4][5]
A small fraction of the substance is metabolized to 6-hydroxy- and 20-dihydro-triamcinolone; most of it probably undergoes glucuronidation, and a smaller part sulfation. Three quarters are excreted via the urine, and the rest via the faeces.[4][17]
Due to corticoids’ mechanism of action, the effects are delayed as compared to plasma concentrations. Depending on the route of administration and the treated condition, the onset of action can be from two hours up to one or two days after application; and the drug can act much longer than its elimination half-life would suggest.[4][5]
Chemistry
Triamcinolone is a syntheticpregnanecorticosteroid and derivative of cortisol (hydrocortisone) and is also known as 1-dehydro-9α-fluoro-16α-hydroxyhydrocortisone or 9α-fluoro-16α-hydroxyprednisolone as well as 9α-fluoro-11β,16α,17α,21-tetrahydroxypregna-1,4-diene-3,20-dione.[20][21]
The substance is a light-sensitive, white to off-white, crystalline powder, or has the form of colourless, matted crystals. It has no odour or is nearly odourless. Information on the melting point varies, partly due to the substance’s polymorphism: 260 to 263 °C (500 to 505 °F), 264 to 268 °C (507 to 514 °F), or 269 to 271 °C (516 to 520 °F) can be found in the literature.[4]
In 2010, TEVA and Perrigo launched the first generic inhalable triamcinolone.[22]
According to Chang et al. (2014), “Triamcinolone acetonide (TA) is classified as an S9 glucocorticoid in the 2014 Prohibited List published by the World Anti-Doping Agency, which caused it to be prohibited in international athletic competition when administered orally, intravenously, intramuscularly or rectally”.[23]
See also
Glucocorticoid (a chart comparing various glucocorticoids)
Ciltacabtagene autoleucel is a BCMA-directed CAR T-cell therapy used in the treatment of relapsed or refractory multiple myeloma in previously treated patients.
U.S. FDA Approves CARVYKTI (ciltacabtagene autoleucel), Janssen’s First Cell Therapy, a BCMA-Directed CAR-T Immunotherapy for the Treatment of Patients with Relapsed or Refractory Multiple Myeloma
In the pivotal clinical study, 98 percent of patients with relapsed or refractory multiple myeloma responded to a one-time treatment with ciltacabtagene autoleucel and 78 percent of patients who responded experienced a stringent complete response
HORSHAM, Pa., February 28, 2022 – The Janssen Pharmaceutical Companies of Johnson & Johnson announced today the U.S. Food and Drug Administration (FDA) has approved CARVYKTI (ciltacabtagene autoleucel; cilta-cel) for the treatment of adults with relapsed or refractory multiple myeloma (RRMM) after four or more prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.1 The approval is based on data from the pivotal CARTITUDE-1 study, which included patients who had received a median of six prior treatment regimens (range, 3-18), and had previously received a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody.1 In December 2017, Janssen entered into an exclusive worldwide license and collaboration agreement with Legend Biotech USA, Inc. to develop and commercialize ciltacabtagene autoleucel.
CARVYKTI is a chimeric antigen receptor T-cell (CAR-T) therapy featuring two B-cell maturation antigen (BCMA)-targeting single domain antibodies.1 In the pivotal CARTITUDE-1 study, one-time treatment with ciltacabtagene autoleucel resulted in deep and durable responses, with 98 percent (95 percent Confidence Interval [CI], 92.7-99.7) of patients with RRMM responding to therapy (98 percent overall response rate [ORR] (n=97).1 Notably, 78 percent (95 percent CI, 68.8-86.1) of the patients achieving this level of response (n=76) experienced a stringent complete response (sCR), a measure in which a physician is unable to observe any signs or symptoms of disease via imaging or other tests after treatment.1 At a median of 18 months follow-up, median duration of response (DOR) was 21.8 months.1
CARVYKTI is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the CARVYKTI REMS Program.1 The Safety Information for CARVYKTI includes a Boxed Warning regarding Cytokine Release Syndrome (CRS), Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS), Parkinsonism and Guillain-Barré syndrome, hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS), and prolonged and/or recurrent cytopenias.1 Warnings and Precautions include prolonged and recurrent cytopenias, infections, hypogammaglobulinemia, hypersensitivity reactions, secondary malignancies, and effects on ability to drive and use machines.1 The most common adverse reactions (≥20 percent) are pyrexia, CRS, hypogammaglobulinemia, hypotension, musculoskeletal pain, fatigue, infections-pathogens unspecified, cough, chills, diarrhea, nausea, encephalopathy, decreased appetite, upper respiratory tract infection, headache, tachycardia, dizziness, dyspnea, edema, viral infections, coagulopathy, constipation, and vomiting.1
“We are committed to harnessing our science, deep disease understanding and capabilities to bring forward cell therapies like CARVYKTI as we continue to focus on our ultimate goal of delivering a cure for multiple myeloma,” said Peter Lebowitz, M.D., Ph.D., Global Therapeutic Area Head, Oncology, Janssen Research & Development, LLC. “We extend our sincere gratitude to the patients, their families and the teams of researchers and study centers who have participated in the clinical study of CARVYKTI and enabled today’s approval.”
Multiple myeloma is an incurable blood cancer that affects a type of white blood cell called plasma cells, which are found in the bone marrow. 2 Despite the development of additional treatment options in recent years, most people living with multiple myeloma face poor prognoses after experiencing disease progression following treatment with three major therapy classes, which include an immunomodulatory agent, a proteasome inhibitor and an anti-CD38 monoclonal antibody. 3
“The responses in the CARTITUDE-1 study showed durability over time and resulted in the majority of heavily pretreated patients achieving deep responses after 18-month follow-up,” said Sundar Jagannath, M.D.†, Director of the Center of Excellence for Multiple Myeloma and Professor of Medicine, Hematology and Medical Oncology, at The Tisch Cancer Institute at the Icahn School of Medicine at Mount Sinai, and principal study investigator. “The approval of cilta-cel provides physicians an immunotherapy treatment option that offers patients an opportunity to be free from anti-myeloma therapies for a period of time.”
As a personalized medicine, CARVYKTI treatment requires extensive training, preparation, and certification to ensure a positive experience for patients. Through a phased approach, Janssen and Legend Biotech will activate a limited network of certified treatment centers as the company works to scale its production capacity and increase the availability of CARVYKTI throughout the U.S. in 2022 and beyond, to ensure that we can provide CARVYKTI treatment to oncologists and their patients in a reliable and timely manner.
“This approval of Janssen’s first cell therapy is a testament to our continuing commitment in oncology to deliver new therapeutic options and drive toward our vision of the elimination of cancer,” said Mathai Mammen, M.D., Ph.D., Executive Vice President, Pharmaceuticals, Janssen Research & Development, LLC, Johnson & Johnson. “Today’s approval underscores our determination to develop therapies that can help patients living with what remains an intractable blood cancer today and at the same time offer hope for the future.”
The longer-term efficacy and safety profile of ciltacabtagene autoleucel is being assessed in the ongoing CARTITUDE-1 study. Two-year follow-up results recently presented at the American Society of Hematology (ASH) 2021 Annual Meeting showed that 98 percent of patients treated with ciltacabtagene autoleucel for RRMM responded to therapy (98 percent overall response rate [ORR] (n=97), and a majority of patients achieving sustained depth of response with 83 percent of patients achieving an sCR at the 22-month follow-up.4
About CARVYKTI(ciltacabtagene autoleucel) CARVYKTI is a BCMA-directed, genetically modified autologous T-cell immunotherapy, which involves reprogramming a patient’s own T-cells with a transgene encoding a chimeric antigen receptor (CAR) that identifies and eliminates cells that express the B-cell maturation antigen (BCMA). BCMA is primarily expressed on the surface of malignant multiple myeloma B-lineage cells, as well as late-stage B-cells and plasma cells. The CARVYKTI CAR protein features two BCMA-targeting single domain antibodies designed to confer high avidity against human BCMA. Upon binding to BCMA-expressing cells, the CAR promotes T-cell activation, expansion, and elimination of target cells.1
In December 2017, Janssen Biotech, Inc. entered into an exclusive worldwide license and collaboration agreement with Legend Biotech USA, Inc. to develop and commercialize ciltacabtagene autoleucel.
In April 2021, Janssen announced the submission of a Marketing Authorisation Application to the European Medicines Agency seeking approval of CARVYKTI for the treatment of patients with relapsed and/or refractory multiple myeloma. In addition to a U.S. Breakthrough Therapy Designation granted in December 2019, ciltacabtagene autoleucel received a Breakthrough Therapy Designation in China in August 2020. Janssen also received an Orphan Drug Designation for CARVYKTI from the U.S. FDA in February 2019, and from the European Commission in February 2020.
About the CARTITUDE-1 Study CARTITUDE-1 (NCT03548207) is an ongoing Phase 1b/2, open-label, multi-center study evaluating ciltacabtagene autoleucel for the treatment of patients with relapsed or refractory multiple myeloma, who previously received a proteasome inhibitor (PI), an immunomodulatory agent (IMiD) and an anti-CD38 monoclonal antibody, and who had disease progression on or after the last regimen. All patients in the study had received a median of six prior treatment regimens (range, 3-18). Of the 97 patients enrolled in the trial, 99 percent were refractory to the last line of treatment and 88 percent were triple-class refractory, meaning their cancer did not respond, or no longer responds, to an IMiD, a PI and an anti-CD38 monoclonal antibody.1
About Multiple Myeloma Multiple myeloma is an incurable blood cancer that affects some white blood cells called plasma cells, which are found in the bone marrow.3 When damaged, these plasma cells rapidly spread and replace normal cells in the bone marrow with tumors. In 2022, it is estimated that more than 34,000 people will be diagnosed with multiple myeloma, and more than 12,000 people will die from the disease in the U.S.5 While some people diagnosed with multiple myeloma initially have no symptoms, most patients are diagnosed due to symptoms that can include bone fracture or pain, low red blood cell counts, tiredness, high calcium levels, kidney problems or infections.2
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The most common adverse reactions include pyrexia, cytokine release syndrome, hypogammaglobulinemia, musculoskeletal pain, fatigue, infections, diarrhea, nausea, encephalopathy, headache, coagulopathy, constipation, and vomiting.[2]
Ciltacabtagene autoleucel is a B-cell maturation antigen (BCMA)-directed genetically modified autologous chimeric antigen receptor (CAR) T-cell therapy.[1][2] Each dose is customized using the recipient’s own T-cells, which are collected and genetically modified, and infused back into the recipient.[1][2]
Ciltacabtagene autoleucel was approved for medical use in the United States in February 2022.[2][3][4]
Medical uses
Ciltacabtagene autoleucel is indicated for the treatment of adults with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.[1][2]
History
The safety and efficacy of ciltacabtagene autoleucel were evaluated in CARTITUDE-1 (NCT03548207), an open label, multicenter clinical trial evaluating ciltacabtagene autoleucel in 97 participants with relapsed or refractory multiple myeloma who received at least three prior lines of therapy which included a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody and who had disease progression on or after the last chemotherapy regimen; 82% had received four or more prior lines of antimyeloma therapy.[1][2]
To treat seizures in cyclin-dependent kinase-like 5 deficiency disorder
Ganaxolone, sold under the brand name Ztalmy, is a medication used to treat seizures associated with cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD).[1][2]
Ganaxolone was approved for medical use in the United States in March 2022.[1]
Ganaxolone is the 3β-methylated synthetic analog of allopregnanolone; it belongs to a class of compounds referred to as neurosteroids. Ganaxolone is an allosteric modulator of GABAA receptors acting through binding sites which are distinct from the benzodiazepine binding site. It has activity in a broad range of animal models of epilepsy. Ganaxolone has been shown to be well tolerated in adults and children. In early phase II studies, Ganaxolone has been shown to have activity in adult patients with partial-onset seizures and epileptic children with history of infantile spasms. It is currently undergoing further development in infants with newly diagnosed infantile spasms, in women with catamenial epilepsy, and in adults with refractory partial-onset seizures.
Ganaxolone is in phase III clinical studies for the treatment of partial seizures in adults. Phase II clinical trials is ongoing for treatment of uncontrolled seizures in PCDH19 female pediatric epilepsy and Fragile X syndrome.
Ganaxolone was originally developed by CoCensys (aquired by Purdue Pharma). In 2003, Marinus Pharmaceuticals obtained the compound from Purdue Pharma.
In 2015, it was granted as orphan drug designation for the treatment of PCDH19 female epilepsy.
In an embodiment, the disclosure provides a method for using pregnenolone to make 21-OH ganaxolone and other intermediary compounds which are useful for preparing neurosteroid derivatives. The method of making 21-OH ganaxolone is shown below in Route 1.
Route 1
Referring to Route 1, Synthesis of 1-((3S,8R,10S,13S,14S,17S)-3-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethenone :
Pregnenolone (3.17 g, 10 mmol) was dissolved in 30 mL of THF and 5 mL of acetic acid. To it, 10% W/C (0.3 g) was added. The resulting mixture was shaken under 60 psi hydrogen at 60°C overnight. It was filtered through a Celite ® pad and concentrated to give 3.2 g of the desired product (100%). 1 H NMR (400 MHz, CDCl3) δ 3.58 (tt, J = 11.0, 4.8 Hz, 1H), 2.50 (t, J = 9.0 Hz, 1H), 2.19 – 2.11 (m, 2H), 2.09 (s, 3H ), 2.06 – 1.93 (m, 2H), 1.85 – 1.75 (m, 1H), 1.74 -1.50 (m, 6H), 1.47 – 1.04 (m, 9H), 1.04 – 0.82 (m, 2H), 0.79 (s , 3H), 0.72 – 0.61 (m, 1H), 0.58 (d, J = 2.4 Hz, 3H).
[0107] Synthesis of (8R,10S,13S,14S,17S)-l7-acetyl-l0,l3-dimethyltetradecahydro-1H-cyclopenta[a]phenanthren-3(2H)-one:
To a solution of the above product (1-((3S,8R,10S,13S,14S,17S)-3-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethanone, 3.2 g, 10 mmol) in 40 mL of THF and 10 mL of acetic acid was added NaBr (1.03 g, 0.1 eq.). It was cooled in an ice bath and was followed by the dropwise addition of NaOCl (82 mL, 10-15%, 18 eq.) at such a rate that the internal temperature was maintained <40 °C. After addition, it was stirred at room temperature for 2h. Thin layer chromatography (TLC) indicated it was complete. The mixture was diluted with dichloromethane and layers were separated. The organic layer was washed with Na 2 S 2 O 3 (10% aq.), H 2 O, NaHCO 3 (sat.) and NaCl (sat.). Drying over Na 2SO 4 and concentration afforded 3.8 g of the crude product, which was recrystallized from CH 2 Cl 2 /Hex to give 2.57 g of the desired product (81%). 1 H NMR (400 MHz, CDC13): 2.51 (t, 1H), 2.2-2.4 (m, 3H), 2.1-2.2 (m, 1H), 2.10 (s, 3H), 1.98-2.01 (m, 2H) , 1.6-1.7 (m, 4H), 1.55-1.6 (m, 1H), 1.3-1.4 (m, 7H), 1.1-1.2 (m, 2H), 0.99 (s, 3H), 0.95-0.98 (m, 1H), 0.75-0.78 (m, 1H), 0.62 (s, 3H).
Synthesis of 1-((2’R,8R,10S,13S,14S,17S)-10,11-dimethylhexadecahydrospiro[cyclopenta[a]phenanthrene-3,2′-oxiran]-17-yl)ethanone.
Under argon, trimethyl sulfoxonium iodide (2.6 g, 1.7 eq.) and sodium t-butoxide (1.18 g, 1.75 eq.) in DMSO (20 mL) was heated at 65 °C for 2h. After it was cooled to RT, the above di-ketone ((8R, 10S, 13 S, 14S, 17S)-17-acetyl- 10,13 -dimethyl tetradecahy dro-1H-cyclopenta[a]phenanthren-3(2H) -one, 2.2 g, 7 mmol) was added scoop-wise so that the internal temperature was maintained between 25-35 °C. The resulting mixture was stirred at RT for 2h. After TLC indicated it was complete, it was quenched with 30 mL of H 2 O, stirred for 10 min and was kept in fridge overnight. The precipitate was filtered, washed with 20 mL of (4:1 of H 2 O /MeOH), dried to give 94% of the desired product (W = 2.17 g). 1H NMR (400 MHz, CDC13) δ 2.63 (s, 2H), 2.53 (t, J = 8.9 Hz, 1H), 2.20 – 2.13 (m, 1H), 2.11 (s, 3H), 2.10 – 1.95 (m, 2H), 1.87 (dd, J = 13.9, 13.1 Hz, 1H), 1.76 – 1.59 (m, 4H), 1.58 – 1.48 (m, 1H), 1.48 – 1.24 (m, 5H), 1.24 – 1.07 (m, 3H), 1.02 – 0.87 (m, 2H), 0.86 (dd, J = 3.7, 2.2 Hz, 1H), 0.84 (s, 3H), 0.81 – 0.74 (m, 1H), 0.61 (s, 3H).
[0109] Synthesis of 1-((3R,8R,10S,13S,14S,17S)-3-hydroxy-3,10,13-trimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethanone (ganaxolone) .
To a solution of the above epoxide (1.5 g, 4.56 mmol) in 15 mL of THF and 15 mL of MeOH were added Nal (1.02 g, 1.5 eq.) and HO Ac (0.6 mL, 2.2 eq.). The resulting mixture was heated at 65°C for 2h. After TLC indicated that the epoxy was completely converted to an iodo compound, it was cooled to RT. Sodium acetate (1.02 g, 2.7 eq.) and 150 mg of 10% Pd/C were added and the mixture was transferred to a hydrogenation bottle with the aid of MeOH (10 mL) and was hydrogenated under 50 psi hydrogen over the weekend. It was filtered throughCelite ® and the filtrate was concentrated. The residue was then partitioned between dichloromethane and water. The aqueous solution was extracted twice with CH 2 Cl 2 and the combined organic layers were washed with brine, dried over Na 2 SO 4 and concentrated. The Biotage flash purification with 10-35% EtOAc in hexane to give 0.5 g of the desired product (33%).
The synthesis was repeated with 1.1 g of the epoxy and 1 g of the product was obtained (90%).
Both lots of product were combined and recrystallized with CH 2 Cl 2 and hexane to give 0.522 g of the product with 96.6% purity by HPLC. 1 H NMR (400 MHz, Chloroform-d) δ 2.51 (t, J = 8.9 Hz, 1H), 2.18 – 2.10 (m, 1H), 2.09 (s, 3H), 2.01 – 1.93 (m, 1H), 1.72 – 1.57 (m, 4H), 1.57 – 1.41 (m, 5H), 1.41 – 1.30 (m, 3H), 1.30 – 1.20 (m, 3H), 1.18 (s, 3H), 1.17 – 1.09 (m, 2H) , 1.00 – 0.85 (m, 1H), 0.78 (ddd, J = 10.6, 7.7, 5.4 Hz, 1H), 0.73 (d, J = 0.6 Hz, 3H), 0.58 (s, 3H). UV: Absorbances at 206.2 nm. TLC: (Silica Gel plates) 20% EtOAc/Hexane; R f = 0.50. HPLC: Sunfire C18 5m 250 x 4.6mm; flow 1.0 mL/min; Waters 996 PDA detection at 210 nm; solvent 80% Acetonitrile in H 2 O (0.1% formic acid) over 30 min; retention time 8.24 min; 96.6%.
Two naturally occurring metabolites of progesterone, 3α-hydroxy-5α- and 5β-pregnan-20-one (1 and 2), are potent allosteric modulators of the GABAA receptor. Their therapeutic potential as anxiolytics, anticonvulsants, and sedative/hypnotics is limited by rapid metabolism. To avoid these shortcomings, a series of 3β-substituted derivatives of 1 and 2 was prepared. Small lipophilic groups generally maintain potency in both the 5α- and 5β-series as determined by inhibition of [35S]TBPS binding. In the 5α-series, 3β-ethyl, -propyl, -trifluoromethyl and -(benzyloxy)methyl, as well as substituents of the form 3β-XCH2, where X is Cl, Br, or I or contains unsaturation, show limited efficacy in inhibiting [35S]TBPS binding. In the 5β-series, the unsubstituted parent 2 is a two-component inhibitor, whereas all of the 3β-substituted derivatives of 2 inhibit TBPS via a single class of binding sites. In addition, all of the 3-substituted 5β-sterols tested are full inhibitors of [35S]TBPS binding. Electrophysiological measurements using α1β2γ2L receptors expressed in oocytes show that 3β-methyl- and 3β-(azidomethyl)-3α-hydroxy-5α-pregnan-20-one (6 and 22, respectively) are potent full efficacy modulators and that 3α-hydroxy-3β-(trifluoromethyl)-5α-pregnan-20-one (24) is a low-efficacy modulator, confirming the results obtained from [35S]TBPS binding. These results indicate that modification of the 3β-position in 1 and 2 maintains activity at the neuroactive steroid site on the GABAA receptor. In animal studies, compound 6 (CCD 1042) is an orally active anticonvulsant, while the naturally occurring progesterone metabolites 1 and 2 are inactive when administered orally, suggesting that 3β-substitution slows metabolism of the 3-hydroxyl, resulting in orally bioavailable steroid modulators of the GABAA receptor.
Addition of the sulfur ylide generated from trimethylsulfoxonium iodide and NaH to the 20-ethylene ketal of pregnane-3,20-dione (I) furnished the spiro oxirane derivative (II). This was reduced to the tertiary alcohol (III) by means of LiAlH4 in refluxing THF. Then, acid hydrolysis of the ethylene ketal function of (III) provided the title compound. Alternatively, the intermediate ketal (III) was prepared by addition of methylmagnesium bromide to ketone (I), followed by chromatographic separation of the resultant mixture of 3-alpha and 3-beta methyl adducts.
Starting from the unprotected diketone (IV), selective addition of dimethyloxosulfonium methylide to the 3 keto group furnished oxirane (V). This was then reduced to the title alcohol by treatment with tributylstannyl hydride and AIBN.
Regioselective addition of dimethylsulfoxonium methylide to 5-alpha-pregnane-3,20-dione (I) gave the epoxide (II). Opening of the epoxide ring of (II) with sodium methoxide produced the hydroxy ether (III). Bromination of (III) with Br2 in the presence of a catalytic amount of HBr afforded bromo ketone (IV). This was then condensed with imidazole (V) in refluxing acetonitrile to furnish the title compound.
Regioselective addition of dimethylsulfoxonium methylide to 5-alpha-pregnane-3,20-dione (I) gave the epoxide (II). Opening of the epoxide ring of (II) with sodium methoxide produced the hydroxy ether (III). Bromination of (III) with Br2 in the presence of a catalytic amount of HBr afforded bromo ketone (IV). This was then condensed with 6-hydroxyquinoline (V) in the presence of potassium tert-butoxide to furnish the quinolinyl ether (VI). The quinoline ring was then oxidized with m-chloroperbenzoic acid, yielding the title N-oxide.
3. WO9318053A1.
4. WO9427608A1.
WO2011019821A2 / US8362286B2.
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The exact mechanism of action for ganaxolone is unknown; however, results from animal studies suggest that it acts by blocking seizure propagation and elevating seizure thresholds.[3][4]
Ganaxolone is thought to modulate both synaptic and extrasynaptic GABAA receptors to normalize over-excited neurons.[2] Ganaxolone’s activation of the extrasynaptic receptor is an additional mechanism that provides stabilizing effects that potentially differentiates it from other drugs that increase GABA signaling.[2]
The most common adverse events reported across clinical trials have been somnolence (sleepiness), dizziness, and fatigue.[5] In 2015, the MIND Institute at the University of California, Davis, announced that it was conducting, in collaboration with Marinus Pharmaceuticals, a randomized, placebo-controlled, Phase 2 clinical trial evaluating the effect of ganaxolone on behaviors associated with Fragile X syndrome in children and adolescents.[9][10][11]
^ Jump up to:abcdefCarter RB, Wood PL, Wieland S, Hawkinson JE, Belelli D, Lambert JJ, White HS, Wolf HH, Mirsadeghi S, Tahir SH, Bolger MB, Lan NC, Gee KW (March 1997). “Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3alpha-hydroxy-3beta-methyl-5alpha-pregnan-20-one), a selective, high-affinity, steroid modulator of the gamma-aminobutyric acid(A) receptor”. The Journal of Pharmacology and Experimental Therapeutics. 280 (3): 1284–95. PMID9067315.
^ Jump up to:abcKaminski RM, Livingood MR, Rogawski MA (July 2004). “Allopregnanolone analogs that positively modulate GABA receptors protect against partial seizures induced by 6-Hz electrical stimulation in mice”. Epilepsia. 45 (7): 864–7. doi:10.1111/j.0013-9580.2004.04504.x. PMID15230714. S2CID21974013.
^Reddy DS, Rogawski MA (December 2000). “Chronic treatment with the neuroactive steroid ganaxolone in the rat induces anticonvulsant tolerance to diazepam but not to itself”. The Journal of Pharmacology and Experimental Therapeutics. 295 (3): 1241–8. PMID11082461.
Crenolanib besylate CAS#: 670220-93-6 (besylate) Chemical Formula: C32H35N5O5S
Molecular Weight: 601.72
Crenolanib besylate (CP-868,596-26 or AR-868,596-26, 4-piperidinamine, 1-[2-[5-[(3-Methyl-3-oxetanyl) methoxy]-1H-benzimidazol-1-yl]- 8-quinolinyl]-, monobenzenesulfonate) is an investigational inhibitor being developed by AROG Pharmaceuticals, LLC. The compound is currently being evaluated for safety and efficacy in clinical trials for various types of cancer, including acute myeloid leukemia (AML),[1][2]gastrointestinal stromal tumor (GIST),[3] and glioma.[4] Crenolanib is an orally bioavailable benzamidazole that selectively and potently inhibits signaling of wild-type and mutant isoforms of class III receptor tyrosine kinases (RTK) FLT3 (FMS-like Tyrosine Kinase 3), PDGFR α (Platelet-Derived Growth Factor Receptor), and PDGFR β. Unlike most RTK inhibitors, crenolanib is a type I mutant-specific inhibitor that preferentially binds to phosphorylated active kinases with the ‘DFG in’ conformation motif.[5]
CN 109678849
PATENT
WO/2022/060421CRENOLANIB FOR TREATING TRK KINASE ASSOCIATED PROLIFERATIVE DISORDERS
The synthesis of the chloro- 8- trifluoromethanesulfonic acids base quinoline (Ι) of 2-
Compound 2- chloro-8-hydroxyquinolines 50g, DMF150ml, trifluoromethanesulfchloride chloride 53g, triethylamine 25g are added to 250ml In three-necked bottle, stir.Temperature control reacts 20~30h at 25~30 DEG C.After reaction completely, the solid of precipitation is filtered, filter cake is used Wash washing, 40 DEG C of forced air dryings, the chloro- 8- trifluoromethanesulfonic acids base benzimidazoles of gained off-white powder 2- in n-hexane 20ml × 3 83.39g yield 95.78%.
