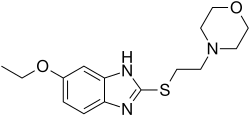
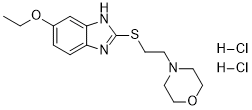
19388-87-5[RN] 243-016-5[EINECS] 2H-1,2,4-Thiadiazine, 4,4′-methylenebis[tetrahydro-, 1,1,1′,1′-tetraoxide 4,4′-methanediylbis(1,2,4-thiadiazinane) 1,1,1′,1′-tetraoxide UNII-8OBZ1M4V3V тауролидин توروليدين 牛磺利定 NMR https://www.apexbt.com/downloader/document/C4559/NMR-2.pdf MS https://www.apexbt.com/downloader/document/C4559/MS-2.pdf Taurolidine CAS Registry Number: 19388-87-5 CAS Name: 4,4¢-Methylenebis(tetrahydro-1,2,4-thiadiazine) 1,1,1¢,1¢-tetraoxide Additional Names: 4,4¢-methylenebis(perhydro-1,2,4-thiadiazine 1,1-dioxide); bis(1,1-dioxoperhydro-1,2,4-thiadiazin-4-yl)methane Trademarks: Drainasept (Geistlich); Taurolin (HMR); Tauroflex (Geistlich) Molecular Formula: C7H16N4O4S2, Molecular Weight: 284.36 Percent Composition: C 29.57%, H 5.67%, N 19.70%, O 22.51%, S 22.55% Literature References: Broad spectrum, synthetic formaldehyde carrier formed by the condensation of two molecules of taurine and three molecules of formaldehyde. Prepn: FR1458701; R. W. Pfirrmann, US3423408 (1966, 1969 both to Ed. Geistlich Söhne). Antibacterial activity in mice: M. K. Browne et al.,J. Appl. Bacteriol.41, 363 (1976). Anti-endotoxin activity in lab animals: R. W. Pfirrmann, G. B. Leslie, ibid.46, 97 (1979). Mechanism of action: E. Myers et al.,ibid.48, 89 (1980). HPLC determn of metabolites in plasma: A. D. Woolfson et al.,Int. J. Pharm.49, 135 (1989). Pharmacokinetics: C. Steinbach-Lebbin et al.,Arzneim.-Forsch.32, 1542 (1982). Metabolism in humans: B. I. Knight et al.,Br. J. Clin. Pharmacol.12, 695 (1981). Clinical trials in peritonitis: M. K. Browne et al.,Surg. Gynecol. Obstet.146, 721 (1978); G. Wesch et al.,Fortschr. Med.101, 545 (1983); in wound sepsis: A. K. Halsall et al.,Pharmatherapeutica2, 673 (1981); in pleural infection: A. A. Conlan et al.,S. Afr. Med. J.64, 653 (1983). Properties: White crystals, mp 154-158°. Sol in water. Melting point: mp 154-158° Therap-Cat: Antibacterial. Keywords: Antibacterial (Synthetic).
It is derived from the endogenous amino acidtaurine. Taurolidine’s putative mechanism of action is based on a chemical reaction. During the metabolism of taurolidine to taurinamide and ultimately taurine and water, methylol groups are liberated that chemically react with the mureins in the bacterial cell wall and with the amino and hydroxyl groups of endotoxins and exotoxins. This results in denaturing of the complex polysaccharide and lipopolysaccharide components of the bacterial cell wall and of the endotoxin and in the inactivation of susceptible exotoxins.[3]
Taurolidine is an antibacterial drug and also has antiendotoxic substance, which is used as an antiseptic solution in surgery for washing out the abdominal cavity and it also prevents septic shock. It is commercially sold as Taurolidine (Formula I). The present invention relates to a process for the preparation of Taurolidine which provides significant advantages over the existing processes.
Formula I
The current process for the preparation of Taurolidine is depicted in Scheme 1
Formula II
Formula IVFormula IThe present inventors thus propose an industrially viable procedure for isolation of Taurolidine in substantially pure form.Taurolidine is dissolved in a suitable solvent to obtain a clear solution. The product starts to precipitate and an anti solvent is added optionally to maximize the precipitation procedure. The solvents employed for the purification are non -aqueous aprotic solvents comprising DMSO, DMAc, DMF, Acetonitirle, DMSO being the most preferred solvent. The antisolvents employed are toluene, ethyl acetate, dichloromethane, ether; toluene being the most preferred.Taurolidine obtained by the instant procedure has purity greater than or equal to 99.5 %. The process of the invention is illustrated by the following examples to obtain Taurolidine. Example ICbz-Taurine sodium salt (Formula II)To 1000ml of water in the RBF charge 192gm of (3.0 eq) of sodium hydroxide under cooling followed by 200gm of Taurine and dissolve it until clear solution is obtained. Cool to 0°C to 5°C, and Charge 50% CBZ-C1 in toluene at 0°C to 5°C. After completion of addition, maintain at room temperature for 14h. Separate the toluene layer and wash the aqueous layer with 2x200ml of ethyl acetate. Add slowly 27gm of sodium hydroxide in 60ml of water to the aqueous layer and adjust pH to 12- 14. Cool to 0°C to 5°C and a white solid separates from the solution. Filter the solid and dry the solid at 60 -70 °C. Weight of the solid: 320 gExample 2Cbz-Taurinamide (Formula III)To a clean dry flask charge 1500ml of toluene and charge 320gm of Formula II and cool to 0°C to 5°C. Charge 308 gm of PC15 slowly at 0°C to 5°C for 2hrs. Maintain at 0°C to 5°C up to completion of reaction. Quench the RM into another flask containing 2 ltr of water at 0°C to 5°C. Separate the organic layer, wash and extract the aqueous layer with toluene. Dry the organic layer with sodium sulphate and cool to 0°C to 5°C. Purge ammonia gas into the reaction mass till the reaction is complete. Filter the solid and dissolve the solid in 21tr of water and extract the aqueous layer with 2x600ml of ethyl acetate. Dry the organic layer with sodium sulphate and concentrate it under reduced pressure to obtain a white solid. Weight of the solid: 150 gExample 3Taurinamide Succinate (Formula IV)Take a suspension of 100 g of Cbz-Taurinamide in 1000 ml methanol, and 10% Pd/C (1 .0 g) and subject to hydrogenation at 45-50 psi. Upon completion of the reaction filter the catalyst and add succinic acid (1 .0 eq) to the solvent and distill off the solvent under vacuum to provide the title compound in about 90% yield as a white solid.Example 4Taurolidine (Formula I)To a solution of 100 g Taurinamide succinate in water is added sat sodium bicarbonate solution and pH adjusted to 7-8. To the solution was added formaldehyde (50 ml) and allowed to stir for 4 h. The solid obtained was filtered and washed with water to give Taurolidine. The title compound was obtained in about 70% yield and about 98% purity.Example 5Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. The solid is filtered and washed with toluene and dried to give a white solid in 40 % yield. The product obtained was >99.5% pure.Example 6Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. To the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70 % yield. The product obtained was >99.5% pure by HPLC and passed elemental analysis within 0.4% of the theoretical values.Example 7Purification of TaurolidineTaurolidine (100 g) was dissolved in DMAc (800 ml) and to the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70% yield.