The synthesis of (base of piperidines -4) the quinoline t-butyl carbamates of 2- chloro- 8 (II)
BINAP 0.2g, toluene 70ml are added into 250ml three-necked bottles, temperature control stirs 1h at 20~25 DEG C.Added again into bottle The chloro- 8- trifluoromethanesulfonic acids base quinoline 10g of 2-, piperidin-4-yl t-butyl carbamate 6.41g, potassium carbonate 7.8g, stir lower by instead Answer liquid to be heated to 80 DEG C~100 DEG C, keep 20~30h of this thermotonus.TLC is detected, and whether reaction is complete.Reaction is complete, stops Only heat.20~30 DEG C are cooled to, dichloroethanes 50ml is added, adds diatomite to filter out the solid in reaction solution, filter cake second Acetoacetic ester 150ml is washed., 20~25 DEG C of stirring 8h.The solid separated out in solution is filtered out, filtrate is molten with 5% disodium hydrogen phosphate Liquid 2x50ml is washed.Organic phase is concentrated to dryness again, adds acetonitrile 50ml, 20~25 DEG C of 10~20h of stirring and crystallizing.Filtering analysis The solid gone out, 40 DEG C of forced air dryings obtain the tertiary fourth of yellow solid 10.69g, 2- chloro- 8 (piperidin-4-yl) benzimidazole carbamic acid Ester, yield 92.3%.
The synthesis of 5- (3- methy oxetane -3- methoxyl groups) benzimidazole (III)
Compound 3- methyl -3- oxetane methanols 30.77g, THF140ml, metallic sodium 6.95g are added to the necks of 250ml tri- In bottle, 66 DEG C of backflow 4h are heated under stirring, 55 DEG C is cooled to, then adds 5- hydroxybenzimidazole 40.4g, stir lower heat Backflow, react 20~24h.
Ethyl acetate 100ml is added into reaction bulb, 0.5h dissolvings are stirred at 30~50 DEG C, are then reduced to -5 DEG C, are added dropwise just Hexane 30ml, stirs 1h, and suction filtration obtains light yellow solid, 40 DEG C of dryings to constant weight, obtains 56.41 grams, yield 85.8%.
(1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline-8-yl }-piperazine The synthesis of pyridine -4- bases-t-butyl carbamate (IV)
II (50 grams), III (30.14 grams), potassium carbonate 80g, DIPHOS 4.3g, toluene 700ml, are added in 2L three-necked bottles, add Enter acid chloride 0.9g, stir.Stirring is lower to heat up, and temperature control reacts 24~30h at 80~100 DEG C.After the completion of reaction, it is cooled to 55 DEG C add dichloroethanes 700ml.10min is stirred, adds the solid in diatomite filtering reacting liquid, the filter cake chloroethenes of 500ml bis- Alkane rinses.Concentrate the filtrate to it is dry, add ethyl acetate 480ml, be heated to flowing back, be cooled to 20~25 DEG C of 10~20h of crystallization. The solid separated out is filtered, 50 DEG C of forced air dryings, obtains white solid, the amount of obtaining 70.51g, yield 93.90%.
(1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline-8-yl } -4- The synthesis of amino piperidine (V)
By compound (1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline -8- Base }-piperidin-4-yl-t-butyl carbamate 5g, caustic alcohol 2.8g, 2- methyltetrahydrofuran 30ml and water 0.08ml be added to In 100ml three-necked bottles, stir.The mixture is heated to flowing back, and stirs 3~4h under reflux.
TLC is detected, and reaction is complete.Stop heating, add purified water 60ml, extracting and demixing.Aqueous phase is extracted with 2 × 20ml of ethyl acetate Take, merge organic phase, washed with saturated nacl aqueous solution 20ml.Be concentrated under reduced pressure organic phase, and 30ml is added into condensate residue Ethyl acetate, in 20~25 DEG C of stirring and crystallizing 6h.The solid separated out is filtered, filtrate decompression is concentrated to dryness.Added into residue 24ml ethyl acetate, in 20~25 DEG C of 10~12h of stirring and crystallizing.Filter the solid separated out, dry white solid product, the amount of obtaining 3.68g, yield 90.3%.
Mutations of FLT3 comprise one of the most frequently identified types of genetic alterations in Angiomyolipoma.[7][8] Approximately one-third of AML patients present with a mutation in this gene.[9] The majority of these mutations result in constitutive activation of downstream signaling pathways and aberrant cell growth.[7] Mutations in FLT3 have also been reported in acute lymphoblastic leukemia (ALL)[10] and myelodysplastic syndrome (MDS).[11]
Crenolanib inhibits both wild type FLT3 and its constitutively active mutations. In vitro studies have shown that crenolanib has low Kd for the FLT3 enzyme with constitutively activating internal tandem duplication (ITD) mutations and tyrosine kinase domain (TKD) mutations, D835H and D835Y, as compared to wild type. Crenolanib tightly binds to FLT3-ITD, FLT3-D835H and FLT3-D835Y with Kd of 0.74 nM, 0.4 nM, and 0.18 nM, respectively.[14] Crenolanib inhibits the phosphorylation of the FLT3-ITD receptor in transfected TF-1 cells and the FLT3-D835Y TKD mutation in transfected Ba/F3 cells at nanomolar IC50 concentrations of 1.3 nM and 8.8 nM, respectively.[15]Immunoblot experiments performed in the Molm14FLT3-ITD positive cell line show that crenolanib inhibits downstream signaling of FLT3 at a concentration of 10 nM.[15]MTT assay measurements of crenolanib cytotoxicity evaluated in the FLT3-ITD expressing cell lines Molm14 and MV411, showed that crenolanib is toxic at IC50 concentrations of 7 nM and 8 nM, respectively.[15]
PDGFRα: wild-type and mutant
Crenolanib has been shown to inhibit PDGFRα with an IC50 of 0.4 ng/mL in porcine aortic epithelial cell lines. In Chinese hamster ovary (CHO) cells expressing PDGFRα, crenolanib inhibited the phosphorylation of wild type PDGFRα at an IC50 of 10 nM.[16] Additionally, crenolanib completely blocked PDGFRα phosphorylation and downstream AKT signaling at a concentration between 0.1 and 1 uM in Ink4a/Arf-/- mouse astrocytes transfected to stably co-express both human PDGFRα and PDGF AA.[17] The lung cancer cell line H1703, which is reported to have amplification of both PDGFRA (4q12) and PDGFC (4q32) genes on chromosome 4, and also overexpress PDGFRα, was sensitive to crenolanib with an IC50 of ~80 nM.[18] In CHO cells expressing an activating exon 18 (D842V) PDGFRα mutation, crenolanib was effective at an IC50 of 6nM and IC90 of 25nM. In addition, crenolanib also inhibited phosphorylation of the double mutants PDGFRα (V561D + D842V and T674I + D842V).[16]
PDGFRβ: wild-type
Crenolanib has been shown to inhibit PDGFRβ with an IC50 of 0.8 ng/mL in porcine aortic epithelial cell lines. Crenolanib inhibits the ability of recombinant PDGFRβ to phosphorylate a synthetic tyrosine substrate (poly-glutamic acid-tyrosine), with an IC50 of 0.4 ng/mL. Evaluation of the antitumor activity of crenolanib in a genetically engineered BSG DIPG mouse model showed that it is highly selective for PDGFRβ with an IC50 of 10 nM when measured by BrdU assay and 1.25 uM by MTT assay.
C-Kit: wild-type and mutant
Crenolanib has been shown to have IC50 and Kd values of 67 nM and 78 nM, respectively, for wild type c-KIT in in vitro assays[citation needed]. Similar assays show that crenolanib inhibits c-KIT activating mutations D816H and D816V with IC50 concentrations of 5.4 and 2.5 nM, respectively.[14][citation needed] Human bone marrowprogenitor cell growth assays showed that crenolanib has modest effects on GM-CSF and BFUE driven colony formation at the IC50 concentration of 20 nM.[15]
Clinical
Phase I single-agent[19] and Phase Ib combination[20] studies have investigated the clinical pharmacology of crenolanib in patients with cancer. Pharmacokinetic and safety studies of Crenolanib administered alone or in combination with docetaxel with or without axitinib have been completed. Results suggest that Crenolanib is well tolerated as a single agent, and can also be safely combined with docetaxel and axitinib due to their non-overlapping toxicity profiles.
Clinical trials
Clinical trial number NCT01229644 for “A Phase II Study of Crenolanib (CP-868,596), a Selective and Potent Inhibitor of PDGFR, for the Treatment of Adult Gliomas” at ClinicalTrials.gov
Clinical trial number NCT01243346 for “Phase II Study of Crenolanib (CP-868,596), for the Treatment of Patients With Advanced Gastrointestinal Stromal Tumors With the D842-related Mutations and Deletions in the PDGFRA Gene” at ClinicalTrials.gov
Clinical trial number NCT01393912 for “PDGFR Inhibitor Crenolanib in Children/Young Adults With Diffuse Intrinsic Pontine Glioma or Recurrent High-Grade Glioma” at ClinicalTrials.gov
Clinical trial number NCT01522469 for “Phase II Study of Crenolanib in Subjects With Relapsed/Refractory AML With FLT3 Activating Mutations” at ClinicalTrials.gov
Clinical trial number NCT01657682 for “A Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients With FLT3 Activating Mutations” at ClinicalTrials.gov
^ Cancer Genome Atlas Research Network; Ley, T. J.; Miller, C.; Ding, L.; Raphael, B. J.; Mungall, A. J.; Robertson, A.; Hoadley, K.; Triche Jr, T. J.; Laird, P. W.; Baty, J. D.; Fulton, L. L.; Fulton, R.; Heath, S. E.; Kalicki-Veizer, J.; Kandoth, C.; Klco, J. M.; Koboldt, D. C.; Kanchi, K. L.; Kulkarni, S.; Lamprecht, T. L.; Larson, D. E.; Lin, L.; Lu, C.; McLellan, M. D.; McMichael, J. F.; Payton, J.; Schmidt, H.; Spencer, D. H.; et al. (2013). “Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia”. New England Journal of Medicine. 368 (22): 2059–2074. doi:10.1056/NEJMoa1301689. ISSN0028-4793. PMC3767041. PMID23634996.
^ Jump up to:ab Muralidhara, C.; Ramachandran, A.; Jain, V. K. (2012). “Abstract 3683: Crenolanib, a novel Type I, mutant-specific inhibitor of Class III receptor tyrosine kinases, preferentially binds to phosphorylated kinases”. Cancer Research. 72 (8 Supplement): 3683. doi:10.1158/1538-7445.AM2012-3683.
^ Jump up to:abcd Galanis, A.; Rajkhowa, T.; Muralidhara, C.; Ramachandran, A.; Levis, M. (2012). “Abstract 3660: Crenolanib: A next generation FLT3 inhibitor”. Cancer Research. 72 (8 Supplement): 3660. doi:10.1158/1538-7445.am2012-3660.
^ Yang, X.-L.; Mashimo, T.; Su, Y.; Vemireddy, V.; Guntipalli, P.; Ramachandran, A.; Chaudhary, P.; Mickey, B.; Hatanpaa, K.; Maher, E.; Bachoo, R. M. (2011). “Abstract 1111: Preclinical evaluation of CP868,596, a novel PDGFR Inhibitor for treatment of glioblastoma”. Cancer Research. 71 (8 Supplement): 1111. doi:10.1158/1538-7445.am2011-1111.
^ Peyton, M.; Chaudhary, P.; Ramachandran, A.; Minna, J. (2011). “Abstract 3601: CP-868,596, a highly potent and selective PDGFR TKI inhibits growth of PDGFR -driven lung cancer cells”. Cancer Research. 71 (8 Supplement): 3601. doi:10.1158/1538-7445.am2011-3601.
LutetiumLu 177Vipivotide Tetraxetan is a radioconjugate composed of PSMA-617, a human prostate-specific membrane antigen (PSMA)-targeting ligand, conjugated to the beta-emitting radioisotope lutetiumLu 177 (177Lu), with potential antineoplastic activity against PSMA-expressing tumor cells. Upon intravenous administration of lutetiumLu 177vipivotide tetraxetan, vipivotide tetraxetan targets and binds to PSMA-expressing tumor cells. Upon binding, PSMA-expressing tumor cells are destroyed by 177Lu through the specific delivery of beta particle radiation. PSMA, a tumor-associated antigen and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells.
The most common adverse reactions include fatigue, dry mouth, nausea, anemia, decreased appetite, and constipation.[2]
Lutetium (177Lu) vipivotide tetraxetan is a radioconjugate composed of PSMA-617, a human prostate-specific membrane antigen (PSMA)-targeting ligand, conjugated to the beta-emitting radioisotope lutetium Lu 177 (177Lu), with potential antineoplastic activity against PSMA-expressing tumor cells.[4] Upon intravenous administration of lutetium Lu 177 vipivotide tetraxetan, vipivotide tetraxetan targets and binds to PSMA-expressing tumor cells.[4] Upon binding, PSMA-expressing tumor cells are destroyed by 177Lu through the specific delivery of beta particle radiation.[4] PSMA, a tumor-associated antigen and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells.[4]
Lutetium (177Lu) vipivotide tetraxetan was approved for medical use in the United States in March 2022.[2][5]
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Efficacy was evaluated in VISION (NCT03511664), a randomized (2:1), multicenter, open-label trial that evaluated lutetium (177Lu) vipivotide tetraxetan plus best standard of care (BSoC) (n=551) or BSoC alone (n=280) in men with progressive, prostate-specific membrane antigen (PSMA)-positive metastatic castration-resistant prostate cancer (mCRPC).[2] All participants received a GnRH analog or had prior bilateral orchiectomy.[2] Participants were required to have received at least one androgen receptor pathway inhibitor, and 1 or 2 prior taxane-based chemotherapy regimens.[2] Participants received lutetium (177Lu) vipivotide tetraxetan 7.4 GBq (200 mCi) every 6 weeks for up to a total of 6 doses plus BSoC or BSoC alone.[2]
The U.S. Food and Drug Administration granted the application for lutetium (177lu) vipivotide tetraxetan priority review and breakthrough therapy designations.[2]
Percent Composition: C 67.30%, H 6.63%, F 4.63%, N 13.65%, O 7.80%
Literature References: Combined serotonin (5-HT2) and dopamine (D2) receptor antagonist. Prepn: L. E. J. Kennis, J. Vandenberk, EP196132; eidem,US4804663 (1986, 1989 both to Janssen). Pharmacology: P. A. J. Janssen et al.,J. Pharmacol. Exp. Ther.244, 685 (1988). Receptor binding studies: J. E. Leysen et al.,ibid.247, 661 (1988). HPLC determn in plasma: A. Avenoso et al.,J. Chromatogr. B746, 173 (2000). Clinical study in psychoses: Y. G. Gelders et al.,Pharmacopsychiatry23, 206 (1990); in autism: L. Scahill et al., N. Engl. J. Med.347, 314 (2002). Brief review: M. G. Livingston, Lancet343, 457-460 (1994). Review of pharmacology and therapeutic potential: S. Grant, A. Fitton, Drugs48, 253-273 (1994); B. Green, Curr. Med. Res. Opin.16, 57-65 (2000); of clinical experience in schizophrenia: H.-J. Möller, Expert Opin. Pharmacother.6, 803-818 (2005),
Risperidone, sold under the brand name Risperdal among others, is an atypical antipsychotic[2] used to treat schizophrenia and bipolar disorder.[2] It is taken either by mouth or by injection (subcutaneous or intramuscular).[2] The injectable versions are long-acting and last for 2-4 weeks.[6]
The Friedel-Crafts condensation of 1,3-difluorobenzene (I) with 1-acetylpiperidine-4-carbonyl chloride (II) by means of AlCl3 in dichloromethane gives 1-acetyl-4-(2,4-difluorobenzoyl)piperidine (III), which is hydrotyzed with refluxing 6N HCl to yield 4-(2,4-difluorobenzoyl)piperidine (IV). The reaction of (IV) with hydroxylamine in refluxing ethanol affords the corresponding oxime (V), which is cyclized by means of KOH in boiling water giving 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole (VI). Finally, this compound is condensed with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (VII) by means of K2CO3 and Kl in a variety of solvents.
SYN
ES 2050069
The intermediate 3-(2-chloroethyl)-2-methyl-6, 7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (V) has been obtained as follows: The cyclization of 2-aminopyridine (I) with 3-acetyltetrahydrofuran-2-one (II) by means of polyphosphoric acid (PPA) at 160 C gives 3-(2-hydroxyethyl)-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (III), which is hydrogenated with H2 over Pd/C in ethanol/water to yield the tetrahydro derivative (IV). Finally, the OH group of (IV) is treated with SOCl2 in dichloromethane to afford the target 2-chloroethyl intermediate (V).
SYN
The condensation of piperidine-4-carboxylic acid (VI) with ethyl chloroformate (VII) by means of Na2CO3 in toluene/water gives 1-(ethoxycarbonyl)piperidine-4-carboxylic acid (VIII), which is treated with SOCl2 to yield the corresponding acyl chloride (IX). The Friedel-Crafts condensation of (IX) with refluxing 1,3-difluorobenzene (X) by means of AlCl3 gives 4-(2,4-difluorobenzoyl)piperidine-1-carboxylic acid ethyl ester (XI), which is treated with concentrated HCl at 100 C to yield 4-(2,4-difluorobenzoyl)piperidine (XII). The condensation of piperidine (XII) with the 2-chloroethyl intermediate (V) by means of KI and NaHCO3 in refluxing acetonitrile affords the adduct (XIII), which is treated with hydroxylamine hydrochloride and KOH in refluxing pyridine/ethanol to provide the corresponding oxime (XIV). Finally, this compound is cyclized by means of KOH in refluxing water or with NaH in refluxing THF to afford in both cases the target 1,2-benzisoxazole.
SYN
The intermediate 3-(2-aminoethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (IV) has been obtained as follows: The condensation of 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (I) with dibenzylamine (II) by means of NaHCO3 in refluxing acetonitrile gives the tertiary amine (III), which is debenzylated by hydrogenation with H2 over Pd/C in warm ethanol to afford the target intermediate (IV).
SYN
The condensation of tetrahydropyran-4-carbonyl chloride (V) with refluxing 1,3-difluorobenzene (VI) by means of AlCl3 gives 1-(2,4-difluorophenyl)-1-(tetrahydropyran-4-yl)methanone (VII), which is treated with hydroxylamine hydrochloride and sodium acetate in refluxing ethanol/water to yield the corresponding oxime (VIII). The cyclization of (VIII) by means of KOH in refluxing methanol affords 6-fluoro-3-(tetrahydropyran-4-yl)-1,2-benzisoxazole (IX), which is treated with NaI and Ac-Cl and then with K2CO3 in refluxing acetonitrile to provide the 5-iodopentanol derivative (X). The reaction of the OH group of (X) with Ms-Cl and TEA in dichloromethane gives the corresponding mesylate (XI), which is finally cyclized with the intermediate amine (IV) by means of NaHCO3 in refluxing acetonitrile to yield the target piperidine.
SYN
SYN
Eur. Pat. Appl. 196132
SYN
Production Route of Risperidone
(CAS NO.: ), with other name of 4H-Pyrido(1,2-a)pyrimidin-4-one, 6,7,8,9-tetrahydro-3-(2-(4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl)ethyl)-2-methyl-, could be produced through many synthetic methods.Following is one of the synthesis routes: The Friedel-Crafts condensation of 1,3-di (I) with 1-acetylpiperidine-4-carbonyl chloride (II) by means of AlCl3 in dichloromethane gives 1-acetyl-4-(2,4-difluorobenzoyl)piperidine (III), which is hydrotyzed with refluxing 6N HCl to yield 4-(2,4-difluorobenzoyl)piperidine (IV). The reaction of (IV) with hydroxylamine in refluxing ethanol affords the corresponding oxime (V), which is cyclized by means of KOH in boiling water giving 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole (VI). Finally, this compound is condensed with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (VII) by means of K2CO3 and Kl in a variety of solvents.
Risperidone (7.2.1) (Risperdal) is the first second-generation antipsychotic that was specifically designed as a combined D2 and serotonin 5-HT(2A) receptor antagonist, thus following the pharmacological mechanism thought to be responsible for the antipsychotic effects. After its advent in the 1990s as the first novel second-generation antipsychotic, risperidone has achieved worldwide acceptance. It was initially approved for use in schizophrenia, mania of bipolar disorder, and irritability and aggression of autism. But it is also effectively used in other instances of psychosis, including schizoaffective disorder, depression with psychotic features, and psychosis secondary to general medical conditions. Risperidone may be effective in other conditions such as major depression, various anxiety disorders, delirium, dementia, for Alzheimer’s dementia, which occurs in 6–8% of persons older than 65 and increases to 30% among those 85 years or older, and substance abuse disorders [84–113].
Risperidone is proposed for inclusion in the WHO Model List of Essential Medications for treatment of schizophrenia, mania, and autism.
Risperidone (7.2.1) was synthesized starting from 1-acetyl-4-piperidine-carbonyl chloride (7.2.4), which was used to acylate 1,3-difluorobenzene (7.2.5) in dichloromethane using aluminum chloride as Lewis acid. The reaction gave 1-(4-(2,4-difluorobenzoyl)piperidin-1-yl)ethan-1-one (7.2.6). The protecting acetyl group of the last was removed off by hydrolysis in 6 N hydrochloric acid on reflux, which gave (2,4-difluorophenyl)(piperidin-4-yl)methanone (7.2.7). The obtained product was converted further to corresponding oxime (7.2.8) on reaction with hydroxylamine hydrochloride in ethanol in the presence of N,N-diethylenethanamine. Synthesized oxime (7.2.8) was cyclized to 6-fluoro-3-(piperidin-4-yl)benzo[d]isoxazole (7.2.9) on reflux with 50% potassium hydroxide solution in water. At the final stage the obtained product (7.2.9) was alkylated with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (7.2.10) on heating at 85–90°C in dimethylformamide in the presence of sodium carbonate and potassium iodide, which gave the desired product, risperidone (7.2.1) [114,115]. Later, another method of (7.2.7) → (7.2.1) transformation was proposed, which involved the reductive alkylation of (2,4-difluorophenyl)(piperidin-4-yl)methanone (7.2.7) with aldehyde (7.2.11) and sodium cyanoborohydride, which gave compound (7.2.12), coherently converted to oxime (7.2.13) and further to the desired compound, risperidone (7.2.1) [116] (Scheme 7.7).
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Studies evaluating the utility of risperidone by mouth for maintenance therapy have reached varying conclusions. A 2012 systematic review concluded that evidence is strong that risperidone is more effective than all first-generation antipsychotics other than haloperidol, but that evidence directly supporting its superiority to placebo is equivocal.[15] A 2011 review concluded that risperidone is more effective in relapse prevention than other first- and second-generation antipsychotics with the exception of olanzapine and clozapine.[16] A 2016 Cochrane review suggests that risperidone reduces the overall symptoms of schizophrenia, but firm conclusions are difficult to make due to very low-quality evidence. Data and information are scarce, poorly reported, and probably biased in favour of risperidone, with about half of the included trials developed by drug companies. The article raises concerns regarding the serious side effects of risperidone, such as parkinsonism.[17] A 2011 Cochrane review compared risperidone with other atypical antipsychotics such as olanzapine for schizophrenia:[18]
Summary
Risperidone seems to produce somewhat more extrapyramidal side effects and clearly more prolactin increase than most other atypical antipsychotics. It may also differ from other compounds in the occurrence of other adverse effects such as weight gain, metabolic problems, cardiac effects, sedation, and seizures. Nevertheless, the large proportion of participants leaving studies early and incomplete reporting of outcomes makes drawing firm conclusions difficult.[18]
showOutcomeFindings in wordsFindings in numbersQuality of evidence
Long-acting injectable formulations of antipsychotic drugs provide improved compliance with therapy and reduce relapse rates relative to oral formulations.[19][20] The efficacy of risperidone long-acting injection appears to be similar to that of long acting injectable forms of first generation antipsychotics.[21]
Bipolar disorder
Second-generation antipsychotics, including risperidone, are effective in the treatment of manic symptoms in acute manic or mixed exacerbations of bipolar disorder.[22][23][24] In children and adolescents, risperidone may be more effective than lithium or divalproex, but has more metabolic side effects.[25] As maintenance therapy, long-acting injectable risperidone is effective for the prevention of manic episodes but not depressive episodes.[26] The long-acting injectable form of risperidone may be advantageous over long acting first generation antipsychotics, as it is better tolerated (fewer extrapyramidal effects) and because long acting injectable formulations of first generation antipsychotics may increase the risk of depression.[27]
Autism
Compared to placebo, risperidone treatment reduces certain problematic behaviors in autistic children, including aggression toward others, self-injury, challenging behaviour, and rapid mood changes.[28] The evidence for its efficacy appears to be greater than that for alternative pharmacological treatments.[29] Weight gain is an important adverse effect.[4][30] Some authors recommend limiting the use of risperidone and aripiprazole to those with the most challenging behavioral disturbances in order to minimize the risk of drug-induced adverse effects.[31] Evidence for the efficacy of risperidone in autistic adolescents and young adults is less persuasive.[32]
Risperidone has not demonstrated a benefit in the treatment of eating disorders or personality disorders, except for limited evidence in schizotypal personality disorder.[34]
While antipsychotic medications such as risperidone have a slight benefit in people with dementia, they have been linked to higher incidence of death and stroke.[34] Because of this increased risk of death, treatment of dementia-related psychosis with risperidone is not FDA approved and carries a black box warning.[4]
Forms
Available forms of risperidone include tablet, oral dissolving tablet, oral solution, and powder and solvent for suspension for injection.[35]
While atypical antipsychotics appear to have a lower rate of movement problems as compared to typical antipsychotics, risperidone has a high risk of movement problems among the atypicals.[36][37] Atypical antipsychotics however are associated with a greater amount of weight gain.[37]
Drug interactions
Carbamazepine and other enzyme inducers may reduce plasma levels of risperidone.[4] If a person is taking both carbamazepine and risperidone, the dose of risperidone will likely need to be increased. The new dose should not be more than twice the patient’s original dose.[4]
CYP2D6 inhibitors, such as SSRI medications, may increase plasma levels of risperidone and those medications.[4]
Since risperidone can cause hypotension, its use should be monitored closely when a patient is also taking antihypertensive medicines to avoid severe low blood pressure.[4]
The British National Formulary recommends a gradual withdrawal when discontinuing antipsychotic treatment to avoid acute withdrawal syndrome or rapid relapse.[40] Some have argued the additional somatic and psychiatric symptoms associated with dopaminergic super-sensitivity, including dyskinesia and acute psychosis, are common features of withdrawal in individuals treated with neuroleptics.[41][42][43][44] This has led some to suggest the withdrawal process might itself be schizomimetic, producing schizophrenia-like symptoms even in previously healthy patients, indicating a possible pharmacological origin of mental illness in a yet unknown percentage of patients currently and previously treated with antipsychotics. This question is unresolved, and remains a highly controversial issue among professionals in the medical and mental health communities, as well as the public.[45]
Dementia
Older people with dementia-related psychosis are at a higher risk of death if they take risperidone compared to those who do not. Most deaths are related to heart problems or infections.[4]
Risperidone has been classified as a “qualitatively atypical” antipsychotic agent with a relatively low incidence of extrapyramidal side effects (when given at low doses) that has more pronounced serotonin antagonism than dopamine antagonism. Risperidone contains the functional groups of benzisoxazole and piperidine as part of its molecular structure. Although not a butyrophenone, it was developed with the structures of benperidol and ketanserin as a basis. It has actions at several 5-HT (serotonin) receptor subtypes. These are 5-HT2C, linked to weight gain, 5-HT2A, linked to its antipsychotic action and relief of some of the extrapyramidal side effects experienced with the typical neuroleptics.[48]
It has been found that D-amino acid oxidase, the enzyme that catalyses the breakdown of D-amino acids (e.g. D-alanine and D-serine — the neurotransmitters) is inhibited by risperidone.[49]
Risperidone acts on the following receptors:
Dopamine receptors: This drug is an antagonist of the D1 (D1, and D5) as well as the D2 family (D2, D3 and D4) receptors, with 70-fold selectivity for the D2 family. This drug has “tight binding” properties, which means it has a long half-life and like other antipsychotics, risperidone blocks the mesolimbic pathway, the prefrontal cortex limbic pathway, and the tuberoinfundibular pathway in the central nervous system. Risperidone may induce extrapyramidal side effects, akathisia and tremors, associated with diminished dopaminergic activity in the striatum. It can also cause sexual side effects, galactorrhoea, infertility, gynecomastia and, with chronic use reduced bone mineral density leading to breaks, all of which are associated with increased prolactin secretion.[48]
Serotonin receptors: Its action at these receptors may be responsible for its lower extrapyramidal side effect liability (via the 5-HT2A/2C receptors) and improved negative symptom control compared to typical antipsychotics such as haloperidol for instance. Its antagonistic actions at the 5-HT2C receptor may account, in part, for its weight gain liability.[medical citation needed]
Alpha α1 adrenergic receptors: This action accounts for its orthostatic hypotensive effects and perhaps some of the sedating effects of risperidone.[48]
Histamine H1 receptors: effects on these receptors account for its sedation and reduction in vigilance. This may also lead to drowsiness and weight gain.[48]
Voltage-gated sodium channels: Because it accumulates in synaptic vesicles, Risperidone inhibits voltage-gated sodium channels at clinically used concentrations.[51]
Risperidone undergoes hepatic metabolism and renal excretion. Lower doses are recommended for patients with severe liver and kidney disease.[4] The active metabolite of risperidone, paliperidone, is also used as an antipsychotic.[52]
Risperidone was approved by the United States Food and Drug Administration (FDA) in 1993 for the treatment of schizophrenia.[63] In 2003, the FDA approved risperidone for the short-term treatment of the mixed and manic states associated with bipolar disorder. In 2006, the FDA approved risperidone for the treatment of irritability in autistic children and adolescents.[64] The FDA’s decision was based in part on a study of autistic people with severe and enduring problems of violent meltdowns, aggression, and self-injury; risperidone is not recommended for autistic people with mild aggression and explosive behavior without an enduring pattern.[65] On 22 August 2007, risperidone was approved as the only drug agent available for treatment of schizophrenia in youths, ages 13–17; it was also approved that same day for treatment of bipolar disorder in youths and children, ages 10–17, joining lithium.
On 16 December 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Okedi, intended for the treatment of schizophrenia in adults for whom tolerability and effectiveness has been established with oral risperidone.[66] The applicant for this medicinal product is Laboratorios Farmacéuticos Rovi, S.A.[66]
Availability
Janssen’s patent on risperidone expired on 29 December 2003, opening the market for cheaper generic versions from other companies, and Janssen’s exclusive marketing rights expired on 29 June 2004 (the result of a pediatric extension). It is available under many brand names worldwide.[1]
Risperidone is available as a tablet, an oral solution, and an ampule, which is a depot injection.[1]
In November 2013, J&J was fined $2.2 billion for illegally marketing risperidone for use in people with dementia.[70]
In 2015, Steven Brill posted a 15-part investigative journalism piece on J&J in The Huffington Post, called “America’s most admired lawbreaker”, which was focused on J&J’s marketing of risperidone.[71][72]
J&J has faced numerous civil lawsuits on behalf of children who were prescribed risperidone who grew breasts (a condition called gynecomastia); as of July 2016 there were about 1,500 cases in Pennsylvania state court in Philadelphia, and there had been a February 2015 verdict against J&J with $2.5 million awarded to a man from Alabama, a $1.75M verdict against J&J that November, and in 2016 a $70 million verdict against J&J.[73] In October 2019, a jury awarded a Pennsylvania man $8 billion in a verdict against J&J.[74]
Names
Brand names include Risperdal, Risperdal Consta, Risperdal M-Tab, Risperdal Quicklets, Risperlet, Okedi, and Perseris.[75]
^ Jump up to:abcd Hamilton R (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. pp. 434–435. ISBN9781284057560.
^ Jump up to:ab Hasnain M, Vieweg WV, Hollett B (July 2012). “Weight gain and glucose dysregulation with second-generation antipsychotics and antidepressants: a review for primary care physicians”. Postgraduate Medicine. 124 (4): 154–67. doi:10.3810/pgm.2012.07.2577. PMID22913904. S2CID39697130.
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^“Respiridone”. The American Society of Health-System Pharmacists. Archived from the original on 13 April 2011. Retrieved 3 April 2011.
^ Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, et al. (September 2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis”. Lancet. 382 (9896): 951–62. doi:10.1016/S0140-6736(13)60733-3. PMID23810019. S2CID32085212.
^ Osser DN, Roudsari MJ, Manschreck T (2013). “The psychopharmacology algorithm project at the Harvard South Shore Program: an update on schizophrenia”. Harvard Review of Psychiatry. 21 (1): 18–40. doi:10.1097/HRP.0b013e31827fd915. PMID23656760. S2CID22523977.
^ Leucht C, Heres S, Kane JM, Kissling W, Davis JM, Leucht S (April 2011). “Oral versus depot antipsychotic drugs for schizophrenia–a critical systematic review and meta-analysis of randomised long-term trials”. Schizophrenia Research. 127 (13): 83–92. doi:10.1016/j.schres.2010.11.020. PMID21257294. S2CID2386150.
^ Lafeuille MH, Dean J, Carter V, Duh MS, Fastenau J, Dirani R, Lefebvre P (August 2014). “Systematic review of long-acting injectables versus oral atypical antipsychotics on hospitalization in schizophrenia”. Current Medical Research and Opinion. 30 (8): 1643–55. doi:10.1185/03007995.2014.915211. PMID24730586. S2CID24814527.
^ Muralidharan K, Ali M, Silveira LE, Bond DJ, Fountoulakis KN, Lam RW, Yatham LN (September 2013). “Efficacy of second generation antipsychotics in treating acute mixed episodes in bipolar disorder: a meta-analysis of placebo-controlled trials”. Journal of Affective Disorders. 150 (2): 408–14. doi:10.1016/j.jad.2013.04.032. PMID23735211.
^ Nivoli AM, Murru A, Goikolea JM, Crespo JM, Montes JM, González-Pinto A, et al. (October 2012). “New treatment guidelines for acute bipolar mania: a critical review”. Journal of Affective Disorders. 140 (2): 125–41. doi:10.1016/j.jad.2011.10.015. PMID22100133.
^ Jesner OS, Aref-Adib M, Coren E (January 2007). “Risperidone for autism spectrum disorder”. The Cochrane Database of Systematic Reviews (1): CD005040. doi:10.1002/14651858.CD005040.pub2. PMID17253538.
^ Sharma A, Shaw SR (2012). “Efficacy of risperidone in managing maladaptive behaviors for children with autistic spectrum disorder: a meta-analysis”. Journal of Pediatric Health Care. 26 (4): 291–9. doi:10.1016/j.pedhc.2011.02.008. PMID22726714.
^ McPheeters ML, Warren Z, Sathe N, Bruzek JL, Krishnaswami S, Jerome RN, Veenstra-Vanderweele J (May 2011). “A systematic review of medical treatments for children with autism spectrum disorders”. Pediatrics. 127 (5): e1312–21. doi:10.1542/peds.2011-0427. PMID21464191. S2CID2903864.
^ Joint Formulary Committee. British National Formulary (online) London: BMJ Group and Pharmaceutical Press http://www.medicinescomplete.com [Accessed on 2 February 2020]
^ Jump up to:ab Pillay J, Boylan K, Carrey N, Newton A, Vandermeer B, Nuspl M, MacGregor T, Jafri SH, Featherstone R, Hartling L (March 2017). “First- and Second-Generation Antipsychotics in Children and Young Adults: Systematic Review Update”. Comparative Effectiveness Reviews (184): ES–24. PMID28749632. Report 17-EHC001-EF. Bookshelf ID: NBK442352. Compared with FGAs, SGAs may decrease the risk for experiencing any extrapyramidal symptom (EPS). FGAs probably cause lower gains in weight and BMI.
^ BMJ Group, ed. (March 2009). “4.2.1”. British National Formulary (57 ed.). United Kingdom: Royal Pharmaceutical Society of Great Britain. p. 192. ISSN0260-535X. Withdrawal of antipsychotic drugs after long-term therapy should always be gradual and closely monitored to avoid the risk of acute withdrawal syndromes or rapid relapse.
^ Chouinard G, Jones BD (January 1980). “Neuroleptic-induced supersensitivity psychosis: clinical and pharmacologic characteristics”. The American Journal of Psychiatry. 137 (1): 16–21. doi:10.1176/ajp.137.1.16. PMID6101522.
^ Miller R, Chouinard G (November 1993). “Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias, neuroleptic-induced supersensitivity psychosis and refractory schizophrenia”. Biological Psychiatry. 34 (10): 713–38. doi:10.1016/0006-3223(93)90044-E. PMID7904833. S2CID2405709.
^ Chouinard G, Jones BD, Annable L (November 1978). “Neuroleptic-induced supersensitivity psychosis”. The American Journal of Psychiatry. 135 (11): 1409–10. doi:10.1176/ajp.135.11.1409. PMID30291.
^ Moncrieff J (July 2006). “Does antipsychotic withdrawal provoke psychosis? Review of the literature on rapid onset psychosis (supersensitivity psychosis) and withdrawal-related relapse”. Acta Psychiatrica Scandinavica. 114 (1): 3–13. doi:10.1111/j.1600-0447.2006.00787.x. PMID16774655. S2CID6267180.
^ National Institute of Mental Health. PDSD Ki Database (Internet) [cited 2013 Aug 10]. ChapelHill (NC): University of North Carolina. 1998-2013. Available from: “Archived copy”. Archived from the original on 8 November 2013. Retrieved 16 May 2016.
^ Smith C, Rahman T, Toohey N, Mazurkiewicz J, Herrick-Davis K, Teitler M (October 2006). “Risperidone irreversibly binds to and inactivates the h5-HT7 serotonin receptor”. Molecular Pharmacology. 70 (4): 1264–70. doi:10.1124/mol.106.024612. PMID16870886. S2CID1678887.
^ Jump up to:abcd Brunton L, Chabner B, Knollman B. Goodman and Gilman’s The Pharmacological Basis of Therapeutics, Twelfth Edition. McGraw Hill Professional; 2010.
^ Abou El-Magd RM, Park HK, Kawazoe T, Iwana S, Ono K, Chung SP, et al. (July 2010). “The effect of risperidone on D-amino acid oxidase activity as a hypothesis for a novel mechanism of action in the treatment of schizophrenia”. Journal of Psychopharmacology. 24 (7): 1055–67. doi:10.1177/0269881109102644. PMID19329549. S2CID39050369.
^ Hecht EM, Landy DC (February 2012). “Alpha-2 receptor antagonist add-on therapy in the treatment of schizophrenia; a meta-analysis”. Schizophrenia Research. 134 (2–3): 202–6. doi:10.1016/j.schres.2011.11.030. PMID22169246. S2CID36119981.
^ Brauner, Jan M.; Hessler, Sabine; Groemer, Teja W.; Alzheimer, Christian; Huth, Tobias (2014). “Risperidone inhibits voltage-gated sodium channels”. European Journal of Pharmacology. 728: 100–106. doi:10.1016/j.ejphar.2014.01.062. PMID24508524.
^ Parent M, Toussaint C, Gilson H (1983). “Long-term treatment of chronic psychotics with bromperidol decanoate: clinical and pharmacokinetic evaluation”. Current Therapeutic Research. 34 (1): 1–6.
^ Jump up to:ab Jørgensen A, Overø KF (1980). “Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. III. Serum levels”. Acta Psychiatrica Scandinavica. Supplementum. 279: 41–54. doi:10.1111/j.1600-0447.1980.tb07082.x. PMID6931472.
^ Jump up to:ab Reynolds JE (1993). “Anxiolytic sedatives, hypnotics and neuroleptics.”. Martindale: The Extra Pharmacopoeia (30th ed.). London: Pharmaceutical Press. pp. 364–623.
^ Ereshefsky L, Saklad SR, Jann MW, Davis CM, Richards A, Seidel DR (May 1984). “Future of depot neuroleptic therapy: pharmacokinetic and pharmacodynamic approaches”. The Journal of Clinical Psychiatry. 45 (5 Pt 2): 50–9. PMID6143748.
^ Young D, Ereshefsky L, Saklad SR, Jann MW, Garcia N (1984). Explaining the pharmacokinetics of fluphenazine through computer simulations. (Abstract.). 19th Annual Midyear Clinical Meeting of the American Society of Hospital Pharmacists. Dallas, Texas.
^ Janssen PA, Niemegeers CJ, Schellekens KH, Lenaerts FM, Verbruggen FJ, van Nueten JM, et al. (November 1970). “The pharmacology of fluspirilene (R 6218), a potent, long-acting and injectable neuroleptic drug”. Arzneimittel-Forschung. 20 (11): 1689–98. PMID4992598.
^ Beresford R, Ward A (January 1987). “Haloperidol decanoate. A preliminary review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in psychosis”. Drugs. 33 (1): 31–49. doi:10.2165/00003495-198733010-00002. PMID3545764.
^ Reyntigens AJ, Heykants JJ, Woestenborghs RJ, Gelders YG, Aerts TJ (1982). “Pharmacokinetics of haloperidol decanoate. A 2-year follow-up”. International Pharmacopsychiatry. 17 (4): 238–46. doi:10.1159/000468580. PMID7185768.
^ Larsson M, Axelsson R, Forsman A (1984). “On the pharmacokinetics of perphenazine: a clinical study of perphenazine enanthate and decanoate”. Current Therapeutic Research. 36 (6): 1071–88.
^“Electronic Orange Book”. Food and Drug Administration. April 2007. Archived from the original on 19 August 2007. Retrieved 24 May 2007.
^ Scahill L (July 2008). “How do I decide whether or not to use medication for my child with autism? Should I try behavior therapy first?”. Journal of Autism and Developmental Disorders. 38 (6): 1197–8. doi:10.1007/s10803-008-0573-7. PMID18463973. S2CID20767044.
^ Jump up to:ab“Okedi: Pending EC decision”. European Medicines Agency. 15 December 2021. Retrieved 18 December 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
(Method 2): The title compound was prepared starting from 2-amino-5-dimethylamino- benzoic acid methyl ester dihydrochloride through the hydrolysis under basic condition To 5.0 g of 2-amino-5-dimethylamino-benzoic acid methyl ester di-hydrochloride, there were added 15 mL of water and 15.6 mL of a 6M aqueous solution of sodium hydroxide and the resulting mixture was heated to 40°C for 2 hours. After the confirmation of the progress of the reaction according to HPLC, the reaction system was cooled to room temperature, a 6M hydrochloric acid aqueous solution was dropwise added to the reaction system to thus neutralize the same and to separate out crystals (pH 4.9) and then the reaction system was stirred at 10°C for 2 hours. The solid thus obtained was isolated through the filtration under reduced pressure, washed with 30 mL of water and then dried under reduced pressure at 60°C for 14 hours. Title compound 3.14 g was obtained as gray-colored solid. The physical properties determined were almost identical to those observed for the same compound prepared in the above-mentioned synthesis example. H-NMR (400MHz, DMSO-d6): δ 8.21 (bs, 3H), 7.10 (d, 1H, J=2.8Hz), 6.97 (dd, 1H, J=9.1, 2.8Hz), 6.70 (d, 1H, J=9.1 Hz), 2.72 (s, 6H); 13C-NMR (100MHz, DMSO-d6): δ168.89, 144.55, 141.61, 123.29, 117.90, 114.78, 110.11,41.95; MS (ESI+): m/z 181.3 (MH+), (ESI-): m/z 179.2 (M-H–).
Step 2
Step 1: Synthesis of Nα-(2,6-dichlorobenzoyl) -4-{2-ethoxycarbonylamino-5-dimethyl- amino-benzoylamino}-L-phenylalanine methyl ester To 1.96 g of 2-amino-5-dimethylaminobenzoic acid, there were added 12 mL of acetonitrile and 5.29 mL of pyridine to form a suspension and then the resulting suspension was cooled to 4°C. To this suspension there was dropwise added 4.17 mL of ethyl chloroformate over 5 minutes and then the mixture was stirred at 25°C for one hour. After confirming the disappearance of the starting material by HPLC, 0.7 mL of ethanol was added to the mixture to thus decompose the excess ethyl chloroformate and the mixture was further stirred for additional one hour. To this reaction solution there were added 4.0 g of 4-amino-Nα-(2,6-dichlorobenzoyl)-L-phenylalanine methyl ester and 12 mL of N,Ndimethylformamide, and the resulting mixture was stirred overnight. Subsequently, 48 mL of methanol was drop-wise added, the resulting mixture was stirred at 10°C overnight and then the solid separated from the mixture was isolated through filtration under reduced pressure. The solid was then washed with 8 mL of methanol and dried at 70°C for 5 hours under reduced pressure. Title compound 5.50 g was obtained as pale yellow solid. 1H-NMR (400MHz, DMSO-d6): δ 10.29 (s, 1H), 9.42 (bs, 1H), 9.24 (d, 1H, J=7.9Hz), 7.73 (bs, 1H), 7.62 (d, 2H, J=8.4Hz), 7.48-7.44 (m, 2H), 7.41 (dd, 1H, J=9.5, 6.2Hz), 7.27 (d, 2H,J=8.4Hz), 7.01 (d, 1H, J=2.7Hz), 6.93 (dd, 1H, J=9.1, 2.9Hz), 4.71 (ddd, 1H, J=9.2, 8.1, 5.7Hz), 4.05 (q, 2H, J=7.0Hz), 3.66 (s, 3H), 3.10 (dd, 1H, J=14.0, 5.6Hz), 2.96 (dd, 1H, J=14.0, 9.2Hz), 2.93 (s, 6H), 1.18 (t, 3H, J=7.2Hz); MS (ESI+): m/z 601.2 (MH+) and 623.2 (M+Na), (ESI–): m/z 599.1 (M-H–).
Step 3
Step2: Synthesis of Na-(2,6-dichlorobenzoyl)-4-{6-dimethylamino-1-methylquinazoline-2,4[1H,3H]-dion-3-yl}-L-phenylalanine methyl ester To 2.0 g of Na-(2,6-dichlorobenzoyl)-4-{2-ethoxycarbonylamino -5-dimethyl- amino-benzoylamino}-L-phenylalanine methyl ester prepared in above-mentioned step 1, were added 16 mL of N,N-dimethylfbrmamide, 0.8 mL of methanol and 0.91 g of potassium carbonate, followed by the stirring of the resulting mixture at 25°C overnight. To this reaction solution, there was added 0.75 mL of methyl p-toluenesulfonate for subjecting the methyl ester to alkylation at 25~40°C. After confirming the disappearance of the starting material by HPLC, 0.75 mL of acetic acid was added to quench the reaction, 16 mL of water was dropped and the solid was separated. Further, 8 mLof N,N-dimethylformamide/water = 1/1 mixed liquid was added to the resulting mixture, followed by the stirring of the mixture at 25°C. Then the solid thus separated was isolated through filtration under reduced pressure and then washed with 8 mL of water. Thereafter, the isolated solid was dried at 70°C for 4 hours under reduced pressure. Desired compound 1.77 g was obtained as pale yellow solid. 1H-NMR (400MHz, DMSO-d6): δ 9.28 (d, 1H, J=8.1 Hz), 7.48-7.36 (m, 6H), 7.31 (dd, 1H, J=3.0, 9.0Hz), 7.24 (d, 1H, J=3.0Hz), 7.20-7.15 (m, 2H), 4.18 (ddd, 1H, J=10.2, 8.1,4.8Hz), 3.69 (s, 3H), 3.49 (s, 3H), 3.22 (dd, 1H, J=14.1, 4.8Hz), 3.02 (dd, 1H, J=14.2, 10.5Hz), 2.94 (s, 6H); MS (ESI+): m/z 569.2 (MH+) and 591.1 (M+Na), (ESI-): m/z 567.2 (M-H–).
Toldimfos is an aromatic phosphorus compound which falls between phosphorous itself and phosphoric acid in the stages of oxidation. Toldimfos sodium is the sodium salt of 2- methyl-4-(dimethylamino)phenylphosphinic acid. It is used to treat and prevent diseases associated with parturition and peri-partum period, developmental and nutritional disorders in young animals, and bone growth disorders and tetany or paresis caused by calcium, magnesium, and phosphorus metabolism disorders. Toldimfos has been used as a human medicine since 1920. While it is no longer indicated for human use, it is used in horses, cattle, sheep, pigs, and goats, and administered by intravenous, intramuscular, or subcutaneous injection. No specific data on the pharmacodynamic action of toldimfos was submitted. The precise mode of action of toldimfos is unknown and it is questionable whether the effect of toldimfos is simply a matter of the substitution of deficient phosphorus. It appears more likely that the effect of toldimfos arises due to multiple stimulation of metabolism with the body.
Toldimfos sodium trihydrate
5787-63-3
Toldimfos [INN:BAN] 57808-64-7
Toldimfos Sodium
CAS Registry Number: 575-75-7
CAS Name: (4-Dimethylamino-o-tolyl)phosphonous acid sodium salt
Additional Names: sodium (4-dimethylamino-o-tolyl)phosphonate; p-dimethylamino-o-toluenephosphonous acid sodium salt
China’s animal husbandry fast development is the important motivity that promotes China’s agricultural and rural economy development, improves farmers’ income.The disease relevant with Nutrition and Metabolism of serious harm animal health is in rising trend in recent years, the direct economic loss that raising poultry nutritive metabolic disease causes over ten billion to China’s animal husbandry every year, and indirect economic loss is difficult to estimate.
Due to the life-time service of chemicals, will cause some poultrys, poultry product drug residue is serious, this is harm humans healthy not only, also affecting the export of farm produce earns foreign exchange, therefore, tackle this problem, research and development are efficient, the new Nutrition and Metabolism medicine of low toxicity, wide spectrum will have huge market.
Toldimfos (Toldimfos Sodium) belongs to the nutritional supplementation medicine of phosphorus supplement, can be used as benzenephosphonic acid (Phenylphosphinicacid, BPA) succedaneum uses, can be used for treating the disease relevant with childbirth and perinatal stage of the food animals such as horse, cattle, pig, sheep, the diseases such as the bone lengthening obstacle being caused by calcium, magnesium, phosphorus metabolism obstacle.
Toldimfos has higher water solublity, mainly excretes through urine rapidly with the former medicine form of not metabolism in animal body, and the half-life is short, can in tissue, not accumulate.
Toldimfos is developed by German Hoechst company, the symptom such as since nineteen twenty, once physical weakness, chronic stress, depression, mental muscle power postoperative for human treatment, that catch was overtired.Now be not used in the mankind, be mainly used in animal.Its commodity are called onofosfan.
In sum, toldimfos, as a kind of nutritional supplementation medicine of new and effective noresidue, has wide market prospect in China.The development of this product will be made outstanding contributions to the sound development of China’s animal husbandry, remarkable economic and social benefits with application.
Mechanism of ActionType 4 cyclic nucleotide phosphodiesterase inhibitors
RegisteredAtopic dermatitis
27 Sep 2021Registered for Atopic dermatitis (In adolescents, In children, In adults) in Japan (Topical)
11 Nov 2020Otsuka Pharmaceutical completes a phase III trial in Atopic dermatitis (In children, In adolescents, In adults) in Japan (Topical) (NCT03961529)
28 Sep 2020Preregistration for Atopic dermatitis in Japan (In children, In adolescents, In adults) (Topical)
Difamilast is under investigation in clinical trial NCT01702181 (A Safety Study to Evaluate the Use and Effectiveness of a Topical Ointment to Treat Adults With Atopic Dermatitis).
Patent Documents 1 and 2 report an oxazole compound having a specific inhibitory action on phosphodiesterase 4 (PDE4) and a method for producing the same. PDE4 is the predominant PDE in inflammatory cells, inhibition of PDE4 increases intracellular cAMP concentration, and the increase in this concentration downregulates the inflammatory response through regulation of the expression of TNF-α, IL-23, and other inflammatory cytokines. .. Elevated cAMP levels also increase anti-inflammatory cytokines such as IL-10. Therefore, it is considered that the oxazole compound is suitable for use as an anti-inflammatory agent. For example, it may be useful for controlling skin eczema and dermatitis, including atopic dermatitis. Patent Document 3 describes an ointment that stably contains an oxazole compound having a specific inhibitory effect on PDE4 and can be efficiently absorbed into the skin. The contents of Patent Documents 1 to 3 are incorporated in the present specification by reference.
[Synthesis of Oxazole Compound (Type A Crystal)]
Compound (5) (white powder) was prepared by the method described in Example 352 of Patent Document 1 (International Publication No. 2007/088383).
[Preparation of B-type crystal 2]
Using the obtained B-type crystal as a seed crystal, it was examined to further prepare a B-type crystal. Specifically,
B-type crystals were prepared as follows according to the method described in Patent Document 3 (International Publication No. 2017/115780).
[0072]
[Chem. 6]
[0073]
Compound (1) 20.00 g (66.8 mmol) and 17.28 g (134 mmol) of diisopropylethylamine were added to 300 mL of ethyl acetate to cool the mixture, and 11.48 g (100 mmol) of methanesulfonyl chloride was introduced into the compound (1) at 10 to 30 ° C. Stir for hours. Subsequently, 17.41 g (200 mmol) of lithium bromide was added, and the mixture was stirred at 20 to 35 ° C. for 1 hour. 100 mL of water was added to the reaction solution to separate the layers, and the organic layer was concentrated under reduced pressure. 300 mL of ethyl acetate was added to the concentrated residue to dissolve it, and the mixture was concentrated again under reduced pressure. 200 mL of N, N-dimethylformamide and 17.33 g (93.6 mmol) of phthalimide potassium were added to the concentrated residue, and the mixture was reacted at 75 to 85 ° C. for 1 hour. 200 mL of water was added to the reaction solution to precipitate crystals, and the precipitated crystals were collected by filtration and dried at 80 ° C. to obtain 27.20 g (yield 95.01%) of compound (3).