Taurolidine has anti-endotoxin, anti-bacterial and anti-adherent properties. In terms of bacteria, taurolidine can chemically react with cell walls, endotoxins and exotoxins to inhibit microbial adhesion and play an antibacterial role. In addition, in terms of anti-tumor, taurolidine can induce cytotoxicity of tumor cells by inducing apoptosis, autophagy and necrosis. The extent to which these processes are involved may vary with the type of tumor cell. Until July 2020, there were more than 260 foreign literature searches on taurolidine research reports, most of which focused on the exploration of the effect of taurolidine on tumor-related signaling pathways, while the application of taurolidine in antiviral activity was not yet available. See research reports. PATENThttps://patents.google.com/patent/CN101274921B/en Taurolidine synthetic operation step:1. the preparation of tauryl villaumite hydrochlorate In being housed, ventpipe, escape pipe, thermometer and churned mechanically 300ml four-necked bottle add Mercaptamine 25g, 200ml methylene dichloride and 32ml dehydrated alcohol, under ice-water bath (below the 10 ℃) mechanical stirring, feed dry appropriate chlorine, reaction begins and heat release immediately, the thick solid of adularescent generates, and temperature remains on below 50 ℃ and stirs, reaction 5h.The whole process HCl gas and the monochloroethane gas of alkali lye absorption reaction process.Stop logical chlorine after reacting end, get yellow mercury oxide, suction filtration is used washed with dichloromethane four times, and vacuum-drying gets white solid 50g, 152~154 ℃ of fusing points.2. the preparation of tauryl azide salt hydrochlorate The reaction flask ice-water bath of containing 45ml water is cooled to-15 ℃, adds NaN 3(2g), after stirring is molten entirely, add slightly pinkiness of tauryl villaumite hydrochlorate (9g) solution in batches, the water-bath of 20 minutes recession deicings, room temperature continues stirring 60 minutes.3. the preparation of tauryl amine hydrochlorate Above-mentioned reaction solution is joined in the 500ml autoclave, add 0.5g 5%Pd/C, feed hydrogen, pressure is 7Mpa, stirring at room 6h.Turn off hydrogen, pour out reaction solution, elimination Pd/C gets colourless reaction solution.The reaction solution that takes a morsel adds in the nuclear-magnetism pipe, adds deuterated reagent D 2O, with 1HNMR determines the transformation efficiency of hydrogenation reaction.Two kinds of CH of tauryl amine hydrochlorate 2D 1The HNMR peak is 3.29~3.31 and 3.40~3.42ppm place, and two kinds of CH of reactant tauryl azide salt hydrochlorate 2D 1The HNMR peak is 3.37~3.38 and 3.82~3.84ppm place.Determine that with the peak height ratio of two kinds of compounds the 4th step added the amount of formaldehyde.4. the preparation of taurolidine With the above-mentioned reaction solution that removes by filter Pd/C, add 5g NaHCO 3, be stirred to molten entirely, frozen water cooling, stir slowly splash into down formaldehyde solution (37%, 2ml), have milky white precipitate to produce after 30 minutes, continue to stir 1h, suction filtration, filter cake is washed 3 times with frozen water.Vacuum-drying gets white powdery solid 2.3g, 170~174 ℃ of fusing points.Embodiment 2:Making with extra care of taurolidine:The above-mentioned taurolidine white powder 5~10g that obtains adds 50~200ml acetonitrile, and heating for dissolving removes by filter a small amount of insolubles, concentrates, and cooling below 10 ℃ gets white powder 5~10g, 172~174 ℃ of fusing points.Embodiment 3:Proton nmr spectra ( 1H-NMR) data are as follows:1HNMR(DMSO-D6,TMS7.26-7.28(t,2H,NH),4.09-4.10(d,4H,N-CH 2),3.53(s,2H,N-CH 2-N),3.28-3.29(t,4H,N-CH 2-CH 2),2.96-2.97(t,4H,S-CH 2-CH 2)。The infrared absorption spectrum data are as follows:IR (KBr compressing tablet cm -1): 3425,3263,1633,1450,1404,1317,1278,1228,1160,1134,1073,1026,993,958,924,830,757,667,532,511.See Fig. 3.The ultimate analysis analytical value:C, 29.04%, N, 18.55%, H, 5.85%; Calculated value: C, 29.57%, N, 19.71%, H, 5.67%Embodiment 4:Taurolidine formulation optimizing injection type of the present invention, as: infusion solution, injection liquid, freeze-dried powder injection or powder ampoule agent for injection etc., more preferably infusion solution.The preparation of infusion solution[prescription 1] taurolidine 10.0~30.0gPVP 40.0~80.0gNaCl 2~5gAdd water to 1000ml[method for making] takes by weighing taurolidine, is dissolved in water, and stirs, and adds the PVP dissolving, and adjust pH to 7.0 is crossed the moisture film of 0.22 μ m, packing, and 121 ℃ of sterilizations 20 minutes, promptly.[prescription 2] taurolidine 10.0~30.0gCitric acid 0.1~1.0gLemon enzyme sodium 10.0~20.0gAdd water to 1000ml[method for making] takes by weighing taurolidine, is dissolved in water, stir, and the dissolving of adding citric acid sodium, adjust pH to 7.0, the moisture film of mistake 0.22 μ m, packing was sterilized 20 minutes for 121 ℃, promptly. PATENThttps://patents.google.com/patent/US8952148B2/en
Example ICbz-Taurine Sodium Salt (Formula II)To 1000 ml of water in the RBF charge 192 gm of (3.0 eq) of sodium hydroxide under cooling followed by 200 gm of Taurine and dissolve it until clear solution is obtained. Cool to 0° C. to 5° C., and Charge 50% CBZ-Cl in toluene at 0° C. to 5° C. After completion of addition, maintain at room temperature for 14 h. Separate the toluene layer and wash the aqueous layer with 2×200 ml of ethyl acetate. Add slowly 27 gm of sodium hydroxide in 60 ml of water to the aqueous layer and adjust pH to 12-14. Cool to 0° C. to 5° C. and a white solid separates from the solution. Filter the solid and dry the solid at 60-70° C. Weight of the solid: 320 g
Example 2Cbz-Taurinamide (Formula III)To a clean dry flask charge 1500 ml of toluene and charge 320 gm of Formula II and cool to 0° C. to 5° C. Charge 308 gm of PCl5 slowly at 0° C. to 5° C. for 2 hrs. Maintain at 0° C. to 5° C. up to completion of reaction. Quench the RM into another flask containing 2 ltr of water at 0° C. to 5° C. Separate the organic layer, wash and extract the aqueous layer with toluene. Dry the organic layer with sodium sulphate and cool to 0° C. to 5° C. Purge ammonia gas into the reaction mass till the reaction is complete. Filter the solid and dissolve the solid in 2 ltr of water and extract the aqueous layer with 2×600 ml of ethyl acetate. Dry the organic layer with sodium sulphate and concentrate it under reduced pressure to obtain a white solid. Weight of the solid: 150 g
Example 3Taurinamide Succinate (Formula IV)Take a suspension of 100 g of Cbz-Taurinamide in 1000 ml methanol, and 10% Pd/C (1.0 g) and subject to hydrogenation at 45-50 psi. Upon completion of the reaction filter the catalyst and add succinic acid (1.0 eq) to the solvent and distill off the solvent under vacuum to provide the title compound in about 90% yield as a white solid.