[0074]
[Chem. 7]
[0075]
Compound (3) 20.00 g (46.7 mmol), 40 mL of a 40% aqueous methylamine solution, 40 mL of methanol, and 100 mL of water were mixed and reacted under reflux for 30 minutes. 200 mL of cyclopentyl methyl ether (CPME) and 20 mL of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75 ° C. to separate the liquids. A mixed solution of 100 mL of water and 20.00 g of sodium chloride was added to the organic layer, and the temperature was adjusted again to 65 to 75 ° C. to separate the liquids. 5 mL of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. Precipitated crystals were collected by filtration to obtain 27.58 g of wet crystals of compound (4).
[0076]
Wet crystals (46.7 mmol) of compound (4) were mixed with 120 mL of ethyl acetate and 7.1 mL (51.4 mmol) of triethylamine, and the mixture was stirred at 20 to 30 ° C. for 1 hour. To the reaction solution, 10.09 g (60.7 mmol) of 2-ethoxybenzoic acid and 11.63 g (60.7 mmol) of 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (WSC) were added, and 20 to 30 were added. The reaction was carried out at ° C. for 1 hour. 60 mL of water and 6 mL of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50 ° C. to separate the solutions. 60 mL of water and 6 mL of a 25%
aqueous sodium hydroxide solution were added to the organic layer, the temperature was adjusted again to 40 to 50 ° C., the liquid was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% aqueous sodium hydroxide solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. Activated carbon was removed by filtration, washed with 12 mL of ethanol, the filtrate was cooled, and 10 mg of B-type crystals (seed crystals) were added to precipitate crystals. Precipitated crystals were collected by filtration and dried at 60 ° C. to obtain 18.38 g (yield 88.18%) of crystals of compound (5).
Production Example 1: Production 1 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
[0146]
[Chem. 11]
[0147]
10.00 g (55.5 mmol) of compound (1a) and 9.20 g (66.6 mmol) of potassium carbonate were added to 40 ml of N,N-dimethylformamide and 6 ml of water, and the mixture was stirred until exotherm subsided. 16.92 g (111 mmol) of sodium chlorodifluoroacetate was added thereto, and the mixture was reacted at 95 to 110°C for 3 hours. 80 ml of butyl acetate and 80 ml of water were added to the reaction solution, and the solution was partitioned. 80 ml of water was added again to the organic layer, followed by partitioning. 3 ml of concentrated hydrochloric acid was added to the organic layer, and the mixture was stirred at 60 to 70°C for 30 minutes. 40 ml of water and 10 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the mixture was partitioned. 5.93 g (61.1 mmol) of sulfamic acid and 10 ml of water were added to the organic layer, and 22.08 g (61.0 mmol) of a 25% sodium chlorite aqueous solution was added dropwise thereto at a temperature of 20°C or below. The mixture was reacted at 20°C or below for 15 minutes, and 10 ml of a 25% sodium hydroxide aqueous solution was added dropwise thereto at a temperature of 20°C or below, followed by pouring in 83.95 g (66.6 mmol) of a 10% sodium sulfite aqueous solution. Additionally, 2 ml of concentrated hydrochloric acid was added and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 40 ml of methanol, 80 ml of water, and 10 ml of a 25% sodium hydroxide aqueous solution were added to the concentrated residue to dissolve the residue, and 5 ml of concentrated hydrochloric acid was added dropwise thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 11.81 g (yield: 86.4%) of compound (3) as a white powder.
Production Example 2: Production 2 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
[0149]
[Chem. 12]
[0150]
10.00 g (53.2 mmol) of compound (1b), 9.55 g (69.1 mmol) of potassium carbonate, and 8.50 g (69.1 mmol) of isopropyl bromide were added to 40 ml of N,N-dimethylformamide, and the mixture was reacted at 75 to 85°C for 2 hours. 80 ml of butyl acetate and 80 ml of water were added to the reaction solution, and the mixture was partitioned. 5.68 g (58.5 mmol) of sulfamic acid and 10 ml of water were added to the organic layer, and 21.15 g (58.5 mmol) of a 25% sodium chlorite aqueous solution was added dropwise thereto at 20°C or below, followed by reaction for 15 minutes. 10 ml of a 25% sodium hydroxide aqueous solution was added thereto at 20°C or below, and subsequently 80.41 g (63.8 mmol) of a 10% sodium sulfite aqueous solution was poured in. Additionally, 2 ml of concentrated hydrochloric acid was added, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 40 ml of methanol, 80 ml of water, and 10 ml of a 25% sodium hydroxide aqueous solution were added to the concentrated residue, and the residue was dissolved, followed by dropwise addition of 5 ml of concentrated hydrochloric acid to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 12.09 g (yield: 92.4%) of compound (3) as a white powder.
[0151]
Production Example 3: Production of Compound (7)
Compound (7) was produced in accordance with the following reaction scheme.
[0152]
[Chem. 13]
Production Example 4: Production of Compound (11)
Compound (11) was produced in accordance with the following reaction scheme.
[0160]
[Chem. 14]
[0161]
Synthesis of Compound (9)
20.00 g (66.8 mmol) of compound (7) and 17.28 g (134 mmol) of N,N-diisopropylethylamine were added to 300 ml of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto and reacted at 20 to 35°C for 1 hour. 100 ml of water was added to the reaction solution, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 300 ml of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 ml of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue and reacted at 75 to 85°C for 1 hour. 200 ml of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 25.90 g (yield: 90.5%) of compound (9) as a white powder.
Synthesis of Compound (10)
15.00 g (35.0 mmol) of compound (9) was mixed with 30 ml of a 40% methylamine aqueous solution, 30 ml of methanol, and 75 ml of water, and reacted under reflux for 30 minutes. 150 ml of cyclopentyl methyl ether (CPME) and 15 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by partitioning. A mixture of 150 ml of water and 7.50 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by partitioning. 3.75 ml of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 11.95 g (yield: quant.) of compound (10) as a white powder.
Synthesis of Compound (11)
13.30 g (39.7 mmol) of compound (10) was mixed with 3.83 g (37.8 mmol) of triethylamine and 108 ml of ethyl acetate, and stirred at 20 to 30°C for 1 hour. 9.78 g (58.9 mmol) of 2-ethoxybenzoic acid and 11.28 g (58.8 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 54 ml of water and 5.4 ml of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by partitioning. 54 ml of water and 5.4 ml of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was partitioned, and the organic layer was concentrated under reduced pressure. 45 ml of ethanol, 18 ml of water, 5.4 ml of a 25% sodium hydroxide aqueous solution, and 0.54 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 11 ml of ethanol. The filtrate was cooled, and a seed crystal was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 35°C, thereby obtaining 12.88 g (72.6%) of compound (11) as a white powder.
Type B Crystal Preparation 2
Analysis was conducted to further prepare the type B crystal using the obtained type B crystal as a seed crystal. More specifically, the type B crystal was prepared as follows, in accordance with the method disclosed in PTL 3 (WO2017/115780).
[0072]
[0073]
20.00 g (66.8 mmol) of compound (1) and 17.28 g (134 mmol) of diisopropylethylamine were added to 300 mL of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto, and the mixture was stirred at 20 to 35°C for 1 hour. 100 mL of water was added to the reaction solution, and the mixture was separated, followed by concentration of the organic layer under reduced pressure. 300 mL of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 mL of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue, and reacted at 75 to 85°C for 1 hour. 200 mL of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 27.20 g (yield: 95.01%) of compound (3).
[0074]
[0075]
20.00 g (46.7 mmol) of compound (3), 40 mL of a 40% methylamine aqueous solution, 40 mL of methanol, and 100 mL of water were mixed and reacted for 30 minutes under reflux. 200 mL of cyclopentyl methyl ether (CPME) and 20 mL of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by separation. A mixture of 100 mL of water and 20.00 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by separation. 5 mL of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration, thereby obtaining 27.58 g of compound (4) as a wet crystal.
[0076]
The wet crystal (46.7 mmol) of compound (4) was mixed with 120 mL of ethyl acetate and 7.1 mL (51.4 mmol) of triethylamine, and stirred at 20 to 30°C for 1 hour. 10.09 g (60.7 mmol) of 2-ethoxybenzoic acid and 11.63 g (60.7 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 60 mL of water and 6 mL of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by separation. 60 mL of water and 6 mL of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% sodium hydroxide aqueous solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 12 mL of ethanol. The filtrate was cooled, and 10 mg of the type B crystal (a seed crystal) was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 18.38 g (88.18%) of compound (5).
PATENT
WO2014034958A1
WO2007058338A2
WO2007058338A9
WO2007058338A3
US9181205B2
US2015239855A1
USRE46792E
US2020078340A1
US2017216260A1
US2019070151A1
US2009221586A1
US8637559B2
US2014100226A1
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
alpha-D-Glucan, (1-6)-, reduced, reaction products with iron hydroxide (Fe(OH)3)
Ferric derisomaltose is an iron injection used in the treatment of iron deficiency anemia.
Ferric derisomaltose, sold under the brand name Monoferric, is a medication for the treatment of iron deficiency anemia (IDA) in adults who have intolerance to oral iron or have had unsatisfactory response to oral iron or who have non-hemodialysis dependent chronic kidney disease (NDD-CKD).[1] It was approved for use in the United States in January 2020.[1][2][3] It is given intravenously.[1]
Iron deficiency is an extremely common condition and is the most frequent cause of anemia worldwide. Iron deficiency results when iron intake, iron stores, and loss of iron from the body do not adequately support production of erythrocytes, also known as red blood cells. Though it is generally considered non life-threatening, iron deficiency may considerably affect quality of life.3
Ferric derisomaltose is a form of iron used in the treatment of iron deficiency. This drug is a complex of iron (III) hydroxide and derisomaltose. The latter is an iron carbohydrate oligosaccharide that works to release iron. Ferric derisomaltose was developed by Pharmacosmos Therapeutics ad was granted FDA approval in January 2020.8,9 Clinical trials show that it is non-inferior to iron sucrose, another form of iron that is often administered in iron deficiency, and less likely to cause serious hypersensitivity that is associated with other forms of injectable iron.1,4
This drug is indicated for the treatment of iron deficiency anemia in adult patients who have experienced intolerance to oral iron preparations or insufficient clinical response to orally administered iron. Ferric derisomaltase is also indicated for patients with non-hemodialysis dependent chronic kidney disease.8 In Australia and United Kingdom, ferric derisomaltase is indicated for cases in which rapid delivery of iron is required.10,11
Iron deficiency is an extremely common condition and is the most frequent cause of anemia worldwide. Iron deficiency results when iron intake, iron stores, and loss of iron from the body do not adequately support production of erythrocytes, also known as red blood cells. Though it is generally considered non life-threatening, iron deficiency may considerably affect quality of life. Ferric derisomaltose is a form of iron used in the treatment of iron deficiency. This drug is a complex of iron (III) hydroxide and derisomaltose. The latter is an iron carbohydrate oligosaccharide that works to release iron. Ferric derisomaltose was developed by Pharmacosmos Therapeutics ad was granted FDA approval in January 2020. Clinical trials show that it is non-inferior to [iron sucrose], another form of iron that is often administered in iron deficiency, and less likely to cause serious hypersensitivity that is associated with other forms of injectable iron.
Monoferric is an iron replacement product containing ferric derisomaltose for intravenous infusion. Ferric derisomaltose is an iron carbohydrate complex with a matrix structure composed of interchanging layers of ferric hydroxide and the carbohydrate derisomaltose. Derisomaltose consists of linear, hydrogenated isomaltooligosaccharides with an average molecular weight of 1000 Da and a narrow molecular weight distribution that is almost devoid of mono-and disaccharides.
Ferric derisomaltose has an average molecular weight of 155,000 Da and has the following empirical formula:
Iron atoms placed in the electronegative cavities of the 3-D structure between and within the derisomaltose molecules. A schematic representation is presented below
Monoferric is a sterile, dark brown, non-transparent aqueous solution with pH 5.0-7.0, containing ferric derisomaltose dissolved in water for injections and filled into Type I glass vials.
Each 1 mL of solution contains 100 mg of elemental iron as ferric derisomaltose in water for injection.
Each 1 mL of solution contains 100 mg of elemental iron as ferric derisomaltose in water for injection.
It was approved for medical use in the United States in May 2018.[1] It was developed by Portola Pharmaceuticals.[3]
ndexanet alfa is a recombinant human coagulation Factor Xa that promotes blood coagulation. It was developed by Portola Pharmaceuticals and was approved in in May 2018. It is marketed as Andexxa for intravenous injection or infusion and is indicated for the reversal of anticoagulation in combination with rivaroxaban and apixaban in cases of life-threatening or uncontrolled bleeding. Rivaroxaban and apixaban are Factor Xa inhibitors that promote anticoagulation in situations where blood clotting is unfavourable, such as in deep vein thrombosis and pulmonary embolism. However, the use of these agents is associated with a risk for uncontrollable bleeding episodes that can lead to can cause serious or fatal bleeding. Andexanet alfa is currently under regulatory review by the European Union and is undergoing clinical development in Japan 1.
Andexanet alfa works by binding to Factor Xa inhibitors and prevent them from interacting with endogenous Factor Xa. It displayed high affinity (0.53–1.53 nmol/L) to apixaban, betrixaban, edoxaban and rivaroxaban 1. However, the effectiveness of andexanet alfa on treating bleeding related to any FXa inhibitors other than apixaban and rivaroxaban was not demonstrated, thus such use is limited 7. Its pharmacokinetic properties are not reported to be affected by factor Xa inhibitors 1. Andexanet alfa retains the structural similarity to that of endogenous human factor Xa, but exists in its mature functional form without the need for activation via the intrinsic or extrinsic coagulation pathways 5 and remains catalytically inactive due to structural modification 1. The procoagulation potential of andexanet alfa is eliminated through the removal of a 34-residue fragment containing Gla: via this truncation, andexanet alfa is unable to bind to membrane surfaces and assemble the prothrombinase complex 5. It also prevents andexanet alfa from taking up space on phospholipid surface membranes, so that native FXa may bind and assemble the prothrominase complex 5. The amino acid residue modification from serine to alanine in the binding site of the catalytic domain allows more effective binding to FXa inhibitors and deters the andexanet alfa from converting prothrombin to thrombin 5.
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Structure of andexant alfa. Andexanet alfa is a modified activated human factor Xa (FXa) that binds FXa with high affinity and a 1:1 stoichiometric ratio but does not have intrinsic catalytic activity (the amino acid serine at position 419 is replaced by alanine) and lacks the membrane-binding-carboxyglutamic acid domain (Gla domain) of native FX. The Gla domains are responsible for the binding of FXa to phospholipids
Medical uses
Andexanet alfa is used to stop life threatening or uncontrollable bleeding in people who are taking rivaroxaban or apixaban.[1]
There are no randomised clinical trials as of 2019. Studies in healthy volunteers show that the molecule binds factor Xa inhibitors and counters their anti-Xa-activity.[4] The only published clinical trial is a prospective, open label, single group study.[5] This study reports results on 352 people and demonstrates a reduction of anti-Xa-activity while also showing an excellent or good hemostatic efficacy in 82%. While people who were expected to die in 30 days were excluded from the study, 14% of participants died. There was no relationship between hemostatic efficacy and reduced anti-Xa-activity.[6] The FDA has demanded a randomised clinical trial: the first results are not expected before 2023.[7]
Adverse effects
Common side effects include pneumonia and urinary tract infections.[2] Severe side effects may include blood clots or cardiac arrest.[2]
Andexanet alfa has a boxed warning that it is associated with arterial and venous blood clots, ischemic events, cardiac arrest, and sudden deaths.[1]
Pharmacology
Mechanism of action
Andexanet alfa is a biologic agent, a recombinant modified version of human activated factor X (FXa).[8] Andexanet alfa differs from native FXa due to the removal of a 34 residue fragment that contains the Gla domain. This modification reduces andexanet alfa’s procoagulant potential. Additionally, a serine to alanine (S419A) mutation in the active site eliminates its activity as a prothrombin to thrombin catalyst, but still allows the molecule to bind to FXa inhibitors.[9]FXa inhibitors bind to andexanet alfa with the same affinity as to natural FXa. As a consequence in the presence of andexanet alfa natural FXa is partially freed, which can lead to effective hemostasis.[3][10] In other words, it acts as a decoy receptor. Andexanet alfa reverses effect of all anticoagulants that act directly through FXa or by binding antithrombin III. The drug is not effective against factor IIa inhibitor dabigatran.[11]
It was approved in the United States in 2018 based on data from two phase III studies on reversing the anticoagulant activity of FXa inhibitors rivaroxaban and apixaban in healthy volunteers.[4] As a condition of its accelerated approval there is a study being conducted comparing it to other currently used reversal agents (“usual care”).[5][12]
Society and culture
Economics
Initial pricing (AWP) is $58,000 per reversal (800 mg bolus + 960 mg infusion, $3,300 per 100 mg vial) which is higher than reversal agents for other DOAC agents (idarucizumab for use in dabigatran reversal is $4,200 per reversal).[13]
^ Justin Morgenstern, “Andexanet Alfa: More garbage science in the New England Journal of Medicine”, First10EM blog, February 11, 2019. Available at: https://first10em.com/andexanet-alfa/.
^ Lu, Genmin; DeGuzman, Francis R.; Lakhotia, Sanjay; Hollenbach, Stanley J.; Phillips, David R.; Sinha, Uma (2008-11-16). “Recombinant Antidote for Reversal of Anticoagulation by Factor Xa Inhibitors”. Blood. 112 (11): 983. doi:10.1182/blood.V112.11.983.983. ISSN0006-4971.
^ Lu G, Deguzman FR, Hollenbach SJ, et al. (March 2013). “A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa”. Nature Medicine. 19 (4): 446–51. doi:10.1038/nm.3102. PMID23455714. S2CID11235887.
^ H. Spreitzer (23 December 2013). “Neue Wirkstoffe – Andexanet Alfa”. Österreichische Apothekerzeitung (in German) (26/2013): 40.
Olipudase alfa was associated with significant improvements in clinically relevant disease end points among patients with chronic visceral acid sphingomyelinase (ASM) deficiency (ASMD), according to results from the phase 2/3 ASCEND trial presented at the 17th Annual WORLDSymposium.
ASMD is a rare, debilitating lysosomal storage disease characterized by a deficiency of the enzyme acid sphingomyelinase, which results in the accumulation of sphingomyelin in various tissues of the body. Olipudase alfa is an investigational enzyme replacement therapy designed to replace deficient or defective ASM.
The multicenter, randomized, double-blind, placebo-controlled ASCEND trial evaluated the efficacy and safety of olipudase alfa in 36 adults with chronic visceral ASMD. Patients were randomly assigned 1:1 to receive olipudase alfa 3mg/kg intravenously every 2 weeks or placebo for 52 weeks. The coprimary end points were the percent change in spleen volume and percent-predicted diffusing capacity of the lung for carbon monoxide (DLCO).
At week 52, treatment with olipudase alfa resulted in a 39.45% reduction in spleen volume, compared with a 0.5% increase for placebo (P <.0001). A decrease in spleen volume of at least 30% was observed in 17 patients (94%) treated with olipudase afla compared with no patients treated with placebo. Additionally, olipudase alfa significantly improved lung function by 22% from baseline compared with 3% for patients receiving placebo (P =.0004), as measured by percent predicted DLCO.
Olipudase alfa also met key secondary end points including a 31.7% reduction in liver volume (vs a 1.4% reduction for placebo; P <.0001) and a 16.8% improvement in mean platelet counts (vs 2.5% with placebo; P =.019) at week 52. Significant improvements in HDL, LDL, AST, ALT, chitotriosidase (54% vs 12% with placebo; P =.0003), and lyso-sphingomyelin (78% vs 6% with placebo) were also observed in the olipudase alfa group at week 52.
With regard to Splenomegaly Related Score, a patient-reported outcome measurement that evaluates patient symptoms associated with an enlarged spleen, findings showed no meaningful difference between olipudase alfa and placebo (-8 point vs -9.3 points, respectively).
As for safety, olipudase alfa was well tolerated with most adverse events being mild to moderate in severity. There were no treatment-related serious adverse events and no adverse event-related discontinuations.
Disclosure: Some authors have declared affiliations with or received funding from the pharmaceutical industry. Please refer to the original study for a full list of disclosures.
Reference
Wasserstein M, Arash-Kaps L, Barbato A, et al. Adults with chronic acid sphingomyelinase deficiency show significant visceral, pulmonary, and hematologic improvements after enzyme replacement therapy with olipudase-alfa: 1-year results of the ASCEND placebo-controlled trial. Presented at: 17th Annual WORLDSymposium; February 8-12, 2021. Abstract 265.
EMA accepts regulatory submission for olipudase alfa,the first potential therapy for ASMD
Olipudase alfa has been granted PRIority MEdicines (PRIME) designation in Europe, Breakthrough Therapy designation in the United States, and SAKIGAKE designation in Japan
European regulatory decision anticipated second half of 2022
DECEMBER 6, 2021
The European Medicines Agency (EMA) has accepted for review under an accelerated assessment procedure the Marketing Authorization Application (MAA) for olipudase alfa, Sanofi’s investigational enzyme replacement therapy which is being evaluated for the treatment of acid sphingomyelinase deficiency (ASMD). Historically referred to as Niemann-Pick disease (NPD) type A and type B, ASMD is a rare, progressive, and potentially life-threatening disease for which no treatments are currently approved. The estimated prevalence of ASMD is approximately 2,000 patients in the U.S., Europe (EU5 Countries) and Japan. If approved, olipudase alfa will become the first and only therapy for the treatment of ASMD.
“Today’s milestone has been decades in the making and our gratitude goes to the ASMD community who has stood by us with endless patience while olipudase alfa advanced through clinical development,” said Alaa Hamed, MD, MPH, MBA, Global Head of Medical Affairs, Rare Diseases, Sanofi. “Olipudase alfa represents thekind of potentially life-changing innovation that is possible when industry, medical professionals and the patient community work together toward a common goal.”
The MAA is based on positive results from two separate clinical trials (ASCEND and ASCEND-Peds) evaluating olipudase alfa in adult and pediatric patients with non-central nervous system (CNS) manifestations of ASMD type A/B and ASMD type B.
Olipudase alfa has received special designations from regulatory agencies worldwide, recognizing the innovation potential of the investigational therapy.
“Scientific innovation is the greatest source of hope for people living with diseaseslike ASMD where there are no approved treatmentsand is a critical component for ensuring a viable healthcare ecosystem,” said Bill Sibold, Executive Vice President of SanofiGenzyme. “At Sanofi, we have a long history of pioneering scientific innovation,and we remain committed to finding solutions to address unmet medical needs, including those of the rare disease community.”
The EMA awarded olipudase alfa the PRIority MEdicines designation, also known as PRIME, intended to aid and expedite the regulatory process for investigational medicines that may offer a major therapeutic advantage over existing treatments, or benefit patients without treatment options.
The U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy designation to olipudase alfa. This designation is intended to expedite the development and review of drugs intended to treat serious or life-threatening diseases and conditions. The criteria for granting Breakthrough Therapy designation include preliminary clinical evidence indicating that the molecule may demonstrate substantial improvement on a clinically significant endpoint over available therapies.
In Japan, olipudase alfa was awarded the SAKIGAKE designation, which is intended to promote research and development in Japan for innovative new medical products that satisfy certain criteria, such as the severity of the intended indication. In September, Sanofi filed the J-NDA submission for olipudase alfa.
AboutASMD
ASMD results from a deficient activity of the enzyme acid sphingomyelinase (ASM), which is found in special compartments within cells called lysosomes and is required to breakdown lipids called sphingomyelin. If ASM is absent or not functioning as it should, sphingomyelin cannot be metabolized properly and accumulates within cells, eventually causing cell death and the malfunction of major organ systems. The deficiency of the lysosomal enzyme ASM is due to disease-causing variants in the sphingomyelin phosphodiesterase 1 gene (SMPD1). The estimated prevalence of ASMD is approximately 2,000 patients in the U.S., Europe (EU5 Countries) and Japan.
ASMD represents a spectrum of disease caused by the same enzymatic deficiency, with two types that may represent opposite ends of a continuum sometimes referred to as ASMD type A and ASMD type B. ASMD type A is a rapidly progressive neurological form of the disease resulting in death in early childhood due to central nervous system complications. ASMD type B is a serious and potentially life-threatening disease that predominantly impacts the lungs, liver, and spleen, as well as other organs. ASMD type A/B represents an intermediate form that includes varying degrees of neurologic involvement. Patients with ASMD type A/B or ASMD type B were studied in the ASCEND trial program. Another type of NPD is NPD type C, which is unrelated to ASMD.
About olipudase alfa
Olipudase alfa is an investigational enzyme replacement therapy designed to replace deficient or defective ASM, allowing for the breakdown of sphingomyelin. Olipudase alfa is currently being investigated to treat non-CNS manifestations of ASMD. Olipudase alfa has not been studied in ASMD type A patients. Olipudase alfa is an investigational agent and the safety and efficacy have not been evaluated by the FDA, EMA, or any other regulatory authority worldwide.
About Sanofi
Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions.
With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe.
Sibutramine was originally developed in 1988 by Boots in Nottingham, UK,[13] and marketed by Knoll Pharmaceuticals after BASF/Knoll AG purchased the Boots Research Division in 1995, and was most recently manufactured and marketed by Abbott Laboratories before its withdrawal from most markets. It has been sold under a variety of brand names including Reductil, Meridia, Siredia, and Sibutrex. It is classified as a Schedule IVcontrolled substance in the United States.
Sibutramine hydrochloride hydrate was approved by the U.S. Food and Drug Administration (FDA) on Nov 16, 1997. It was developed and marketed as Meridia® by Abbott in the US.
Sibutramine hydrochloride hydrate is a serotonin-norepinephrine reuptake inhibitor, it produces its therapeutic effects by norepinephrine, serotonin and dopamine reuptake inhibition. Meridia® is indicated for the management of obesity, including weight loss and maintenance of weight loss, and should be used in conjunction with a reduced calorie diet.
Meridia® is available as capsule for oral use, containing 5, 10 or 15 mg of Sibutramine hydrochloride hydrate. The recommended dose is initiated at 10 mg once daily with or without food and may increase to 15 mg once daily.
Sibutramine has been withdrawn from the market in several countries and regions since 2010, owning to its side effect that associated with increased cardiovascular events and strokes.Route 1
Sibutramine hydrochloride (Sibutramine HCl: C 17 H 26 CIN HCl) is a chemical name {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} -dimethylamine hydrochloride, and has the structure of Formula 1 .
Sibutramine was originally developed as a drug for the treatment of depression and was found to give weight to patients taking this drug. It was developed as an anti-obesity drug. Let your appetite decrease.
Korean Patent Publication No. 1990-274 (corresponding patent DE3212682 (Boots), filed Oct. 21, 1982; priority GB 1981.4.6.) For the preparation of 1- (1-arylcyclobutyl) alkylamine derivative comprising sibutramine It is described. The method for synthesizing sibutramine described in this document proceeds in a total of five steps as follows.
Step A
1- (4-chlorophenyl) -1-cyclobutyl cyanide is obtained from 4-Chlorobenzyl cyanide. Examples of the actual synthesis method described in this document are as follows.
After dissolving 25 g of 4-chlorobenzyl cyanide and 15 ml of 1,3-dibromopropane in 150 ml of DMSO, the solution was dissolved in nitrogen at room temperature (20-35 ° C.) and 7.5 g of NaH dispersed in mineral oil. And 200 ml of DMSO was added dropwise. The mixture was stirred at room temperature for 2 hours, 8 ml of IPA was added dropwise, and 110 ml of water was added dropwise. The mixture is filtered through CELITE and the solid residue is washed with ether. The ether layer is separated, washed with water, dried, evaporated and vacuum distilled at high temperature to separate the desired 1- (4-chlorophenyl) -1-cyclobutyl cyanide. The total reaction time of this step is 5 hours and the yield from the starting material is 78%.
Step B
1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one is obtained from 1- (4-chlorophenyl) -1-cyclobutyl cyanide. Specific synthesis examples are as follows. 35.2 g of 1- (4-chlorophenyl) -1-cyclobutyl cyanide are dissolved in 100 ml of ether and this solution is added to the product prepared by the reaction of 32 g of propylbromide and 6.36 g of magnesium. The ether is replaced with toluene and the mixture is heated under reflux for 1 hour. After adding water, concentrated hydrochloric acid is added, and the mixture is heated under reflux for 1 hour. The mixture obtained in the same manner as in the previous step was treated with ether, water, dried and evaporated and then vacuum distilled to give the desired 1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one. To separate. The reaction time of this step is a total of 22 hours, the yield is 81%. The target product is bp 100-120 ° C / 0.2 mm / Hg.
Step C
N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl from 1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one } Formamide is obtained. Specific synthesis examples are as follows. To 23.5 ml of formamide, 37 g (0.14 mol) of 1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one and 9 ml of HCOOH were added dropwise at 160-170 ° C. The temperature is maintained at 175 ° C. to 180 ° C. for 24 hours. The mixture is extracted with ether and concentrated to afford an oil, which crystallizes the desired N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} formamide from petroleum ether. The reaction time of this step is a total of 24 hours, the yield is 39%. The target is mp 110-112 ° C.
Step D
1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride from N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} formamide Get Specific synthesis examples are as follows. 4 g of N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} formamide, 25 ml of 2-methoxyethyl ether, 10 ml of water and 22 ml of concentrated hydrochloric acid were refluxed for 18 hours. Stir under. Dilute with water, wash with ether, and add 35 ml of 5M aqueous NaOH solution. After completion of the process by treatment with ether, water and brine, treated with magnesium sulfate, filtered and concentrated. The concentrated crude product is saturated with hydrochloric acid dissolved in 20 ml of ether. The resulting solid is filtered, concentrated and crystallized with petroleum ether to give the desired product 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride. The reaction time of this step is a total of 20 hours, the yield is 96% oil, 46% hydrochloride. The target product is mp 163-165 ° C.
Step E
The final target from 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} -dimethyl Amine hydrochloride, ie sibutramine hydrochloride, is obtained. Specific synthesis examples are as follows. 3.3 g of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride, 2.99 g of HCOOH and 1 ml of water are mixed while cooling. 3.93 ml of 37% aqueous formaldehyde is added and heated at 85-95 ° C. for 18 hours. Excess hydrochloric acid is added and the solution is evaporated to dryness. 5N NaOH solution is added, extracted with ether and concentrated to give a pale yellow oil. This oil is dissolved in a mixture of 4 mL IPA, 20 mL ether and 2 mL hydrochloric acid is added dropwise. Concentrate, repeatedly dissolve in ethanol and concentrate again. Polishing with petroleum ether gives a yellow solid and recrystallizes with acetone to give the final target sibutramine hydrochloride. The reaction time of this step is a total of 18 hours, the yield is 80%. The target product is mp 195-197 ° C. The yield in 5 steps (A to E) is 18.9%.
As described above, the conventional sibutramine synthesis method has a total of five steps, which is complicated and takes a long time, and requires high temperature vacuum distillation (step A). Since the reaction proceeds at the high temperature of the raw material there was a problem that the yield is reduced. In fact, the synthesis was performed by applying the conventional sibutramine synthesis method, the total yield was very low as 18.9%.
First step
4-Chlorobenzyl cyanide is reacted with 1,3-dibromopropane to give 1- (4-chlorophenyl) -1-cyclobutyl cyanide.
In a flask at room temperature (20-35 ° C.), 14.1 g (352 mmol) of NaH dispersed in mineral oil (200%) and 200 ml of DMSO were added. 25 g (160 mmol) of 4-chlorobenzyl cyanide and 1,3- A solution of 36 g (176 mmol) of dibromopropane dissolved in 200 ml of DMSO was added dropwise. The mixture is stirred at room temperature for 2 hours, 10 ml of IPA is added dropwise and 200 ml of water is added dropwise. The mixture is filtered through a CELITE filter and the solid residue is washed with ether. The ether layer is separated, washed with water, filtered, concentrated and dried to give 34.72 g of crude product of 1- (4-chlorophenyl) -1-cyclobutyl cyanide. The yield of crude product is 109.8%.
2nd step
1- (4-chlorophenyl) -1-cyclobutyl cyanide isobutyl magnesium bromide is reacted to obtain 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine.
10 g (52 mmol) of 1- (4-chlorophenyl) -1-cyclobutyl cyanide was dissolved in 25 ml of toluene at room temperature, followed by addition of a 2.0 M solution of isobutyl magnesium bromide dissolved in 40 ml of diethyl ether. The mixture is heated at reflux at a temperature of at least 105 ° C. for 2 hours. After completion of the reaction at 0 ° C. with methanol, 2.4 g of NaBH 4 was added to the mixture at 0-25 ° C. and stirred for 1 hour or more. Concentrate, treat with ether, water, concentrate again, and vacuum dry. The yield of crude product is 91.6%.
3rd step
The amine group of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine was dimethylated to obtain {1- [1- (4-chlorophenyl) -cyclobutyl]-which is the final object of the present invention. 3-methylbutyl} -dimethylamine hydrochloride, ie sibutramine hydrochloride, is obtained.
5.02 g of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine and 10 ml of HCOOH are mixed with cooling. 6 ml of 37% aqueous formaldehyde is added and heated at 85-95 ° C. for 18 hours. Excess 2M HCl is added and the solution is evaporated to dryness. 5N NaOH solution is added, extracted with ether and concentrated to give a pale yellow oil. After dissolving in a small amount of ether, ether saturated with HCl gas is slowly added dropwise at 0 ° C. The solid obtained was filtered and dried in vacuo to give 5.12 g of sibutramine hydrochloride as the final target. Yield of the product is 92%, mp 193.5-194.8 ° C. The total yield of the first to third stages is 52.7%. The H 1 NMR results of the final product are as follows: H 1 NMR (CDCl 3 ) 1.058 (6H, dd), 1.400 (2H, m), 1.508 (2H, m), 2.193 (3H, d), 2.316 (2H , m), 2.784 (2H, m), 2.910 (3H, d), 2.967 (1H, m), 3.568 (1H, m), 7.386 (2H, d), 7.638 (2H, d), 10.771 (1H, s)
The present invention is to shorten the process that was conventionally carried out in five steps to three steps to greatly shorten the process as well as to eliminate the difficult and time-consuming high-temperature vacuum distillation process to enable mass production In addition, it is possible to greatly reduce production time and production costs by improving the process step by step, and to reduce the production cost by showing a yield improvement effect nearly three times that of the conventional synthesis method in terms of overall yield.
Claims (3)
Hide Dependent
In the method for synthesizing {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} -dimethylamine from 4-chlorobenzyl cyanide,(a) reacting 4-chlorobenzyl cyanide with 1,3-dibromopropane to obtain 1- (4-chlorophenyl) -1-cyclobutyl cyanide;(b) To isobutyl magnesium bromide dissolved in diethyl ether is added to 1- (4-chlorophenyl) -1-cyclobutyl cyanide dissolved in toluene, and the mixture is refluxed at a temperature of 105 ° C. or higher at 1-3 ° C. Heating and cooling for a period of time, followed by addition of NaBH 4 at 0-25 ° C., followed by stirring for at least 1 hour to obtain 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine;(c) Dimethylating the amine group of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine to yield {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methyl Improved synthesis method of sibutramine consisting of a three-step reaction comprising the step of obtaining butyl} -dimethylamine.
The method according to claim 1,In step (a), the solution of 4-chlorobenzyl cyanide and 1,3-dibromopropane dissolved in DMSO is added dropwise to the mixture of NaH and DMSO dispersed in mineral oil, followed by filtration. , Washing, concentrating and drying to obtain a crude product of 1- (4-chlorophenyl) -1-cyclobutyl cyanide, and proceeding to the next step (b) as it is without distillation at high temperature. Improved Synthesis of Sibutramine.
The method according to claim 1,In step (c), 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine is mixed with formaldehyde in a free base state, and 37% aqueous formaldehyde is added thereto, and 85 An improved method for synthesizing sibutramine, characterized in that sibutramine hydrochloride is obtained by heating at -95 ° C for 15-22 hours followed by addition of hydrochloric acid.
SYN
DE 3212682; GB 2098602; US 4806570
4-Chlorobenzyl cyanide (I) is cycloalkylated with 1,3-dibromopropane to yield 1-(4-chlorophenyl)cyclobutyl cyanide (II). The cyclobutyl cyanide (II) is treated with 2-methylpropyl magnesium bromide te give the imine salt (III), which may be either hydrolyzed to the ketone (IV), which is then formylaminated with formamide and formic acid and subsequently hydrolyzed, or reduced with sodium borohydride in ethanol to give 1-[1-(4-chlorophenyl)cyclobutyl]-3-methylbutylamine (V). Eschweiler-Clarke methylation and hydrochloride formation yield N-[1-[1-(4-chlorophenyl)cyclo butyl]-3-methylbutyl]-N,N-dimethylamine hydrochloride monohydrate
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
A history of coronary artery disease (e.g., angina, history of myocardial infarction), congestive heart failure, tachycardia, peripheral arterial occlusive disease, arrhythmia or cerebrovascular disease (stroke or transient ischemic attack (TIA))[14]
A higher number of cardiovascular events has been observed in people taking sibutramine versus control (11.4% vs. 10.0%).[15] In 2010 the FDA noted the concerns that sibutramine increases the risk of heart attacks and strokes in patients with a history of cardiovascular disease.[15]
Frequently encountered side effects are: dry mouth, paradoxically increased appetite, nausea, strange taste in the mouth, upset stomach, constipation, trouble sleeping, dizziness, drowsiness, menstrual cramps/pain, headache, flushing, or joint/muscle pain.
In a 2016 Cochrane review sibutramine was found to substantially increase blood pressure and heart rate in some patients, in the updated review in 2021 sibutramine was not included since the drug had been withdrawn from the market.[16] When used, regular blood pressure monitoring needed to be performed.
The following side effects are infrequent but serious and require immediate medical attention: cardiac arrhythmias, paresthesia, mental/mood changes (e.g., excitement, restlessness, confusion, depression, rare thoughts of suicide).
Currently, no case of pulmonary hypertension has been noted. (Fenfluramine, of the 1990s “Fen-Phen” combo, forced excess release of neurotransmitters—a different action. Phentermine was uninvolved in the rare—but clinically significant—heart issues of fenfluramine.)
Interactions
Sibutramine has a number of clinically significant interactions. The concomitant use of sibutramine and monoamine oxidase inhibitors (MAOIs, such as selegiline) is not indicated, as it may increase the risk of serotonin syndrome, a somewhat rare but serious adverse drug reaction.[17] Sibutramine should not be taken within two weeks of stopping or starting an MAOI. Taking both sibutramine and certain medications used in the treatment of migraines—such as ergolines and triptans—as well as opioids, may also increase the risk for serotonin syndrome, as may the use of more than one serotonin reuptake inhibitor at the same time.[17]
Unlike other serotonergic appetite suppressants like fenfluramine, sibutramine and its metabolites have only low and likely inconsequential affinity for the 5-HT2B receptor.[21]
Pharmacokinetics
Sibutramine is well absorbed from the gastrointestinal tract (77%), but undergoes considerable first-pass metabolism, reducing its bioavailability. The drug itself reaches its peak plasma level after 1 hour and has also a half-life of 1 hour. Sibutramine is metabolized by cytochrome P450isozymeCYP3A4 into two pharmacologically-active primary and secondary amines (called active metabolites 1 and 2) with half-lives of 14 and 16 hours, respectively. Peak plasma concentrations of active metabolites 1 and 2 are reached after three to four hours. The following metabolic pathway mainly results in two inactive conjugated and hydroxylated metabolites (called metabolites 5 and 6). Metabolites 5 and 6 are mainly excreted in the urine.
Sibutramine and its two active N-demethylated metabolites may be measured in biofluids by liquid chromatography–mass spectrometry. Plasma levels of these three species are usually in the 1–10 μg/L range in persons undergoing therapy with the drug. The parent compound and norsibutramine are often not detectable in urine, but dinorsibutramine is generally present at concentrations of >200 μg/L.[26][27][28]
Society and culture
Regulatory approval
Studies are ongoing into reports of sudden death, heart failure, renal failure and gastrointestinal problems. Despite a 2002 petition by Ralph Nader-founded NGOPublic Citizen,[29] the FDA made no attempts to withdraw the drug, but was part of a Senate hearing in 2005.[30] Similarly, David Graham, FDA “whistleblower”, testified before a Senate Finance Committee hearing that sibutramine may be more dangerous than the conditions it is used for.[31]
Between January 2003 and November 2005, a large randomized-controlled “Sibutramine Cardiovascular OUTcomes” (SCOUT) study with 10,742 patients examined whether or not sibutramine administered within a weight management program reduces the risk for cardiovascular complications in people at high risk for heart disease and concluded that use of silbutramine had a RR 1.16 for the primary outcome (composit of nonfatal MI, nonfatal CVA, cardiac arrest, and CV death).[32]
In a dissenting article, “Sibutramine: gone, but not forgotten”, David Haslam (chairman of the National Obesity Forum) says that the SCOUT study is flawed as it only covered high-risk patients and did not consider obese patients who do not have cardiovascular complications or similar contraindications [33]
On January 21, 2010, the European Medicines Agency recommended suspension of marketing authorizations for sibutramine based on the SCOUT study results.[34]
In August 2010 the FDA added a new contraindication for patients over 65 years of age due to the fact that clinical studies of sibutramine did not include sufficient numbers of such patients.[14]
Abbott Laboratories announced on October 8, 2010 that it is withdrawing sibutramine from the US market under pressure from the FDA, citing concerns over minimal efficacy coupled with increased risk of adverse cardiovascular events.[35]
Counterfeit weight-loss products
On December 22, 2008, the United States Food and Drug Administration issued an alert to consumers naming 27 different products marketed as “dietary supplements” for weight loss, that illegally contain undisclosed amounts of sibutramine.[36][37] In March 2009, Dieter Müller et al. published a study of sibutramine poisoning cases from similar Chinese “herbal supplements” sold in Europe, containing as much as twice the dosage of the legally licensed drug.[38]
An additional 34 products were recalled by the FDA on April 22, 2009, further underscoring the risks associated with unregulated “herbal supplements” to unsuspecting persons. This concern is especially relevant to those with underlying medical conditions incompatible with undeclared pharmaceutical adulterants.[39] In January 2010, a similar alert was issued for counterfeit versions of the over-the-counter weight loss drug Alli sold over the Internet. Instead of the active ingredient orlistat, the counterfeit drugs contain sibutramine, and at concentrations at least twice the amount recommended for weight loss.[40]
In March 2010 Health Canada advised the public that illegal “Herbal Diet Natural” had been found on the market, containing sibutramine, which is a prescription drug in Canada, without listing sibutramine as an ingredient.[41] In October 2010 FDA notified consumers that “Slimming Beauty Bitter Orange Slimming Capsules contain the active pharmaceutical ingredient sibutramine, a prescription-only drug which is a stimulant. Sibutramine is not listed on the product label.”[42]
In October 2010 the MHRA in the UK issued a warning regarding “Payouji tea” and “Pai You Guo Slim Capsules” which were found to contain undeclared quantities of sibutramine.[43]
On December 30, 2010 the FDA released a warning regarding “Fruta Planta” dietary products, which were found to contain undeclared amounts of sibutramine. The recall stated that “there is NO SAFE formula on the US market and that all versions of Fruta Planta contain sibutramine. All versions of the formula are UNSAFE and should not be purchased from any source.”[44]
Some illegal weight loss products imported into Ireland have been found to contain sibutramine.[45][46] Similar concerns have been raised in Australia, where illegal imported supplements have been found to contain sibutramine, resulting in public alerts from Australia’s Therapeutic Goods Administration.[47]
In October 2011, the FDA warned that 20 brands of dietary supplements were tainted with sibutramine.[48] In a 2018 study FDA has found synthetic additives including sibutramine in over 700 diet supplements marketed as “natural”, “traditional” or “herbal remedies”.[49]
^ (in German) Sibutramin-Vertrieb in der Europäischen Union ausgesetzt [1]. Abbott Laboratories in Germany. Press Release 2010-01-21. Retrieved 2010-01-27
^ Buckett WR, Thomas PC, Luscombe GP (1988). “The pharmacology of sibutramine hydrochloride (BTS 54 524), a new antidepressant which induces rapid noradrenergic down-regulation”. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 12 (5): 575–84. doi:10.1016/0278-5846(88)90003-6. PMID2851857. S2CID24787523.
^ (in Portuguese) Cloridrato de sibutramina monoidratado. Bula. [Sibutramine hydrochloride monohydrate—label information]. Medley (2007).
^ Nisoli E, Carruba MO (October 2000). “An assessment of the safety and efficacy of sibutramine, an anti-obesity drug with a novel mechanism of action”. Obesity Reviews. 1 (2): 127–39. doi:10.1046/j.1467-789x.2000.00020.x. PMID12119986. S2CID20553857.
^ Heal DJ, Aspley S, Prow MR, Jackson HC, Martin KF, Cheetham SC (August 1998). “Sibutramine: a novel anti-obesity drug. A review of the pharmacological evidence to differentiate it from d-amphetamine and d-fenfluramine”. International Journal of Obesity and Related Metabolic Disorders. 22 Suppl 1: S18–28, discussion S29. PMID9758240.
^ Kim KA, Song WK, Park JY (November 2009). “Association of CYP2B6, CYP3A5, and CYP2C19 genetic polymorphisms with sibutramine pharmacokinetics in healthy Korean subjects”. Clinical Pharmacology and Therapeutics. 86 (5): 511–8. doi:10.1038/clpt.2009.145. PMID19693007. S2CID24789264.
^ Hofbauer K (2004). Pharmacotherapy of obesity : options and alternatives. Boca Raton, Fla: CRC Press. ISBN978-0-415-30321-7.
^ Jain DS, Subbaiah G, Sanyal M, et al. Liquid chromatography/electrospray ionization tandem mass spectrometry validated method for the simultaneous quantification of sibutramine and its primary and secondary amine metabolites in human plasma and its application to a bioequivalence study. Rapid Comm. Mass Spec. 20: 3509-3521, 2006.
^ Thevis M, Sigmund G, Schiffer AK, Schänzer W. Determination of N-desmethyl- and N-bisdesmethyl metabolites of Sibutramine in doping control analysis using liquid chromatography-tandem mass spectrometry. Eur. J. Mass Spec. 12: 129-136, 2006.
^ R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 8th edition, Biomedical Publications, Foster City, CA, 2008, pp. 1426–1427.
^ Japsen B (13 March 2005). “FDA weighs decision on Meridia; Health advisory likely for Abbott obesity drug”. Chicago Tribune. Chicago, Illinois. p. 1.
CAS Name: 2-[5-Ethyltetrahydro-5-[tetrahydro-3-methyl-5-[tetrahydro-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyl-2H-pyran-2-yl]-2-furyl]-2-furyl]-9-hydroxy-b-methoxy-a,g,2,8-tetramethyl-1,6-dioxaspiro[4.5]decane-7-butyric acid
Literature References: Polyether antibiotic. Major factor in antibiotic complex isolated from Streptomyces cinnamonensis. Discovery and isolation: Haney, Hoehn, Antimicrob. Agents Chemother.1967, 349. Production: Haney, Hoehn, US3501568 (1970 to Lilly). Structure: Agtarap et al.,J. Am. Chem. Soc.89, 5737 (1967). Crystal structure studies: Lutz et al.,Helv. Chim. Acta53, 1732 (1970); ibid.54, 1103 (1971). Fermentation studies: Stark et al.,Antimicrob. Agents Chemother.1967, 353. Chemistry: Agtarap, Chamberlin, ibid. 359. Stereocontrolled total synthesis: T. Fukuyama et al.,J. Am. Chem. Soc.101, 262 (1979); D. B. Collum et al.,ibid.102, 2117, 2118, 2120 (1980). 13C-NMR study: J. A. Robinson, D. L. Turner, Chem. Commun.1982, 148. Biosynthesis: Day et al.,Antimicrob. Agents Chemother.4, 410 (1973). Review: Stark, “Monensin, A New Biologically Active Compound Produced by a Fermentation Process”, in Fermentation Advances, Pap. Int. Ferment. Symp., 3rd, 1968, D. Perlman, Ed. (Academic Press, New York, 1969) pp 517-540.