Example 4Taurolidine (Formula I)To a solution of 100 g Taurinamide succinate in water is added sat sodium bicarbonate solution and pH adjusted to 7-8. To the solution was added formaldehyde (50 ml) and allowed to stir for 4 h. The solid obtained was filtered and washed with water to give Taurolidine. The title compound was obtained in about 70% yield and about 98% purity.
Example 5Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. The solid is filtered and washed with toluene and dried to give a white solid in 40% yield. The product obtained was >99.5% pure.
Example 6Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. To the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70% yield. The product obtained was >99.5% pure by HPLC and passed elemental analysis within 0.4% of the theoretical values.
Example 7Purification of TaurolidineTaurolidine (100 g) was dissolved in DMAc (800 ml) and to the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70% yield.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Taurolidine is an antimicrobial agent used in an effort to prevent catheter infections. It however is not approved for this use in the United States as of 2011.[4]
Catheter lock solution in home parenteral nutrition (HPN) or total parenteral nutrition (TPN): catheter-related blood stream infections (CRBSI) remains the most common serious complication associated with long-term parenteral nutrition. The use of taurolidine as a catheter lock solution shows a reduction of CRBSI.[1][5] The overall quality of the evidence however is not strong enough to justify routine use.[1][5]
Catheter lock solution: Taurolidine decreases the adherence of bacteria and fungi to host cells by destructing the fimbriae and flagella and thus prevent the biofilm formation.[6][7] Taurolidine is the active ingredient of antimicrobial catheter lock solutions for the prevention and treatment of catheter related bloodstream infections (CRBSIs) and is suitable for use in all catheter based vascular access devices.[8][1]Bacterial resistance against taurolidine has never been observed in various studies.[9][10]
The use of a taurolidine lock solution may decrease the risk of catheter infection in children with cancer but the evidence is tentative.[11]
Side effects
No systemic side effects have been identified. The safety of taurolidine has also been confirmed in clinical studies with long-term intravenous administration of high doses (up to 20 grams daily). In the body, taurolidine is metabolized rapidly via the metabolites taurultam and methylol taurinamide, which also have a bactericidal action, to taurine, an endogenous aminosulphonic acid, carbon dioxide and water. Therefore, no toxic effects are known or expected in the event of accidental injection. Burning sensation while instilling, numbness, erythema, facial flushing, headache, epistaxis, and nausea have been reported.[12]
Toxicology
Taurolidine has a relatively low acute and subacute toxicity.[1] Intravenous injection of 5 grams taurolidine into humans over 0.5–2 hours produce only burning sensation while instilling, numbness, and erythema at the injection sites.[12] For treatment of peritonitis, taurolidine was administered by peritoneal lavage, intraperitoneal instillation or intravenous infusion, or by a combination thereof. The total daily dose ranged widely from 0.5 to 50 g. The total cumulative dose ranged from 0.5 to 721 g. In those patients who received intravenous taurolidine, the daily intravenous dose was usually 15 to 30 g but several patients received up to 40 g/day. Total daily doses of up to 40 g and total cumulative doses exceeding 300 g were safe and well tolerated.[12][13][14][15][16]
Pharmacology
Metabolism: Taurolidine and taurultam are quickly metabolized to taurinamide, taurine, carbon dioxide and water. Taurolidine exists in equilibrium with taurultam and N-methylol-taurultam in aqueous solution.[17]
Pharmacokinetic (elimination): The half-life of the terminal elimination phase of taurultam is about 1.5 hours, and of the taurinamide metabolite about 6 hours. 25% of the taurolidine dose applied is renally eliminated as taurinamide and/or taurine.[13][14][18]
Mechanism of action
Following administration of taurolidine, the antimicrobial and antiendotoxin activity of the taurolidine molecule is conferred by the release of three active methylol (hydroxymethyl) groups as taurolidine is rapidly metabolized by hydrolysis via methylol taurultam to methylol taurinamide and taurine. These labile N-methylol derivatives of taurultam and taurinamide react with the bacterial cell-wall resulting in lysis of the bacteria, and by inter- and intramolecular cross-linking of the lipopolysaccharide-protein complex, neutralization of the bacterial endotoxins which is enhanced by enzymatic activation. This mechanism of action is accelerated and maximised when taurolidine is pre-warmed to 37 °C (99 °F). Microbes are killed and the resulting toxins are inactivated; the destruction time in vitro is 30 minutes.[19]
The chemical mode of action of taurolidine via its reactive methylol groups confers greater potency in vivo than indicated by in vitrominimum inhibitory concentration (MIC) values, and also appears to preclude susceptibility to resistance mechanisms.[14]
Taurolidine binding to lipopolysaccharides (LPS) prevents microbial adherence to host epithelial cells, thereby prevents microbial invasion of uninfected host cells. Although the mechanism underlying its antineoplastic activity has not been fully elucidated, it may be related to this agent’s anti-adherence property.[6][7] Taurolidine has been shown to block interleukin 1 (IL-1) and tumour necrosis factor (TNF) in human peripheral blood mononuclear cells (PBMC).[20] In addition, taurolidine also promotes apoptosis by inducing various apoptotic factors and suppresses the production of vascular endothelial growth factor (VEGF), a protein that plays an important role in angiogenesis.[21]
Taurolidine is highly active against the common infecting pathogens associated with peritonitis and catheter sepsis, this activity extends across a wide-spectrum of aerobic and anaerobic bacteria and fungi (with no diminution of effect in the presence of biological fluids, e.g. blood, serum, pus).[15][16][22]
The chemical name for taurolidine is 4,4′-Methylene-bis(1,2,4-thiadiazinane)-1,1,1’,1′-tetraoxide.