Properties: Crystals, mp 103-105° (monohydrate). [a]D +47.7°. pKa 6.6 (in 66% DMF). Very stable under alkaline conditions. Slightly sol in water; more sol in hydrocarbons; very sol in other organic solvents. LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn).
Melting point: mp 103-105° (monohydrate)
pKa: pKa 6.6 (in 66% DMF)
Optical Rotation: [a]D +47.7°
Toxicity data: LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn)
The structure of monensin was first described by Agtarap et al. in 1967, and was the first polyether antibiotic to have its structure elucidated in this way. The first total synthesis of monensin was reported in 1979 by Kishi et al.[3]
Production / synthesis Monensin is produced in vivo by Streptomyces cinnamonensis as a natural defense against competing bacteria. Monensin presents a formidable challenge to synthetic chemists as it possesses 17 asymmetric centers on a backbone of only 26 carbon atoms. Although its total synthesis has been described (e.g., Kishi et al., 1979), the high complexity of monensin makes an extraction from the bacterium the most economical procedure for its production. The total synthesis has 56 steps and a yield of only 0.26%. The chemical precursors are 2-allyl-1,3-propanediol and 2- (furan-2-yl)acetonitrile. The method used for synthesizing monensin is based on the principle of “absolute asymmetric synthesis”. Molecules are constructed out of prefabricated building blocks in the correct conformation, aiming for higher yields of the desired enantiomer. New stereocenters are also introduced. Using this method, monensin is assembled in two parts, a larger right side and a smaller left one. The penultimate step is connecting the left and the right halves of monensin, which are independently generated, in an Aldol-condensation. The two halves’ keto end groups (C7/ C8) are linked by eliminating a water molecule. The C7 atom is favored over the C1 atom, because it is more reactive. For catalyzing this step, Yoshito Kishi’s group used iPr2NMgBr (Hauser base) and THF to coordinate it at a temperature of − 78°C. Thus, they were able to isolate the molecule in the right conformation at a ratio of 8:1. Due to the low temperature required for a high yield of the correct enantiomer, the reaction is very solw. One of the most difficult steps is the last one: the connection of the spiro center. This is due to a characteristic feature of spiro compounds; they open and close very easily. Therefore, the conditions for forming the right conformation must be optimal in the last step of synthesis. The biosynthesis in a cell culture of Streptomyces cinnamonensis involves a complex medium containing, among other components, glucose, soybean oil, and grit. Cultivation is carried out for a week at a temperature of 30°C and under constant aeration. Product isolation requires filtration, acidification to pH3, extraction with chloroform and purification with activated carbon. In this way, a few grams per liter of monensin are produced and isolated. For crystallization, azeotropic distillation is necessary. In vivo, polyether backbones are assembled by modular polyketide synthases and are modified by two key enzymes, epoxidase and epoxide hydrolase, to generate the product. Precursors of the polyketide pathway are acetate, butyrate and propionate.
A polyether antibiotic, Monensin was the first member of this class of molecules to be structurally characterized.1 The structural features of these polyethers comprise of a terminal carboxylic acid, multiple cyclic ether rings (ex. Tetrahydrofuran and tetrahydropyran), a large amount of stereocenters and (for many of these molecules) one or more spiroketal moieties.2 Monensin was introduced into the market in 1971 and is used to fight coccidial infections in poultry and as an additive in cattle feed.3 Of the 26 carbon atom’s in Monensin’s backbone, 17 are stereogenic and six of those are contiguous. Coupled with a spiroketal moiety, three hydrofuran rings and two hydropyran rings, the molecule was an attractive synthetic target.
1. Agtarap, A.; Chamberlain, J.W.; Pinkerton, M.; Stein-rauf, L. J. Am. Chem. Soc. 1967, 89, 5737 2. Polyether Antibiotics : Naturally Occurring Acid Ionophores. Westley J.W.; Marcel Dekker: New York (1982) Vol. 1-2. 3. Stark, W.M. In Fermentation Advances, Perlman, D., Ed., Academic Press: New York, 1969, 517
Retrosynthetic Analysis of Monensin
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The structure of the sodium (Na+) complex of monensin A.
Monensin A is an ionophore related to the crown ethers with a preference to form complexes with monovalent cations such as: Li+, Na+, K+, Rb+, Ag+, and Tl+.[4][5] Monensin A is able to transport these cations across lipid membranes of cells in an electroneutral (i.e. non-depolarizing) exchange, playing an important role as an Na+/H+antiporter. Recent studies have shown that monensin may transport sodium ion through the membrane in both electrogenic and electroneutral manner.[6] This approach explains ionophoric ability and in consequence antibacterial properties of not only parental monensin, but also its derivatives that do not possess carboxylic groups. It blocks intracellular protein transport, and exhibits antibiotic, antimalarial, and other biological activities.[7] The antibacterial properties of monensin and its derivatives are a result of their ability to transport metal cations through cellular and subcellular membranes.[8]
Uses
Monensin is used extensively in the beef and dairy industries to prevent coccidiosis, increase the production of propionic acid and prevent bloat.[9] Furthermore, monensin, but also its derivatives monensin methyl ester (MME), and particularly monensin decyl ester (MDE) are widely used in ion-selective electrodes.[10][11][12]
In laboratory research, monensin is used extensively to block Golgi transport.[13][14][15]
Toxicity
Monensin has some degree of activity on mammalian cells and thus toxicity is common. This is especially pronounced in horses, where monensin has a median lethal dose 1/100th that of ruminants. Accidental poisoning of equines with monensin is a well-documented occurrence which has resulted in deaths.[16]
^ Kallen, K. J.; Quinn, P.; Allan, D. (1993-02-24). “Monensin inhibits synthesis of plasma membrane sphingomyelin by blocking transport of ceramide through the Golgi: evidence for two sites of sphingomyelin synthesis in BHK cells”. Biochimica et Biophysica Acta (BBA) – Lipids and Lipid Metabolism. 1166 (2–3): 305–308. doi:10.1016/0005-2760(93)90111-l. ISSN0006-3002. PMID8443249.
^ Zhang, G. F.; Driouich, A.; Staehelin, L. A. (December 1996). “Monensin-induced redistribution of enzymes and products from Golgi stacks to swollen vesicles in plant cells”. European Journal of Cell Biology. 71 (4): 332–340. ISSN0171-9335. PMID8980903.
Percent Composition: C 39.85%, H 2.13%, Cl 10.69%, I 38.28%, N 4.22%, O 4.83%
Literature References: Salicylanilide derivative. Prepn: M. A. C. Janssen, V. K. Sipido, BE839481; eidem,US4005218 (1976, 1977 both to Janssen). Effectiveness against Taenia pisiformis in rabbits: R. A. F. Chevis et al.,Vet. Parasitol.7, 333 (1980); against Ancylostoma caninum: J. Guerrero et al.,J. Parasitol.68, 616 (1983); against Fasciola hepatica in sheep: B. E. Stromberg et al.,ibid.70, 446 (1984). Prolonged effect on Haemonchus contortus in sheep: C. A. Hall et al.,Res. Vet. Sci.31, 104 (1981). Acts by uncoupling oxidative phosphorylation: H. Van den Bossche et al.,Arch. Int. Physiol. Biochim.87, 851 (1979); H. J. Kane et al.,Mol. Biochem. Parasitol.1, 347 (1980).
Properties: Crystals from methanol, mp 217.8°.
Melting point: mp 217.8°
Therap-Cat-Vet: Anthelmintic.
N-{5-chloro-4-[(4-chlorophenyl)(cyano)methyl]-2-methylphenyl}-2-hydroxy-3,5-diiodobenzamide is an aromatic amide resulting from the formal condensation of the carboxy group of 3,5-diiodosalicylic acid with the amino group of aniline substituted at positions 2, 4, and 5 by methyl, (4-chlorophenyl)(cyano)methyl, and methyl groups respectively. It is a nitrile, a member of phenols, an organoiodine compound, a monocarboxylic acid amide, an aromatic amide and a member of monochlorobenzenes.
Closantel is a broad-spectrum antiparasitic agent used against
several species and developmental stages of trematodes, nematodes and
arthropods. The anti-trematode activity of closantel is mainly used
against liver fluke. The anti-nematode and anti-arthropod activity is
especially used against those species which feed on blood or plasma.
The drug is widely used in sheep and cattle and can be used
either parenterally (s.c. or i.m.) or orally for both prophylactic and
therapeutic purposes and is available as drench, bolus and injectable
formulations. Closantel has also been combined with mebendazole and
several other benzimidazoles in drench formulations for sheep and with
levamisole in a bolus for cattle (Marsboom et al., 1989).
Closantel has not been evaluated previously by the Joint FAO/WHO
Expert Committee on Food Additives.
Closantel sodium (Closantel Sodium) is a kind of very strong oxidative phosphorylation uncoupler, can suppress the mitochondrial phosphorylation process of polypide, nematode and insect etc. are contacted with blood circulation closely or sucking blood property worm all has and efficiently kills effect, be a kind of broad-spectrum de-worming medicine of efficient, low toxicity, it is huge on market a very large development potentiality.
And 4-chloro-phenyl–(the chloro-4-amino of 2–5-aminomethyl phenyl) cyano group methane is a kind of key intermediate for the synthesis of closantel sodium.But in prior art, the report of the synthetic method of relevant 4-chloro-phenyl–(the chloro-4-amino of 2–5-aminomethyl phenyl) cyano group methane is actually rare, is mainly the iron powder reducing synthetic method.As United States Patent (USP) (US4005218) relates to a kind of with the chloro-α of 4–[the chloro-4-of 2-(hydroxyl imido grpup)-5-methyl-2, the 5-phenylidene] benzyl cyanide (I) is raw material, with excessive iron powder, in ammonium chloride, water and toluene mixing solutions, heating reflux reaction, filter, clean filter cake with a large amount of solvents as tetrahydrofuran (THF) or 4-methyl-2 pentanone, filtrate boils off solvent, then adds the toluene recrystallization to obtain 4-chloro-phenyl–(the chloro-4-amino of 2–5-aminomethyl phenyl) cyano group methane (II).This reaction equation is:
But it is loaded down with trivial details that the shortcoming of the method is operating procedure, the supplementary material consumption is large, and, with producing a large amount of scrap iron powder after iron powder reducing, comparatively thickness, easily comprise product and impurity, and cost recovery is very high, and labour intensity is large, larger to the pollution effect of environment; And product yield and product quality lower.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Tirzepatide is an agonist of human glucose-dependent insulinotropic polypeptide (GIP) and human glucagon-like peptide-1 (GLP-1) receptors, whose amino acid residues at positions 2 and 13 are 2-methylAla, and the C-terminus is amidated Ser. A 1,20-icosanedioic acid is attached to Lys at position 20 via a linker which consists of a Glu and two 8-amino-3,6-dioxaoctanoic acids. Tirzepatide is a synthetic peptide consisting of 39 amino acid residues.
Tirzepatide, sold under the brand name Mounjaro,[1] is a medication used for the treatment type 2 diabetes.[2][3][4] Tirzepatide is given by injection under the skin.[2] Common side effects may include nausea, vomiting, diarrhea, decreased appetite, constipation, upper abdominal discomfort and abdominal pain.[2]
The large-scale manufacture of complex synthetic peptides is challenging due to many factors such as manufacturing risk (including failed product specifications) as well as processes that are often low in both yield and overall purity. To overcome these liabilities, a hybrid solid-phase peptide synthesis/liquid-phase peptide synthesis (SPPS/LPPS) approach was developed for the synthesis of tirzepatide. Continuous manufacturing and real-time analytical monitoring ensured the production of high-quality material, while nanofiltration provided intermediate purification without difficult precipitations. Implementation of the strategy worked very well, resulting in a robust process with high yields and purity.
Preclinical, phase I, and phase II trials have indicated that tirzepatide exhibits similar adverse effects to other established GLP-1 receptor agonists, such as GLP-1 receptor agonist dulaglutide. These effects occur largely within the gastrointestinal tract.[5] The most frequently observed adverse effects are nausea, diarrhoea and vomiting, which increased in incidence with the dosage amount (i.e. higher likelihood the higher the dose). The number of patients who discontinued taking tirzepatide also increased as dosage increased, with patients taking 15 mg having a 25% discontinuation rate vs 5.1% for 5 mg patients and 11.1% for dulaglutide.[6] To a slightly lesser extent, patients also reported reduced appetite.[5] Other side effects reported were dyspepsia, constipation, abdominal pain, dizziness and hypoglycaemia.[7][8]
Tirzepatide has a greater affinity to GIP receptors than to GLP-1 receptors, and this dual agonist behaviour has been shown to produce greater reductions of hyperglycemia compared to a selective GLP-1 receptor agonist.[3]Signaling studies have shown that this is due to tirzepatide mimicking the actions of natural GIP at the GIP receptor.[13] However, at the GLP-1 receptor, tirzepatide shows bias towards cAMP (a messenger associated with regulation of glycogen, sugar and lipid metabolism) generation, rather than β-arrestin recruitment. This combination of preference towards GIP receptor and distinct signaling properties at GLP-1 suggest this biased agonism increases insulin secretion.[13] Tirzepatide has also been shown to increase levels of adiponectin, an adipokine involved in the regulation of both glucose and lipid metabolism, with a maximum increase of 26% from baseline after 26 weeks, at the 10 mg dosage.[3]
Chemistry
Structure
Tirzepatide is an analog of the human GIP hormone with a C20 fatty-diacid portion attached, used to optimise the uptake and metabolism of the compound.[9] The fatty-diacid section (eicosanedioic acid) is linked via a glutamic acid and two (2-(2-aminoethoxy)ethoxy)acetic acid units to the side chain of the lysine residue. This arrangement allows for a much longer half life, extending the time between doses, because of its high affinity to albumin.[14]
Synthesis
The synthesis of tirzepatide was first disclosed in patents filed by Eli Lilly and Company.[15] This uses standard solid phase peptide synthesis, with an allyloxycarbonylprotecting group on the lysine at position 20 of the linear chain of amino acids, allowing a final set of chemical transformations in which the sidechain amine of that lysine is derivatized with the lipid-containing fragment.
Large-scale manufacturing processes have been reported for this compound.[16]
History
Indiana-based pharmaceutical company Eli Lilly and Company first applied for a patent for a method of glycemic control using tirzepatide in early 2016.[15] The patent was published late that year. After passing phase 3 clinical trials, Lilly applied for FDA approval in October 2021 with a priority review voucher.[17]
Following the completion of the pivotal SURPASS-2 trial no. NCT03987919, the company announced on 28 April that tirzepatide had successfully met their endpoints in obese and overweight patients without diabetes.[18] Alongside results from the SURMOUNT-1 trial no. NCT04184622, they suggest that tirzepatide may potentially be a competitor for existing diabetic medication semaglutide, manufactured by Novo Nordisk.[19][20]
In industry-funded preliminary trials comparing tirzepatide to the existing diabetes medication semaglutide (an injected analogue of the hormone GLP-1), tirzepatide showed minor improvement of reductions (2.01%–2.30% depending on dosage) in glycated hemoglobin tests relative to semaglutide (1.86%).[21] A 10 mg dose has also been shown to be effective in reducing insulin resistance, with a reduction of around 8% from baseline, measured using HOMA2-IR (computed with fasting insulin).[3] Fasting levels of IGF binding proteins like IGFBP1 and IGFBP2 increased following tirzepatide treatment, increasing insulin sensitivity.[3] A meta-analysis published by Dutta et al. showed that over 1-year clinical use, tirzepatide was observed to be superior to dulaglutide, semaglutide, degludec, and insulin glargine with regards to glycemic efficacy and obesity reduction. Tirzepatide is perhaps the most potent agent developed to date to tackle the global problem of “diabesity“.[22]
Society and culture
Names
Tirzepatide is the international nonproprietary name (INN).[23]
^ Dahl D, Onishi Y, Norwood P, Huh R, Bray R, Patel H, Rodríguez Á (February 2022). “Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients With Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial”. JAMA. 327 (6): 534–545. doi:10.1001/jama.2022.0078. PMID35133415.
^ Jump up to:abUS patent 9474780, Bokvist BK, Coskun T, Cummins RC, Alsina-Fernandez J, “GIP and GLP-1 co-agonist compounds”, issued 2016-10-25, assigned to Eli Lilly and Co
^ Dutta D, Surana V, Singla R, Aggarwal S, Sharma M (November–December 2021). “Efficacy and safety of novel twincretin tirzepatide a dual GIP and GLP-1 receptor agonist in the management of type-2 diabetes: A Cochrane meta-analysis”. Indian Journal of Endocrinology and Metabolism. 25 (6): 475–489. doi:10.4103/ijem.ijem_423_21.
^World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1). hdl:10665/330896.
Frías JP (November 2020). “Tirzepatide: a glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) dual agonist in development for the treatment of type 2 diabetes”. Expert Rev Endocrinol Metab. 15 (6): 379–394. doi:10.1080/17446651.2020.1830759. PMID33030356.
“Tirzepatide”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03954834 for “A Study of Tirzepatide (LY3298176) in Participants With Type 2 Diabetes Not Controlled With Diet and Exercise Alone (SURPASS-1)” at ClinicalTrials.gov
Clinical trial number NCT03987919 for “A Study of Tirzepatide (LY3298176) Versus Semaglutide Once Weekly as Add-on Therapy to Metformin in Participants With Type 2 Diabetes (SURPASS-2)” at ClinicalTrials.gov
Clinical trial number NCT03882970 for “A Study of Tirzepatide (LY3298176) Versus Insulin Degludec in Participants With Type 2 Diabetes (SURPASS-3)” at ClinicalTrials.gov
Clinical trial number NCT03730662 for “A Study of Tirzepatide (LY3298176) Once a Week Versus Insulin Glargine Once a Day in Participants With Type 2 Diabetes and Increased Cardiovascular Risk (SURPASS-4)” at ClinicalTrials.gov
Clinical trial number NCT04039503 for “A Study of Tirzepatide (LY3298176) Versus Placebo in Participants With Type 2 Diabetes Inadequately Controlled on Insulin Glargine With or Without Metformin (SURPASS-5)” at ClinicalTrials.gov
FDA approves Lilly’s Mounjaro (tirzepatide) injection, the first and only GIP and GLP-1 receptor agonist for the treatment of adults with type 2 diabetes
Mounjaro delivered superior A1C reductions versus all comparators in phase 3 SURPASS clinical trials
While not indicated for weight loss, Mounjaro led to significantly greater weight reductions versus comparators in a key secondary endpoint
Mounjaro represents the first new class of diabetes medicines introduced in nearly a decade and is expected to be available in the U.S. in the coming weeks
INDIANAPOLIS, May 13, 2022 /PRNewswire/ — The U.S. Food and Drug Administration (FDA) approved Mounjaro (tirzepatide) injection, Eli Lilly and Company’s (NYSE: LLY) new once-weekly GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. Mounjaro has not been studied in patients with a history of pancreatitis and is not indicated for use in patients with type 1 diabetes mellitus.
As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP and GLP-1, which are natural incretin hormones.1
“Mounjaro delivered superior and consistent A1C reductions against all of the comparators throughout the SURPASS program, which was designed to assess Mounjaro’s efficacy and safety in a broad range of adults with type 2 diabetes who could be treated in clinical practice. The approval of Mounjaro is an exciting step forward for people living with type 2 diabetes given the results seen in these clinical trials,” said Juan Pablo Frías, M.D., Medical Director, National Research Institute and Investigator in the SURPASS program.
Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
The approval was based on results from the phase 3 SURPASS program, which included active comparators of injectable semaglutide 1 mg, insulin glargine and insulin degludec. Efficacy was evaluated for Mounjaro 5 mg, 10 mg and 15 mg used alone or in combination with commonly prescribed diabetes medications, including metformin, SGLT2 inhibitors, sulfonylureas and insulin glargine. Participants in the SURPASS program achieved average A1C reductions between 1.8% and 2.1% for Mounjaro 5 mg and between 1.7% and 2.4% for both Mounjaro 10 mg and Mounjaro 15 mg. While not indicated for weight loss, mean change in body weight was a key secondary endpoint in all SURPASS studies. Participants treated with Mounjaro lost between 12 lb. (5 mg) and 25 lb. (15 mg) on average.1
Side effects reported in at least 5% of patients treated with Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion (dyspepsia), and stomach (abdominal) pain. The labeling for Mounjaro contains a Boxed Warning regarding thyroid C-cell tumors. Mounjaro is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with Multiple Endocrine Neoplasia syndrome type 2.1
“Lilly has a nearly 100-year heritage of advancing care for people living with diabetes – never settling for current outcomes. We’re not satisfied knowing that half of the more than 30 million Americans living with type 2 diabetes are not reaching their target blood glucose levels,” said Mike Mason, president, Lilly Diabetes. “We are thrilled to introduce Mounjaro, which represents the first new class of type 2 diabetes medication introduced in almost a decade and embodies our mission to bring innovative new therapies to the diabetes community.”
Mounjaro is expected to be available in the United States in the coming weeks. Lilly is committed to helping people access the medicines they are prescribed and will work with insurers, health systems and providers to help enable patient access to Mounjaro. Lilly plans to offer a Mounjaro savings card for people who qualify. Patients or healthcare professionals with questions about Mounjaro can visit www.Mounjaro.com or call The Lilly Answers Center at 1-800-LillyRx (1-800-545-5979).
Tirzepatide is also under regulatory review for the treatment of type 2 diabetes in Europe, Japan and several additional markets. A multimedia gallery is available on Lilly.com.
About the SURPASS clinical trial program The SURPASS phase 3 global clinical development program for tirzepatide began in late 2018 and included five global registration trials and two regional trials in Japan. These studies ranged from 40 to 52 weeks and evaluated the efficacy and safety of Mounjaro 5 mg, 10 mg and 15 mg as a monotherapy and as an add-on to various standard-of-care medications for type 2 diabetes. The active comparators in the studies were injectable semaglutide 1 mg, insulin glargine and insulin degludec. Collectively, the five global registration trials consistently demonstrated A1C reductions for participants taking Mounjaro across multiple stages of their type 2 diabetes journeys, from an average around five to 13 years of having diabetes.2-8
SURPASS-1 (NCT03954834) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=121), 10 mg (N=121) and 15 mg (N=120) as monotherapy to placebo (N=113) in adults with type 2 diabetes inadequately controlled with diet and exercise alone. From a baseline A1C of 7.9%, Mounjaro reduced participants’ A1C by a mean of 1.8%* (5 mg) and 1.7%* (10 mg and 15 mg) compared to 0.1% for placebo. In a key secondary endpoint, from a baseline weight of 189 lb., Mounjaro reduced participants’ weight by a mean of 14 lb.* (5 mg), 15 lb.* (10 mg) and 17 lb.* (15 mg) compared to 2 lb. for placebo.2,3
SURPASS-2 (NCT03987919) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=470), 10 mg (N=469) and 15 mg (N=469) to injectable semaglutide 1 mg (N=468) in adults with type 2 diabetes inadequately controlled with ≥1500 mg/day metformin alone. From a baseline A1C of 8.3%, Mounjaro reduced participants’ A1C by a mean of 2.0%ꝉ (5 mg), 2.2%* (10 mg) and 2.3%* (15 mg) compared to 1.9% for semaglutide. In a key secondary endpoint, from a baseline weight of 207 lb., Mounjaro reduced participants’ weight by a mean of 17 lb.ꝉ (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to 13 lb. for semaglutide.4,5
SURPASS-3 (NCT03882970) was a 52-week study comparing the efficacy of Mounjaro 5 mg (N=358), 10 mg (N=360) and 15 mg (N=358) to titrated insulin degludec (N=359) in adults with type 2 diabetes treated with metformin with or without an SGLT-2 inhibitor. From a baseline A1C of 8.2%, Mounjaro reduced participants’ A1C by a mean of 1.9%* (5 mg), 2.0%* (10 mg) and 2.1%* (15 mg) compared to 1.3% for insulin degludec. From a baseline weight of 208 lb., Mounjaro reduced participants’ weight by a mean of 15 lb.* (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to an increase of 4 lb. for insulin degludec.6
SURPASS-4 (NCT03730662) was a 104-week study comparing the efficacy and safety of Mounjaro 5 mg (N=328), 10 mg (N=326) and 15 mg (N=337) to insulin glargine (N=998) in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The primary endpoint was measured at 52 weeks. From a baseline A1C of 8.5%, Mounjaro reduced participants’ A1C by a mean of 2.1%* (5 mg), 2.3%* (10 mg) and 2.4%* (15 mg) compared to 1.4% for insulin glargine. From a baseline weight of 199 lb., Mounjaro reduced weight by a mean of 14 lb.* (5 mg), 20 lb.* (10 mg) and 23 lb.* (15 mg) compared to an increase of 4 lb. for insulin glargine.7