It is a white to off white odourless crystalline powder. It is practically insoluble in chloroform, slightly soluble in boiling acetone, ethanol, methanol, and ethyl acetate, sparingly soluble in water 8 at 20° and ethyl alcohol, soluble in dilute hydrochloric acid, and dilute sodium hydroxide, and freely soluble in N,N-dimethylformamide (at 60 °C).
History
Taurolidine was first synthesized in the laboratories of Geistlich Pharma AG, Switzerland in 1972. Clinical trials begun in 1975 in patients with severe peritonitis.
Research
Taurolidine demonstrates some anti-tumor properties, with positive results seen in early-stage clinical investigations using the drug to treat gastrointestinal malignancies and tumors of the central nervous system.[23] More recently, it has been found to exert antineoplastic activity. Taurolidine induces cancer cell death through a variety of mechanisms. Even now, all the antineoplastic pathways it employs are not completely elucidated. It has been shown to enhance apoptosis, inhibit angiogenesis, reduce tumor adherence, downregulate pro-inflammatory cytokine release, and stimulate anticancer immune regulation following surgical trauma. Apoptosis is activated through both a mitochondrial cytochrome-c-dependent mechanism and an extrinsic direct pathway. A lot of in vitro and animal data support taurolidine’s tumoricidal action.[24][25][26] Taurolidine has been used as an antimicrobial agent in the clinical setting since the 1970s and thus far appears nontoxic. The nontoxic nature of taurolidine makes it a favorable option compared with current chemotherapeutic regimens. Few published clinical studies exist evaluating the role of taurolidine as a chemotherapeutic agent. The literature lacks a gold-standard level 1 randomized clinical trial to evaluate taurolidine’s potential antineoplastic benefits. However, these trials are currently underway. Such randomized control studies are vital to clarify the role of taurolidine in modern cancer treatment.[21][2]
^ Jump up to:ab Neary PM, Hallihan P, Wang JH, Pfirrmann RW, Bouchier-Hayes DJ, Redmond HP (April 2010). “The evolving role of taurolidine in cancer therapy”. Annals of Surgical Oncology. 17 (4): 1135–43. doi:10.1245/s10434-009-0867-9. PMID20039217. S2CID23807182.
^ Waser PG, Sibler E (1986). “Taurolidine: A new concept in antimicrobial chemotherapy”. In Harms AF (ed.). Innovative Approaches in Drug Research. Elsevier Science Publishers. pp. 155–169.
^ Jump up to:ab Bradshaw JH, Puntis JW (August 2008). “Taurolidine and catheter-related bloodstream infection: a systematic review of the literature”. Journal of Pediatric Gastroenterology and Nutrition. 47 (2): 179–86. doi:10.1097/MPG.0b013e318162c428. PMID18664870. S2CID19136945.
^ Jump up to:ab Gorman SP, McCafferty DF, Woolfson AD, Jones DS (April 1987). “Reduced adherence of micro-organisms to human mucosal epithelial cells following treatment with Taurolin, a novel antimicrobial agent”. The Journal of Applied Bacteriology. 62 (4): 315–20. doi:10.1111/j.1365-2672.1987.tb04926.x. PMID3298185.
^ Jump up to:ab Blenkharn JI (July 1989). “Anti-adherence properties of taurolidine and noxythiolin”. Journal of Chemotherapy. 1 (4 Suppl): 233–4. PMID16312382.
^ Olthof ED, Rentenaar RJ, Rijs AJ, Wanten GJ (August 2013). “Absence of microbial adaptation to taurolidine in patients on home parenteral nutrition who develop catheter related bloodstream infections and use taurolidine locks”. Clinical Nutrition. 32 (4): 538–42. doi:10.1016/j.clnu.2012.11.014. PMID23267744.
^ Jump up to:abc Stendel R, Scheurer L, Schlatterer K, Stalder U, Pfirrmann RW, Fiss I, et al. (2007). “Pharmacokinetics of taurolidine following repeated intravenous infusions measured by HPLC-ESI-MS/MS of the derivatives taurultame and taurinamide in glioblastoma patients”. Clinical Pharmacokinetics. 46 (6): 513–24. doi:10.2165/00003088-200746060-00005. PMID17518510. S2CID33321671.
^ Jump up to:ab Browne MK, MacKenzie M, Doyle PJ (May 1978). “C controlled trial of taurolin in established bacterial peritonitis”. Surgery, Gynecology & Obstetrics. 146 (5): 721–4. PMID347606.
^ Jump up to:ab Browne MK (1981). “The treatment of peritonitis by an antiseptic – taurolin”. Pharmatherapeutica. 2 (8): 517–22. PMID7255507.
^ Browne MK, Leslie GB, Pfirrmann RW (December 1976). “Taurolin, a new chemotherapeutic agent”. The Journal of Applied Bacteriology. 41 (3): 363–8. doi:10.1111/j.1365-2672.1976.tb00647.x. PMID828157.
^ Braumann C, Pfirrman RW, et al. (2013). “Taurolidine, an Effective Multimodal Antimicrobial Drug Versus Traditional Antiseptics and Antibiotics”. In Willy C (ed.). Antiseptics in Surgery – Update 2013. Lindqvist Book Publishing. pp. 119–125.
^ Bedrosian I, Sofia RD, Wolff SM, Dinarello CA (November 1991). “Taurolidine, an analogue of the amino acid taurine, suppresses interleukin 1 and tumor necrosis factor synthesis in human peripheral blood mononuclear cells”. Cytokine. 3 (6): 568–75. doi:10.1016/1043-4666(91)90483-t. PMID1790304.
^ Jump up to:ab Nösner K, Focht J (1994). “In-vitro Wirksamkeit von Taurolidin und 9 Antibiotika gegen klinische Isolate aus chirurgischem Einsendegut sowie gegen Pilze”. Chirurgische Gastroenterologie. 10 (Suppl 2): 10.
^ Stendel R, Picht T, Schilling A, Heidenreich J, Loddenkemper C, Jänisch W, Brock M (2004-04-01). “Treatment of glioblastoma with intravenous taurolidine. First clinical experience”. Anticancer Research. 24 (2C): 1143–7. PMID15154639.
^ Calabresi P, Goulette FA, Darnowski JW (September 2001). “Taurolidine: cytotoxic and mechanistic evaluation of a novel antineoplastic agent”. Cancer Research. 61 (18): 6816–21. PMID11559556.
^ Clarke NW, Wang JH, et al. (2005). “Taurolidine inhibits colorectal adenocarcinoma metastases in vivo and in vitro by inducing apoptosis”. Ir J Med Sci. 174 (Supplement 3): 1.
^ Stendel R, Scheurer L, Stoltenburg-Didinger G, Brock M, Möhler H (2003-06-01). “Enhancement of Fas-ligand-mediated programmed cell death by taurolidine”. Anticancer Research. 23 (3B): 2309–14. PMID12894508.
Daridorexant, formerly known as nemorexant, is a selective dual orexin receptor antagonist used to treat insomnia. Insomnia is characterized by difficulties with sleep onset and/or sleep maintenance and impairment of daytime functioning. It chronically affects the person’s daily functioning and long-term health effects, as insomnia is often associated with comorbidities such as hypertension, diabetes, and depression. Conventional treatments for insomnia include drugs targeting gamma-aminobutyric acid type-A (GABA-A), serotonin, histamine, or melatonin receptors; however, undesirable side effects are frequently reported, such as next-morning residual sleepiness, motor incoordination, falls, memory and cognitive impairment. Novel drugs that target orexin receptors gained increasing attention after discovering the role of orexin signalling pathway in wakefulness and almorexant, an orexin receptor antagonist that improved sleep. Daridorexant was designed via an intensive drug discovery program to improve the potency and maximize the duration of action while minimizing next-morning residual activity.1
Daridorexant works on orexin receptors OX1R and OX2R to block the binding of orexins, which are wake-promoting neuropeptides and endogenous ligands to these receptors. Daridorexant reduces overactive wakefulness: in the investigational trials, daridorexant reportedly improved sleep and daytime functioning in patients with insomnia.1 It was approved by the FDA on January 10, 2022, under the name QUVIVIQ.6 as the second orexin receptor antagonist approved to treat insomnia following suvorexant.2
QUVIVIQ
Generic Name: daridorexant tablets
Brand Name: Quviviq
QUVIVIQ contains daridorexant, an orexin receptor antagonist. The chemical name of daridorexant hydrochloride is (S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl)(5- methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone hydrochloride. The molecular formula is C23H23N6O2Cl * HCl. The molecular weight is 487.38 g/mol.
The structural formula is:
Daridorexant hydrochloride is a white to light yellowish powder that is very slightly soluble in water.
QUVIVIQ tablets are intended for oral administration. Each film-coated tablet contains 27 mg or 54 mg of daridorexant hydrochloride equivalent to 25 mg or 50 mg of daridorexant, respectively. The inactive ingredients are croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, povidone, and silicon dioxide.
In addition, the film coating contains the following inactive ingredients: glycerin, hypromellose, iron oxide black, iron oxide red, microcrystalline cellulose, talc, titanium dioxide, and, in the 50 mg tablet only, iron oxide yellow.
Dosage Forms And Strengths
QUVIVIQ (daridorexant) tablets are available as:
25 mg: light purple, arc-triangle shaped, film-coated tablet debossed with “25” on one side and “i” (Idorsia logo) on the other side, containing 25 mg daridorexant.
50 mg: light orange, arc-triangle shaped, film-coated tablet debossed with “50” on one side and “i” (Idorsia logo) on the other side, containing 50 mg daridorexant.
QUVIVIQ tablets are available as:
25 mg, light purple, arc-triangle shaped film-coated tablets debossed with “25” on one side, and “i” on the other side. NDC 80491-7825-3, bottle of 30 with child-resistant closure
50 mg: light orange, arc-triangle shaped film-coated tablets debossed with “50” on one side, and “i” on the other side. NDC 80491-7850-3, bottle of 30 with child-resistant closure
Since its discovery in 1998, the orexin system has been of interest to the research community as a potential therapeutic target for the treatment of sleep/wake disorders. Herein we describe our efforts leading to the identification of daridorexant, which successfully finished two pivotal phase 3 clinical trials for the treatment of insomnia disorders.
Step 3. Amide (S7) (1000 g, 2.13 mmol) was dissolved in EtOH (5 L) and 32% aqueous HCl (500 mL) was added at 23 °C. The solution was filtered through a Whatman filter (5 µm). The filtrate was heated to 75 °C for 4h. The resulting suspension was cooled to 0 °C and filtered. The product was dried under reduced pressure to yield 93 x HCl (922 g, 89%) as a white solid.
DORA explorers: The orexin system plays an important role in regulating the sleep-wake cycle. Herein we report our optimization efforts toward a novel dual orexin receptor antagonist (DORA) with improved properties over compound 6. Replacing the oxadiazole by a triazole resulted in compounds (e. g. compound 33) with improved properties, such as higher intrinsic metabolic stability, lower plasma protein binding, higher brain free fraction, and increased solubility. Further optimization was needed to decrease the compounds P-glycoprotein susceptibility. Our work led to the identification of compound 42, a potent, brain-penetrating DORA with improved in vivo efficacy in dogs compared with compound 6.
Abstract
The orexin system is responsible for regulating the sleep-wake cycle. Suvorexant, a dual orexin receptor antagonist (DORA) is approved by the FDA for the treatment of insomnia disorders. Herein, we report the optimization efforts toward a DORA, where our starting point was (5-methoxy-4-methyl-2-[1,2,3]triazol-2-yl-phenyl)-{(S)-2-[5-(2-trifluoromethoxy-phenyl)-[1,2,4]oxadiazol-3-yl]-pyrrolidin-1-yl}methanone (6), a compound which emerged from our in-house research program. Compound 6 was shown to be a potent, brain-penetrating DORA with in vivo efficacy similar to suvorexant in rats. However, shortcomings from low metabolic stability, high plasma protein binding (PPB), low brain free fraction (fu brain), and low aqueous solubility, were identified and hence, compound 6 was not an ideal candidate for further development. Our optimization efforts addressing the above-mentioned shortcomings resulted in the identification of (4-chloro-2-[1,2,3]triazol-2-yl-phenyl)-{(S)-2-methyl-2-[5-(2-trifluoromethoxy-phenyl)-4H-[1,2,4]triazol-3-yl]-pyrrolidin-1-yl}l-methanone (42), a DORA with improved in vivo efficacy compared to 6.