SURPASS-5 (NCT04039503) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=116), 10 mg (N=118) and 15 mg (N=118) to placebo (N=119) in adults with inadequately controlled type 2 diabetes already being treated with insulin glargine, with or without metformin. From a baseline A1C of 8.3%, Mounjaro reduced A1C by a mean of 2.1%* (5 mg), 2.4%* (10 mg) and 2.3%* (15 mg) compared to 0.9% for placebo. From a baseline weight of 210 lb., Mounjaro reduced participants’ weight by a mean of 12 lb.* (5 mg), 17 lb.* (10 mg) and 19 lb.* (15 mg) compared to an increase of 4 lb. for placebo.8
*p<0.001 for superiority vs. placebo or active comparator, adjusted for multiplicity ꝉp<0.05 for superiority vs. semaglutide 1 mg, adjusted for multiplicity
About Mounjaro (tirzepatide) injection1 Mounjaro (tirzepatide) injection is FDA-approved as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1). Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
PURPOSE AND SAFETY SUMMARY WITH WARNINGS Important Facts About MounjaroTM (mown-JAHR-OH). It is also known as tirzepatide.
Mounjaro is an injectable prescription medicine for adults with type 2 diabetes used along with diet and exercise to improve blood sugar (glucose).
It is not known if Mounjaro can be used in people who have had inflammation of the pancreas (pancreatitis). Mounjaro is not for use in people with type 1 diabetes. It is not known if Mounjaro is safe and effective for use in children under 18 years of age.
Warnings Mounjaro may cause tumors in the thyroid, including thyroid cancer. Watch for possible symptoms, such as a lump or swelling in the neck, hoarseness, trouble swallowing, or shortness of breath. If you have a symptom, tell your healthcare provider.
Do not use Mounjaro if you or any of your family have ever had a type of thyroid cancer called medullary thyroid carcinoma (MTC).
Do not use Mounjaro if you have Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
Do not use Mounjaro if you are allergic to tirzepatide or any of the ingredients in Mounjaro.
Mounjaro may cause serious side effects, including:
Inflammation of the pancreas (pancreatitis). Stop using Mounjaro and call your healthcare provider right away if you have severe pain in your stomach area (abdomen) that will not go away, with or without vomiting. You may feel the pain from your abdomen to your back.
Low blood sugar (hypoglycemia). Your risk for getting low blood sugar may be higher if you use Mounjaro with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin. Signs and symptoms of low blood sugar may include dizziness or light-headedness, sweating, confusion or drowsiness, headache, blurred vision, slurred speech, shakiness, fast heartbeat, anxiety, irritability, or mood changes, hunger, weakness and feeling jittery.
Serious allergic reactions. Stop using Mounjaro and get medical help right away if you have any symptoms of a serious allergic reaction, including swelling of your face, lips, tongue or throat, problems breathing or swallowing, severe rash or itching, fainting or feeling dizzy, and very rapid heartbeat.
Kidney problems (kidney failure). In people who have kidney problems, diarrhea, nausea, and vomiting may cause a loss of fluids (dehydration), which may cause kidney problems to get worse. It is important for you to drink fluids to help reduce your chance of dehydration.
Severe stomach problems. Stomach problems, sometimes severe, have been reported in people who use Mounjaro. Tell your healthcare provider if you have stomach problems that are severe or will not go away.
Changes in vision. Tell your healthcare provider if you have changes in vision during treatment with Mounjaro.
Gallbladder problems. Gallbladder problems have happened in some people who use Mounjaro. Tell your healthcare provider right away if you get symptoms of gallbladder problems, which may include pain in your upper stomach (abdomen), fever, yellowing of skin or eyes (jaundice), and clay-colored stools.
Common side effects The most common side effects of Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion, and stomach (abdominal) pain. These are not all the possible side effects of Mounjaro. Talk to your healthcare provider about any side effect that bothers you or doesn’t go away.
Tell your healthcare provider if you have any side effects. You can report side effects at 1-800-FDA-1088 or www.fda.gov/medwatch.
Before using
Your healthcare provider should show you how to use Mounjaro before you use it for the first time.
Before you use Mounjaro, talk to your healthcare provider about low blood sugar and how to manage it.
Review these questions with your healthcare provider:
Do you have other medical conditions, including problems with your pancreas or kidneys, or severe problems with your stomach, such as slowed emptying of your stomach (gastroparesis) or problems digesting food?
Do you take other diabetes medicines, such as insulin or sulfonylureas?
Do you have a history of diabetic retinopathy?
Are you pregnant or plan to become pregnant or breastfeeding or plan to breastfeed? It is not known if Mounjaro will harm your unborn baby.
Do you take birth control pills by mouth? These may not work as well while using Mounjaro. Your healthcare provider may recommend another type of birth control when you start Mounjaro or when you increase your dose.
Do you take any other prescription medicines or over-the-counter drugs, vitamins, or herbal supplements?
How to take
Read the Instructions for Use that come with Mounjaro.
Use Mounjaro exactly as your healthcare provider says.
Mounjaro is injected under the skin (subcutaneously) of your stomach (abdomen), thigh, or upper arm.
Use Mounjaro 1 time each week, at any time of the day.
Do not mix insulin and Mounjaro together in the same injection.
If you take too much Mounjaro, call your healthcare provider or seek medical advice promptly.
Learn more For more information, call 1-800-LillyRx (1-800-545-5979) or go to www.mounjaro.com.
This information does not take the place of talking with your healthcare provider. Be sure to talk to your healthcare provider about Mounjaro and how to take it. Your healthcare provider is the best person to help you decide if Mounjaro is right for you.
MounjaroTM and its delivery device base are trademarks owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates.
About Lilly Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
This press release contains forward-looking statements (as that term is defined in the Private Securities Litigation Reform Act of 1995) about Mounjaro (tirzepatide 2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg and 15 mg) injection as a treatment to improve glycemic control in adults with type 2 diabetes, the timeline for supply of Mounjaro to become available, and certain other milestones and ongoing clinical trials of Mounjaro and reflects Lilly’s current beliefs and expectations. However, as with any pharmaceutical product or medical device, there are substantial risks and uncertainties in the process of research, development and commercialization. Among other things, there can be no guarantee that Mounjaro will be commercially successful, that future study results will be consistent with results to date, or that we will meet our anticipated timelines for the commercialization of Mounjaro. For further discussion of these and other risks and uncertainties, see Lilly’s most recent Form 10-K and Form 10-Q filings with the United States Securities and Exchange Commission. Except as required by law, Lilly undertakes no duty to update forward-looking statements to reflect events after the date of this release.
References
Mounjaro. Prescribing Information. Lilly USA, LLC.
Rosenstock, J, et. al. Efficacy and Safety of Once Weekly Tirzepatide, a Dual GIP/GLP-1 Receptor Agonist Versus Placebo as Monotherapy in People with Type 2 Diabetes (SURPASS-1). Abstract 100-OR. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
Rosenstock, J, et. al. (2021). Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. 2021;398(10295):143-155. doi: 10.1016/S0140-6736(21)01324-6.
Frías JP, Davies MJ, Rosenstock J, et al; for the SURPASS-2 Investigators. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385(6)(suppl):503-515. doi: 10.1056/NEJMoa2107519
Frias, J.P. Efficacy and Safety of Tirzepatide vs. Semaglutide Once Weekly as Add-On Therapy to Metformin in Patients with Type 2 Diabetes. Abstract 84-LB. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
Ludvik B, Giorgino F, Jódar E, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet. 2021;398(10300):583-598. doi: 10.1016/S0140-6736(21)01443-4
Del Prato S, Kahn SE, Pavo I, et al; for the SURPASS-4 Investigators. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. 2021;398(10313):1811-1824. doi: 10.1016/S0140-6736(21)02188-7
Dahl D, Onishi Y, Norwood P, et al. Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: the SURPASS-5 randomized clinical trial. JAMA. 2022;327(6):534-545. doi:10.1001/jama.2022.0078
Participants taking tirzepatide lost up to 52 lb. (24 kg) in this 72-week phase 3 study
63% of participants taking tirzepatide 15 mg achieved at least 20% body weight reductions as a key secondary endpoint
INDIANAPOLIS, April 28, 2022 /PRNewswire/ — Tirzepatide (5 mg, 10 mg, 15 mg) achieved superior weight loss compared to placebo at 72 weeks of treatment in topline results from Eli Lilly and Company’s (NYSE: LLY) SURMOUNT-1 clinical trial, with participants losing up to 22.5% (52 lb. or 24 kg) of their body weight for the efficacy estimandi. This study enrolled 2,539 participants and was the first phase 3 global registration trial evaluating the efficacy and safety of tirzepatide in adults with obesity, or overweight with at least one comorbidity, who do not have diabetes. Tirzepatide met both co-primary endpoints of superior mean percent change in body weight from baseline and greater percentage of participants achieving body weight reductions of at least 5% compared to placebo for both estimandsii. The study also achieved all key secondary endpoints at 72 weeks.
For the efficacy estimand, participants taking tirzepatide achieved average weight reductions of 16.0% (35 lb. or 16 kg on 5 mg), 21.4% (49 lb. or 22 kg on 10 mg) and 22.5% (52 lb. or 24 kg on 15 mg), compared to placebo (2.4%, 5 lb. or 2 kg). Additionally, 89% (5 mg) and 96% (10 mg and 15 mg) of people taking tirzepatide achieved at least 5% body weight reductions compared to 28% of those taking placebo.
In a key secondary endpoint, 55% (10 mg) and 63% (15 mg) of people taking tirzepatide achieved at least 20% body weight reductions compared to 1.3% of those taking placebo. In an additional secondary endpoint not controlled for type 1 error, 32% of participants taking tirzepatide 5 mg achieved at least 20% body weight reductions. The mean baseline body weight of participants was 231 lb. (105 kg).
“Obesity is a chronic disease that often does not receive the same standard of care as other conditions, despite its impact on physical, psychological and metabolic health, which can include increased risk of hypertension, heart disease, cancer and decreased survival,” said Louis J. Aronne, MD, FACP, DABOM, director of the Comprehensive Weight Control Center and the Sanford I. Weill Professor of Metabolic Research at Weill Cornell Medicine, obesity expert at NewYork-Presbyterian/Weill Cornell Medical Center and Investigator of SURMOUNT-1. “Tirzepatide delivered impressive body weight reductions in SURMOUNT-1, which could represent an important step forward for helping the patient and physician partnership treat this complex disease.”
For the treatment-regimen estimandiii, results showed:
Average body weight reductions: 15.0% (5 mg), 19.5% (10 mg), 20.9% (15 mg), 3.1% (placebo)
Percentage of participants achieving body weight reductions of ≥5%: 85% (5 mg), 89% (10 mg), 91% (15 mg), 35% (placebo)
Percentage of participants achieving body weight reductions of ≥20%: 30% (5 mg, not controlled for type 1 error), 50% (10 mg), 57% (15 mg), 3.1% (placebo)
The overall safety and tolerability profile of tirzepatide was similar to other incretin-based therapies approved for the treatment of obesity. The most commonly reported adverse events were gastrointestinal-related and generally mild to moderate in severity, usually occurring during the dose escalation period. For those treated with tirzepatide (5 mg, 10 mg and 15 mg, respectively), nausea (24.6%, 33.3%, 31.0%), diarrhea (18.7%, 21.2%, 23.0%), vomiting (8.3%, 10.7%, 12.2%) and constipation (16.8%, 17.1%, 11.7%) were more frequently experienced compared to placebo (9.5% [nausea], 7.3% [diarrhea], 1.7% [vomiting], 5.8% [constipation]).
Treatment discontinuation rates due to adverse events were 4.3% (5 mg), 7.1% (10 mg), 6.2% (15 mg) and 2.6% (placebo). The overall treatment discontinuation rates were 14.3% (5 mg), 16.4% (10 mg), 15.1% (15 mg) and 26.4% (placebo).
Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and the potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
“Tirzepatide is the first investigational medicine to deliver more than 20 percent weight loss on average in a phase 3 study, reinforcing our confidence in its potential to help people living with obesity,” said Jeff Emmick, MD, Ph.D., vice president, product development, Lilly. “Obesity is a chronic disease that requires effective treatment options, and Lilly is working relentlessly to support people with obesity and modernize how this disease is approached. We’re proud to research and develop potentially innovative treatments like tirzepatide, which helped nearly two thirds of participants on the highest dose reduce their body weight by at least 20 percent in SURMOUNT-1.”
Tirzepatide is a novel investigational once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist, representing a new class of medicines being studied for the treatment of obesity. Tirzepatide is a single peptide that activates the body’s receptors for GIP and GLP-1, two natural incretin hormones. Obesity is a chronic, progressive disease caused by disruptions in the mechanisms that control body weight, often leading to an increase in food intake and/or a decrease in energy expenditure. These disruptions are multifactorial and can be related to genetic, developmental, behavioral, environmental and social factors. To learn more, visit Lilly.com/obesity.
Lilly will continue to evaluate the SURMOUNT-1 results, which will be presented at an upcoming medical meeting and submitted to a peer-reviewed journal. Additional studies are ongoing for tirzepatide as a potential treatment for obesity or overweight.
About tirzepatide
Tirzepatide is a once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1 receptor agonists. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with GLP-1 receptor agonism, may result in greater effects on markers of metabolic dysregulation such as body weight, glucose and lipids. Tirzepatide is in phase 3 development for adults with obesity or overweight with weight-related comorbidity and is currently under regulatory review as a treatment for adults with type 2 diabetes. It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH) and heart failure with preserved ejection fraction (HFpEF). Studies of tirzepatide in obstructive sleep apnea (OSA) and in morbidity/mortality in obesity are planned as well.
About SURMOUNT-1 and the SURMOUNT clinical trial program
SURMOUNT-1 (NCT04184622) is a multi-center, randomized, double-blind, parallel, placebo-controlled trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to placebo as an adjunct to a reduced-calorie diet and increased physical activity in adults without type 2 diabetes who have obesity, or overweight with at least one of the following comorbidities: hypertension, dyslipidemia, obstructive sleep apnea or cardiovascular disease. The trial randomized 2,539 participants across the U.S., Argentina, Brazil, China, India, Japan, Mexico, Russia and Taiwan in a 1:1:1:1 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or placebo. The co-primary objectives of the study were to demonstrate that tirzepatide 10 mg and/or 15 mg is superior in percentage of body weight reductions from baseline and percentage of participants achieving ≥5% body weight reduction at 72 weeks compared to placebo. Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg).
The SURMOUNT phase 3 global clinical development program for tirzepatide began in late 2019 and has enrolled more than 5,000 people with obesity or overweight across six clinical trials, four of which are global studies. Results from SURMOUNT-2, -3, and -4 are anticipated in 2023.
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
Tirzepatide results superior A1C and body weight reductions compared to insulin glargine in adults with type 2 diabetes
Newly published data show that participants maintained A1C and weight control up to two years in SURPASS-4, the largest and longest SURPASS trial completed to dateNo increased cardiovascular risk identified with tirzepatide; hazard ratio of 0.74 observed for MACE-4 events
SURPASS-4 is the largest and longest clinical trial completed to date of the phase 3 program studying tirzepatide as a potential treatment for type 2 diabetes. The primary endpoint was measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE) to assess CV risk. In newly published data from the treatment period after 52 weeks, participants taking tirzepatide maintained A1C and weight control for up to two years.
The overall safety profile of tirzepatide, assessed over the full study period, was consistent with the safety results measured at 52 weeks, with no new findings up to 104 weeks. Gastrointestinal side effects were the most commonly reported adverse events, usually occurring during the escalation period and then decreasing over time.
“We are encouraged by the continued A1C and weight control that participants experienced past the initial 52 week treatment period and up to two years as we continue to explore the potential impact of tirzepatide for the treatment of type 2 diabetes,” said John Doupis, M.D., Ph.D., Director, Diabetes Division and Clinical Research Center, Iatriko Paleou Falirou Medical Center, Athens, Greece and Senior Investigator for SURPASS-4.
Tirzepatide is a novel investigational once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single molecule, representing a new class of medicines being studied for the treatment of type 2 diabetes.
SURPASS-4 was an open-label global trial comparing the safety and efficacy of three tirzepatide doses (5 mg, 10 mg and 15 mg) to titrated insulin glargine in 2,002 adults with type 2 diabetes with increased CV risk who were treated with between one and three oral antihyperglycemic medicines (metformin, a sulfonylurea or an SGLT-2 inhibitor). Of the total participants randomized, 1,819 (91%) completed the primary 52-week visit and 1,706 (85%) completed the study on treatment. The median study duration was 85 weeks and 202 participants (10%) completed two years.
Study participants had a mean duration of diabetes of 11.8 years, a baseline A1C of 8.52 percent and a baseline weight of 90.3 kg. More than 85 percent of participants had a history of cardiovascular events. In the insulin glargine arm, the insulin dose was titrated following a treat-to-target algorithm with the goal of fasting blood glucose below 100 mg/dL. The starting dose of insulin glargine was 10 units per day, and the mean dose of insulin glargine at 52 weeks was 43.5 units per day.
About tirzepatide Tirzepatide is a once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with a GLP-1 receptor agonist, may result in greater effects on glucose and body weight. Tirzepatide is in phase 3 development for blood glucose management in adults with type 2 diabetes, for chronic weight management and heart failure with preserved ejection fraction (HFpEF). It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH).
About SURPASS-4 and the SURPASS clinical trial program SURPASS-4 (NCT03730662) is a randomized, parallel, open-label trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to insulin glargine in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The trial randomized 2,002 study participants in a 1:1:1:3 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or insulin glargine. Participants were located in the European Union, North America (Canada and the United States), Australia, Israel, Taiwan and Latin America (Brazil, Argentina and Mexico). The primary objective of the study was to demonstrate that tirzepatide (10 mg and/or 15 mg) is non-inferior to insulin glargine for change from baseline A1C at 52 weeks in people with type 2 diabetes and increased CV risk. The primary and key secondary endpoints were measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE). Study participants enrolled had to have a mean baseline A1C between 7.5 percent and 10.5 percent and a BMI greater than or equal to 25 kg/m2 at baseline. All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg). All participants in the titrated insulin glargine treatment arm started with a baseline dose of 10 units per day and titrated following a treat-to-target algorithm to reach a fasting blood glucose below 100 mg/dL.
The SURPASS phase 3 global clinical development program for tirzepatide has enrolled more than 20,000 people with type 2 diabetes across 10 clinical trials, five of which are global registration studies. The program began in late 2018, and all five global registration trials have been completed.
About Diabetes
Approximately 34 million Americans2 (just over 1 in 10) and an estimated 463 million adults worldwide3 have diabetes. Type 2 diabetes is the most common type internationally, accounting for an estimated 90 to 95 percent of all diabetes cases in the United States alone2. Diabetes is a chronic disease that occurs when the body does not properly produce or use the hormone insulin.
TIC10 (ONC-201) is a potent, orally active, and stable tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) inducer which acts by inhibiting Akt and ERK, consequently activating Foxo3a and significantly inducing cell surface TRAIL. TIC10 can cross the blood-brain barrier.
ONC-201, also known as TIC10, is a potent, orally active, and stable small molecule that transcriptionally induces TRAIL in a p53-independent manner and crosses the blood-brain barrier. TIC10 induces a sustained up-regulation of TRAIL in tumors and normal cells that may contribute to the demonstrable antitumor activity of TIC10. TIC10 inactivates kinases Akt and extracellular signal-regulated kinase (ERK), leading to the translocation of Foxo3a into the nucleus, where it binds to the TRAIL promoter to up-regulate gene transcription. TIC10 is an efficacious antitumor therapeutic agent that acts on tumor cells and their microenvironment to enhance the concentrations of the endogenous tumor suppressor TRAIL.
Akt/ERK Inhibitor ONC201 is a water soluble, orally bioavailable inhibitor of the serine/threonine protein kinase Akt (protein kinase B) and extracellular signal-regulated kinase (ERK), with potential antineoplastic activity. Upon administration, Akt/ERK inhibitor ONC201 binds to and inhibits the activity of Akt and ERK, which may result in inhibition of the phosphatidylinositol 3-kinase (PI3K)/Akt signal transduction pathway as well as the mitogen-activated protein kinase (MAPK)/ERK-mediated pathway. This may lead to the induction of tumor cell apoptosis mediated by tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)/TRAIL death receptor type 5 (DR5) signaling in AKT/ERK-overexpressing tumor cells. The PI3K/Akt signaling pathway and MAPK/ERK pathway are upregulated in a variety of tumor cell types and play a key role in tumor cell proliferation, differentiation and survival by inhibiting apoptosis. In addition, ONC201 is able to cross the blood-brain barrier.
Herein, we present a copper-catalyzed tandem reaction of 2-aminoimidazolines and ortho-halo(hetero)aryl carboxylic acids that causes the regioselective formation of angularly fused tricyclic 1,2-dihydroimidazo[1,2-a]quinazolin-5(4H)-one derivatives. The reaction involved in the construction of the core six-membered pyrimidone moiety proceeded via regioselective N-arylation–condensation. The presented protocol been successfully applied to accomplish the total synthesis of TIC10/ONC201, which is an active angular isomer acting as a tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL): a sought after anticancer clinical agent.
ONC201 is the founding member of a novel class of anti-cancer compounds called imipridones that is currently in Phase II clinical trials in multiple advanced cancers. Since the discovery of ONC201 as a p53-independent inducer of TRAIL gene transcription, preclinical studies have determined that ONC201 has anti-proliferative and pro-apoptotic effects against a broad range of tumor cells but not normal cells. The mechanism of action of ONC201 involves engagement of PERK-independent activation of the integrated stress response, leading to tumor upregulation of DR5 and dual Akt/ERK inactivation, and consequent Foxo3a activation leading to upregulation of the death ligand TRAIL. ONC201 is orally active with infrequent dosing in animals models, causes sustained pharmacodynamic effects, and is not genotoxic. The first-in-human clinical trial of ONC201 in advanced aggressive refractory solid tumors confirmed that ONC201 is exceptionally well-tolerated and established the recommended phase II dose of 625 mg administered orally every three weeks defined by drug exposure comparable to efficacious levels in preclinical models. Clinical trials are evaluating the single agent efficacy of ONC201 in multiple solid tumors and hematological malignancies and exploring alternative dosing regimens. In addition, chemical analogs that have shown promise in other oncology indications are in pre-clinical development. In summary, the imipridone family that comprises ONC201 and its chemical analogs represent a new class of anti-cancer therapy with a unique mechanism of action being translated in ongoing clinical trials.
////////////IMIPRIDONE, TIC 10, ONC 201, NSC 350625, OP 10, Fast Track Designation, Orphan Drug Designation, Rare Pediatric Disease Designation, PHASE 3, GLIOMA, CHIMERIX