1) Synthesis of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-lodo-5-methoxy benzoic acid (15.0 g; 53.9 mmol) is dissolved in anhydrous DMF (45 ml) followed by the addition of 1 H-1 ,2,3-triazole (7.452 g; 108 mmol) and cesium carbonate (35.155 g; 108 mmol). By the addition of cesium carbonate the temperature of the reaction mixture increases to 40°C and gas evolved from the reaction mixture. Copper(l)iodide (514 mg; 2.7 mmol) is added. This triggers a strongly exothermic reaction and the temperature of the reaction mixture reaches 70°C within a few seconds. Stirring is continued for 30 minutes. Then the DMF is evaporated under reduced pressure followed by the addition of water (170 ml) and EtOAc (90 ml). The mixture is vigorously stirred and by the addition of citric acid monohydrate the pH is adjusted to 3-4. The precipitate is filtered off and washed with water and EtOAc and discarded. The filtrate is poured into a separation funnel and the phases are separated. The water phase is extracted again with EtOAc. The combined organic layers are dried over MgS04, filtered and the solvent is evaporated to give 7.1 g of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid as a white powder of 94% purity (6 % impurity is the regioisomerically N1-linked triazolo-derivative); tR [min] = 0.60; [M+H]+ = 220.21
2) Synthesis of (S)-1 -(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid
2-Methyl-L-proline hydrochloride (99.7 g; 602 mmol) is dissolved in a 1/1-mixture of MeCN and water (800 ml) and triethylamine (254 ml; 1810 mmol) is added. The temperature of the reaction mixture slightly rises. The reaction mixture is cooled to 10°C to 15°C followed by careful addition of a solution of Boc20 (145 g; 662 mmol) in MeCN (200 ml) over 10 minutes.
Stirring at RT is continued for 2 hours. The MeCN is evaporated under reduced pressure and aq. NaOH solution (2M; 250 ml) is added to the residual aq. part of the reaction mixture. The water layer is washed with Et20 (2x 300 ml) then cooled to 0°C followed by slow and careful addition of aq. HCI (25%) to adjust the pH to 2. During this procedure a suspension forms.
The precipitate is filtered off and dried at HV to give 1 10.9 g of the title compound as a beige powder; tR [min] = 0.68; [M+H]+ = 230.14
3) Synthesis of (S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-
(S)-1-(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid (60 g; 262 mmol) and HATU (100 g; 264 mmol) is suspended in DCM (600 ml) followed by the addition of DIPEA (84.6 g; 654 mmol) and 6-chloro-2,3-diaminotoluene (41 g; 262 mmol). The reaction mixture is stirred at rt for 14 hours then concentrated under reduced pressure and to the residue is added water followed by the extraction of the product with EtOAc (3x). The combined organic layers are washed with brine, dried over MgS04, filtered and the solvent is evaporated under
reduced pressure to give 185 g of the title compound as a dark brownish oil, which is used in the next step without further purification; tR [min] = 0.89; [M+H]+ = 368.01
4) Synthesis of (S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1 -carboxylate
(S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-methylpyrrolidine-1-carboxylate (185 g; 427 mmol) are dissolved in AcOH (100%; 611 ml), heated to 100°C and stirring continued for 90 minutes. The AcOH is evaporated under reduced pressure and the residue is dissolved in DCM followed by careful addition of saturated sodium bicarbonate solution. The phases are separated, the aq. phase is extracted once more with DCM, the combined aq. phases are dried over MgS04, filtered and the solvent is evaporated under reduced pressure to give 142.92 g of the title compound as a dark brown oil which is used in the next step without further purification; tR [min] = 0.69; [M+H]+ = 350.04
5) Synthesis of (S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride
(S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1-carboxylate (355.53 g; 1.02 mol) are dissolved in dioxane (750 ml) followed by careful addition of HCI solution in dioxane (4M; 750 ml; 3.05 mol). The reaction mixture is stirred for 3 hours followed by the addition of Et20 (800 ml) which triggered precipitation of the product. The solid is filtered off and dried at high vacuum to give 298.84 g of the title compound as a redish powder; tR [min] = 0.59; [M+H]+ = 250.23
6) Synthesis of [(S)-2-(5-chloro-4-methyl-1 H-benzoimidazol-2-yl)-2-methyl-pyrrolidin-1- -(5-methoxy-2-[1,2,3]triazol-2-yl-phenyl)-methanone
(S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride (62.8 g; 121 mmol) is dissolved in DCM (750 ml) followed by the addition of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (62.8 g; 121 mmol) and DIPEA (103 ml; 603 mmol). Stirring is continued for 10 minutes followed by the addition of HATU (47 g; 124 mmol). The reaction mixture is stirred for 16 hours at RT. The solvents are evaporated under reduced pressure and the residue is dissolved in EtOAc (1000 ml) and washed with water (3x 750 ml). The organic phase is dried over MgS04, filtered and the solvent is evaporated under reduced pressure. The residue is purified by CC with EtOAc / hexane = 2 / 1to give 36.68 g of the title compound as an amorphous white powder. tR [min] = 0.73; [M+H]+ = 450.96
Table 1 : Characterisation data for COMPOUND as free base in amorphous form
II. Preparation of crystalline forms of COMPOUND
Example 1 :
Preparation of seeding material of COMPOUND hydrochloride in crystalline Form 1
10 mg COMPOUND is mixed with 0.2 mL 0.1 M aq. HCI and 0.8 mL EtOH. The solvent is fully evaporated and 0.05 mL isopropanol is added. Alternatively 0.05 mL methyl-isobutylketone can be added. The sample is stored closed at room temperature for 4 days and crystalline material of COMPOUND hydrochloride in crystalline Form 1 is obtained. This material can be used as seeding material for further crystallization of COMPOUND hydrochloride in crystalline Form 1.