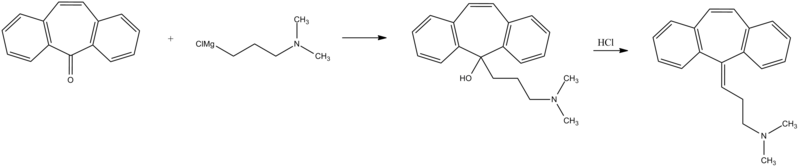












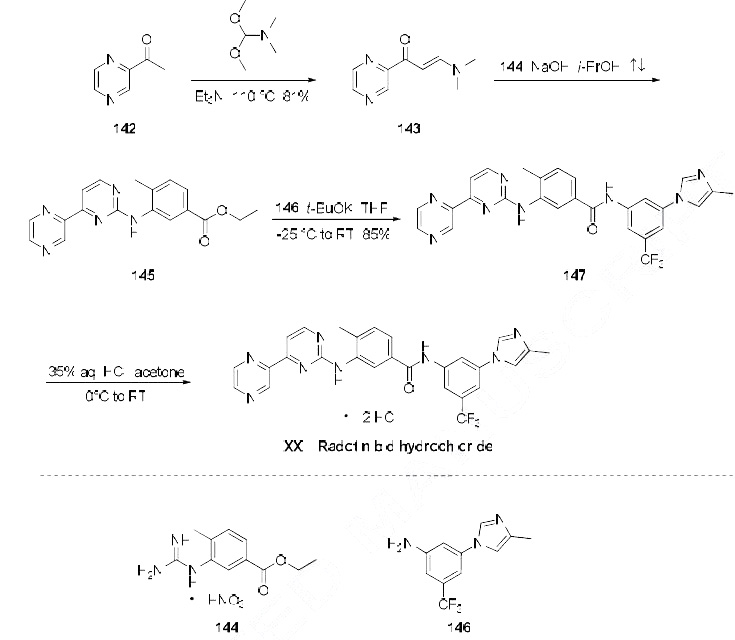










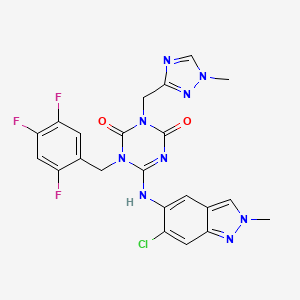
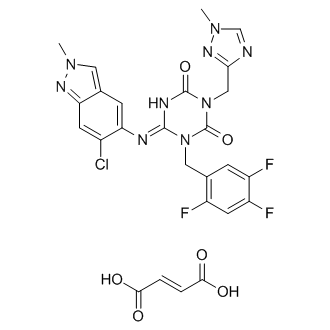










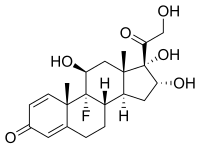
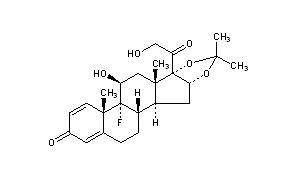

































![{\displaystyle [\alpha ]_{D}^{20}}](http://wikimedia.org/api/rest_v1/media/math/render/svg/b67b300e0d318e964fcdab92c872ed89d1d4f43f) +65° to +72° cm³/dm·g (1% in
+65° to +72° cm³/dm·g (1% in 
 (ciltacabtagene autoleucel), Janssen’s First Cell Therapy, a BCMA-Directed CAR-T Immunotherapy for the Treatment of Patients with Relapsed or Refractory Multiple Myeloma
(ciltacabtagene autoleucel), Janssen’s First Cell Therapy, a BCMA-Directed CAR-T Immunotherapy for the Treatment of Patients with Relapsed or Refractory Multiple Myeloma


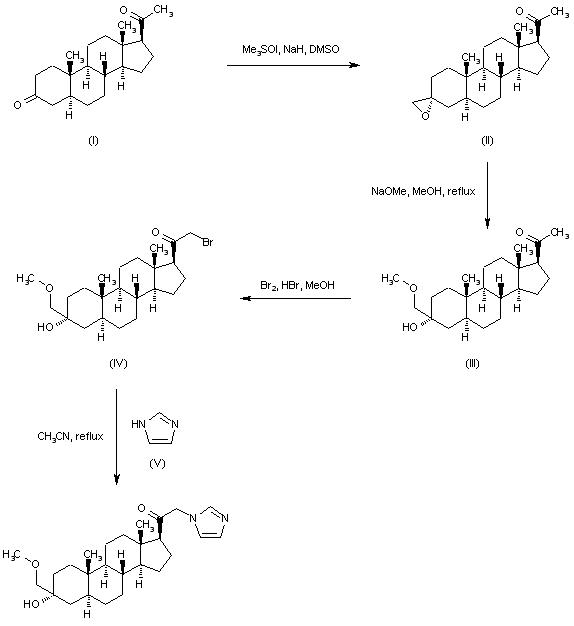
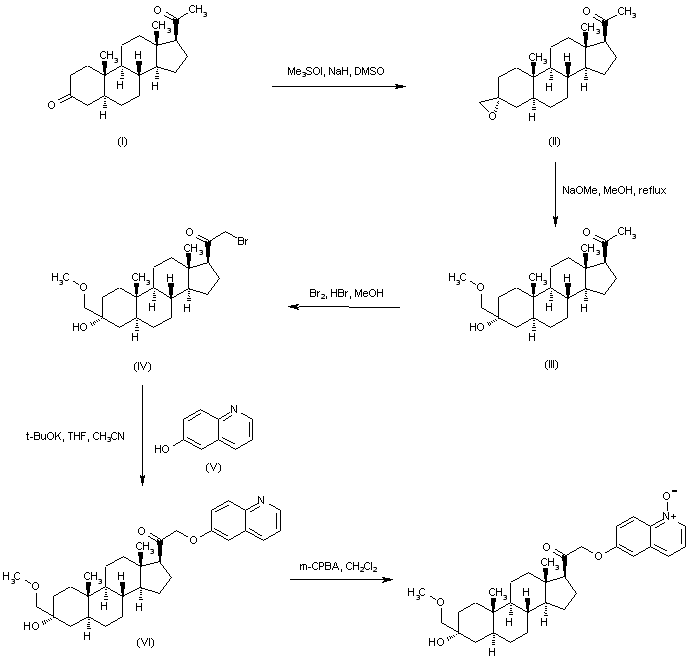





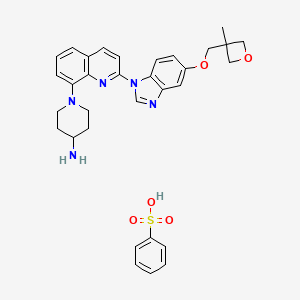

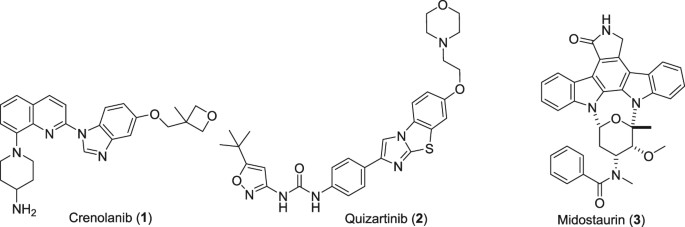











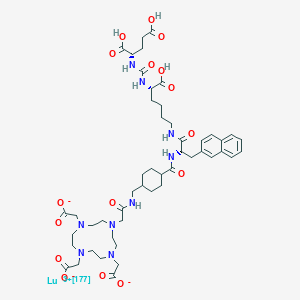



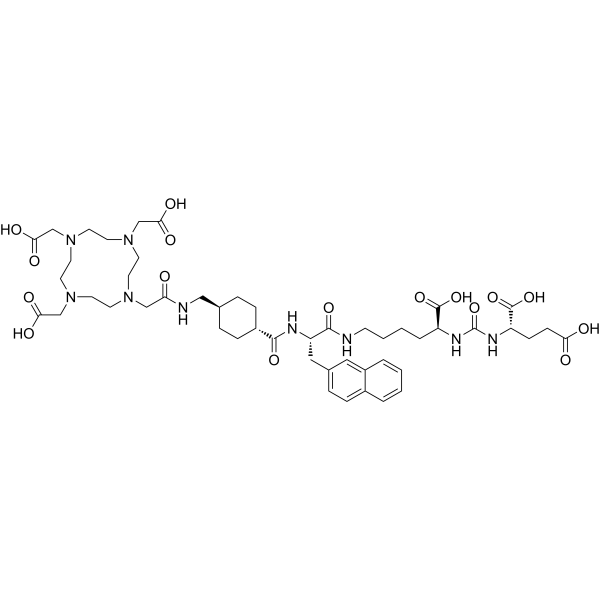


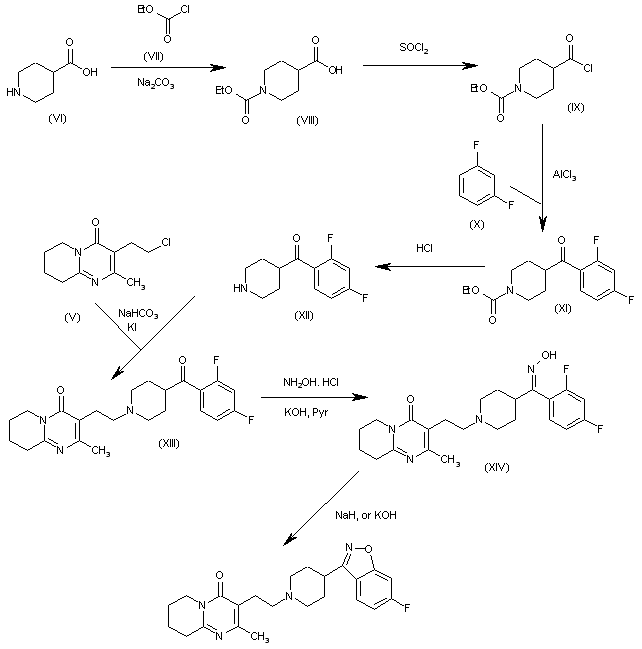
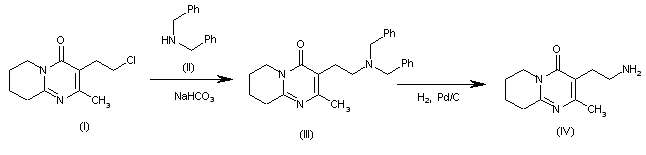
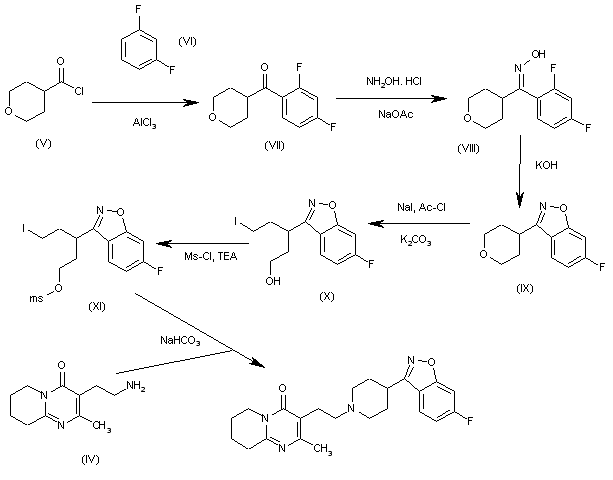






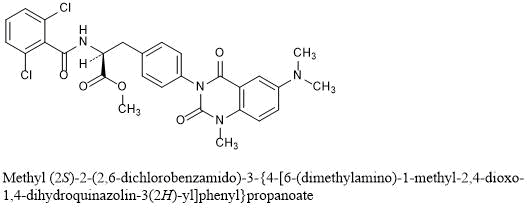
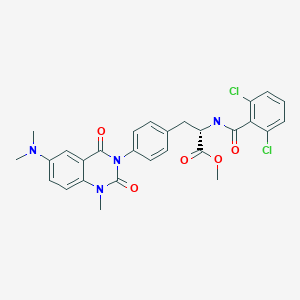









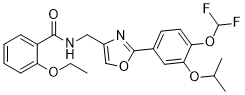
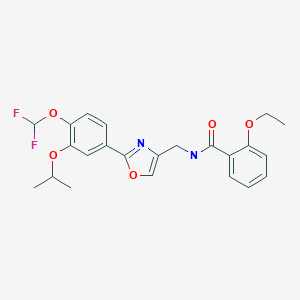













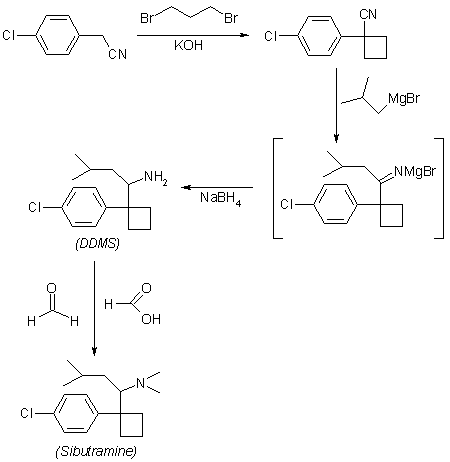
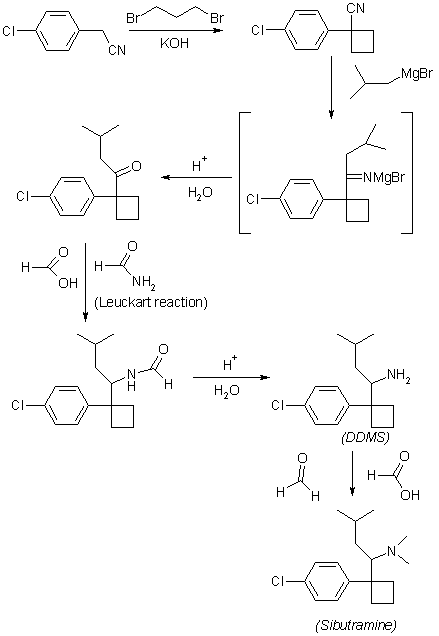









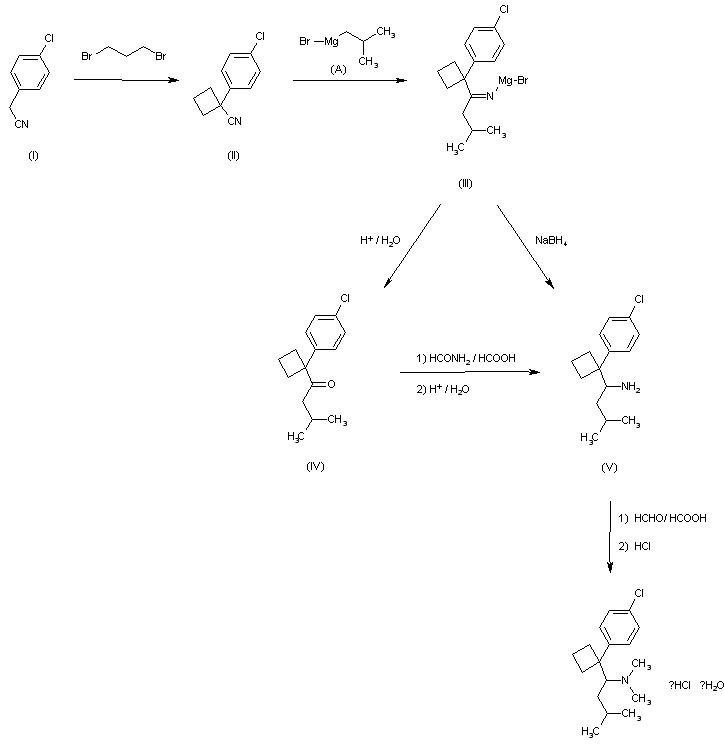


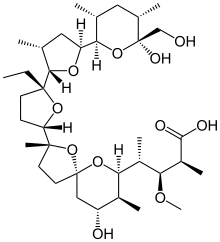
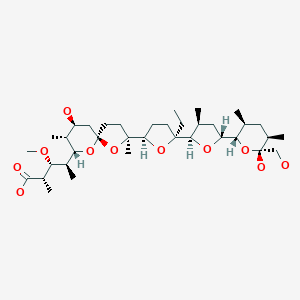









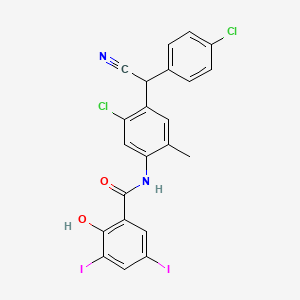
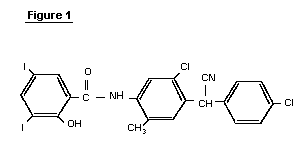












![7-Benzyl-4-(2-methylbenzyl)-1,2,6,7,8,9-hexahydroimidazo[1,2-A]pyrido[3,4-E]pyrimidin-5(4H)-one.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=73777259&t=l)