Example 2: Preparation and characterization of COMPOUND hydrochloride in crystalline form 1
5g COMPOUND is mixed with 0.9 mL 1 M aq. HCI and 20 mL EtOH. The solvent is evaporated and 25 mL isopropanol is added. Seeds of COMPOUND hydrochloride are added and the sample is allowed to stand at room temperature. After about 2 days the suspension is filtered and the solid residue is dried at reduced pressure (2 mbar for 1 hour) and allowed to equilibrate open for 2 hours at 24°C/46% relative humidity. The obtained solid is COMPOUND hydrochloride in crystalline Form 1
Table 2: Characterisation data for COMPOUND hydrochloride in crystalline form 1
4- Fluoro-3-nitroanisole (3.44 g, 1 eq.), 4,5-dibromo-2/-/-1 ,2,3-triazole (4.56 g, 1 eq.)1, K2C03 (2.78 g, 1 eq.) and DMF (30 mL) are heated to 1 10 °C for 32 h. The reaction mixture is cooled to 22 °C and treated with water (70 mL). The resulting suspension is filtered, washed with water (15 mL). The product is slurried in isopropanol (40 mL), filtered and dried under reduced pressure to yield a white solid. Yield: 6.42 g, 84%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, CDCI3) δ: 7.71 (d, J = 8.9 Hz, 1 H), 7.47 (d, J = 2.8 Hz, 1 H), 7.25 (dd, Ji = 2.8 Hz, J2 = 8.9 Hz, 1 H), 3.97 (s, 3 H).
1 X. Wang, L. Zhang, D. Krishnamurthy, C. H. Senanayake, P. Wipf Organic Letters 2010 12 (20), 4632-4635.
5- methoxy-2-(2H-1 ,2,3-triazol-2-yl)aniline
4, 5-Dibromo-2-(4-methoxy-2-nitrophenyl)-2/-/-1 ,2,3-triazole (2 g, 1 eq.), sodium acetate (1.3 g, 3 eq.), and 10% Pd/C 50% water wet (0.3 g) is suspended in EtOAc (10 mL). The mixture is heated to 50 °C and set under hydrogen until conversion is complete. The reaction mixture is filtered over Celite. The filtrate is washed with 1 N NaOH (10 mL) and water (15 mL). The organic layer is concentrated under reduced pressure to yield an oil. Yield: 0.95 g, 94%. Purity: 96% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.05 (s, 2 H), 7.53 (d, J = 8.9 Hz, 1 H), 6.49 (d, J = 2.7 Hz, 1 H), 6.30 (dd, Ji = 2.7 Hz, J2 = 8.9 Hz, 1 H), 5.94 (s, 2 H), 3.74 (s, 3 H).
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)aniline (455 g, 1 eq ) is dissolved in isopropanol (3 L). To the solution is added cone. H2SO4 (235 g, 1 eq.) below 40 °C. The suspension is cooled to
20 °C and filtered. The cake is washed with isopropanol (700 mL) and TBME (1.5 L). The product is dried to obtain a white solid. Yield: 627 g, 91 %. Purity: 100% a/a (LC-MS method 2).
2-(2-iodo-4-methoxyphenyl)-2H-1,2,3-triazole
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)aniline monosulfate (200 g, 1 eq.) is dissolved in 2 M aq. H2SO4 soln. (1.4 L) and cooled to -5 °C. To the solution is added a solution of sodium nitrite (62 g, 1.3 eq.) in water (600 mL) at -5 to 0 °C. The mixture is stirred at 0 °C for 30 min and then added to a preheated mixture of Kl (161 g, 1.4 eq.) in water (700 mL) at 65 °C. The resulting solution is stirred at 60 °C for 20 min, cooled to 20 °C and treated with a soln. of sulfamic acid (27 g, 0.4 eq.) in water (120 mL). The mixture is extracted with isopropyl acetate (2 L). The organic layer is washed with a mixture of 2 N NaOH (500 mL) and 40% NaHS03 soln. (100 mL), and a mixture of 1 N HCI (50 mL) and water (500 mL). The organic layer is concentrated to dryness. The residue is dissolved in isopropanol (700 mL) and cooled to 0 °C. The resulting suspension is filtered. The solid is dried under reduced pressure. Yield: 164 g, 79%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.08 (s, 2 H), 7.57 (d, J = 2.8 Hz, 1 H), 7.43 (d, J = 8.8 Hz, 1 H), 7.13 (dd, Ji = 2.8 Hz, J2 = 8.8 Hz, 1 H), 3.85 (s, 3 H).
5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-(2-lodo-4-methoxyphenyl)-2/-/-1 ,2,3-triazole (200 g, 1 eq.) is dissolved in THF (2 L) and cooled to 0 °C. 2 M iPrMgCI soln. in THF (350 mL, 1.05 eq.) is added at 0 °C. The mixture is cooled to -20 °C and C02 (gas) is bubbled into the solution over 30 min until the exothermicity is ceased. To the mixture is added 2 N HCI (600 mL) at 8 °C and concentrated under reduced pressure to remove 2.4 L solvent. The residue is extracted with TBME (1.6 L). The organic layer is washed with 1 N HCI (200 mL) and extracted with 1 N NaOH (600 mL and 200 mL). The aq. layer is filtered over charcoal (15 g), diluted with water (200 mL) and treated with 32% HCI (160 mL). The resulting suspension is filtered and washed with water (200 mL). Yield: 127 g, 87%. Purity: 100% a/a (LC-MS method 2); MP: 130 °C (DSC goldpan). The obtained product may be re-crystallized from toluene (MP: 130.9 °C) or water (MP: 130 °C).
Table Ref 1 : Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid in crystalline form 2 (recrystallization from toluene)
Technique Data Summary Remarks
XRPD Crystalline see Fig. 8
Reference Example 2
Synthesis of 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
3- Fluoro-4-nitrotoluene (1367 g, 1 eq.), 4,5-dibromo-2/-/-1 ,2,3-triazole (1999 g, 1 eq.), K2C03 (1340 g, 1.1 eq.) and DMF (1 1 L) is heated to 75 °C for 15 h. The reaction mixture is cooled to 22 °C and treated with water (18 L). The resulting suspension is filtered, washed with water (4 L). The product is washed with isopropanol (5 L), and dried under reduced pressure to yield a white solid. Yield: 281 1 g, 88%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.10 (d, J = 8.3 Hz, 1 H), 7.86 (d, J = 1.0 Hz, 1 H), 7.66 (dd, J1 = 0.9 Hz, J2 = 8.3 Hz, 1 H), 2.51 (s, 3 H).
4- Methyl-2-(2H-1 ,2,3-triazol-2-yl)aniline
4, 5-Dibromo-2-(5-methyl-2-nitrophenyl)-2/-/-1 ,2,3-triazole (205 g, 1 eq.), sodium acetate (149 g, 3.2 eq.), and 5% Pd/C 50% water wet (37.8 g) is suspended in EtOAc (0.8 L). The mixture is heated to 40-50 °C and set under hydrogen (2 bar) until conversion is complete. The reaction mixture is filtered over Celite. The filtrate is washed with water (300 mL), 2N NaOH (300 ml_+250 mL) and water (300 mL). The organic layer is concentrated under reduced pressure to yield a yellow oil. Yield: 132 g, 90%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.09 (s, 2 H), 7.48 (d, J = 1.3 Hz, 1 H), 6.98 (dd, J1 = 1.8 Hz, J2 = 8.3 Hz, 1 H), 6.85 (d, J = 8.2 Hz, 1 H), 5.79 (s, 2 H), 2.23 (s, 3 H).
4-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl) aniline (199 g, 1 eq ) is dissolved in isopropanol (1.7 L). To the solution is added cone. H2SO4 (118 g, 1.05 eq.) below 40 °C. The suspension is cooled to 20 °C and filtered. The cake is washed with isopropanol (500 mL). The product is dried to obtain a white solid. Yield: 278 g, 89%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.21 (s, 2 H), 7.70 (s, 1 H), 7.23 (s, 2 H), 2.35 (s, 3 H).
2-(2-iodo-5-methylphenyl)-2H-1 ,2,3-triazole
4-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl)aniline monosulfate (1553 g, 1 eq.) is dissolved in 1 M aq. H2S04 Soln. (1 1 L) and cooled to -5 °C. To the solution is added a solution of sodium nitrite (433 g, 1.1 eq.) in water (4 L) at -5 to 0 °C. The mixture is stirred at 0 °C for 30 min and then added to a preheated mixture of potassium iodide (1325 g, 1.4 eq.) in water (4 L) at 55-70 °C. The resulting solution is stirred at 60 °C for 20 min, cooled to 20 °C and treated with a soln. of sulfamic acid (220 g, 0.4 eq.) in water (900 mL). The mixture is extracted with isopropyl acetate (13 L). The organic layer is washed with a mixture of 2 N NaOH (3.5 L) and 40% NaHSOs soln. (330 g), and a mixture of 1 N HCI (280 mL) and water (3.5 L). The
The crude product, together with a second batch (141 1 g) is purified by distillation on a short path distillation equipment at 120 °C jacket temperature, feeding tank (70 °C), cooling finger (20 °C) and at a pressure of 0.004 mbar. Yield: 2544 g (78%), Purity: 100 % a/a ()LC-MS method 2).
4-Methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-(2-lodo-5-methylphenyl)-2/-/-1 ,2,3-triazole (1250 g, 1 eq.) is dissolved in THF (13 L) and cooled to 0 °C. 2 M iPrMgCI soln. in THF (2.2 L, 1 eq.) is added at 0 °C. The mixture is cooled to -25 °C and CO2 (gas) is bubbled into the solution over 60 min until the exothermicity is ceased. To the mixture is added 2 N HCI (5 L) at 4 °C and concentrated under reduced pressure to remove 14.5 L solvent. The residue is extracted with TBME (10 L). The organic layer is extracted with 1 N NaOH (6 L and 3 L). The aq. layer is filtered over charcoal (15 g), diluted with water (200 mL) and treated with 32% HCI (1 .23 L). The resulting suspension is filtered and washed with water (5 L). Yield: 796 g, 89%. Purity: 100% a/a (LC-MS method 2); MP: 125 °C (DSC goldpan).
The following examples illustrate the invention.
Example 1 :
Example 1.1: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt (potassium 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-5-methoxybenzoic acid (21 .5 g, 0.093 mol, 1 eq.) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.2 g, 2.5 eq.) were suspended in dioxane (600 mL) and water (8.4 mL). To the mixture were added 1 H-1 ,2,3-triazole (10.8 mL, 2 eq.) and trans-/V,/V-dimethylcyclohexane-1 ,2-diamine (1 .32 g, 0.1 eq.). The mixture was heated at reflux for 3.5 h. IPC showed full conversion. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 84: 16. The mixture was cooled to 40 °C and filtered. The cake was washed with dioxane (100 mL). The solid was dried to obtain 50.6 g of a blue solid. The ratio of N{2) to Λ/(1 ) isomer of was 98.6: 1 .4.
Table 1 : Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 1
Example 1.2: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid of Example 1.1 was dissolved in water (300 mL). TBME (200 mL) and 32% aq. HCI (35 mL) was added. The aq. layer was separated and discarded. The organic layer was washed with a mixture of 2N aq. HCI (100 mL) and 32% aq. HCI (20 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 optionally seed crystals were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 12.4 g, 61 %. Purity: 100% a/a, tR 0.63 min. Seed crystals may be obtained by careful crystallization according to the above procedure.
Table 2: Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Example 1.3: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid, e.g. obtained according to the procedure of Reference Example 1 (5 g, 0.0228 mol) and KHCO3 (1.61 g, 0.7 eq) were suspended in dioxane (100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.56 g, 44%. 1H NMR (400 MHz, D20) & 3.80 (s, 3 H), 7.04 (m, 2 H), 7.46 (d, J = 8.7 Hz, 1 H), 7.82 (s, 2 H). MP: 279.5°C (DSC shows additionally a broad endothermic event at about 153 °C to 203 °C which may be attributed to endothermic desolvations; melting is immediately followed by exothermic degradation).
Table 3: Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 2
Example 1.4: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
In an alternative procedure, 2-Bromo-5-methoxybenzoic acid (20 g, 0.086 mol, 1 eq.) copper (I) iodide (0.824 g, 0.05 eq.), and K2C03 powder (26.9 g, 2.25 eq.) were suspended in dioxane (494 mL). To the mixture was added 1 H-1 ,2,3-triazole (12 g, 2 eq.). The mixture was heated at reflux for 1 h. To the mixture was added water (12.5 g, 8 eq.). The mixture was heated at reflux for 2 h. Solvent (100 mL) was removed by distillation. The residue was cooled to 45 °C in 8 min, filtered and washed with dioxane (50 mL).
XRPD corresponds to crystalline form 1 (see Fig. 1 , Example 1.1 ).
Example 1.5: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid of Example 1.4 was dissolved in water (200 mL). The mixture was heated to 50 °C and 20% aq. H2SO4 (40 mL) was added to adjust the pH to 5. The mixture was filtered over Celite. The filtrate was treated at 45 °C with 20% aq. H2S04 (40 mL). At pH 3 seeds (obtained for example using the procedure of reference example 1 ) were added. The suspension was stirred at 45 °C and filtered. The product was washed with water (20 mL) and dried at 60 °C and 10 mbar to yield a white solid. Yield: 10.8 g, 57%. Purity: 100% a/a, tR 0.63 min.
Characterisation of 5-methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid obtained according to Example 1.5:
XRPD corresponds to crystalline form 1 (see Fig. 2, Example 1.2).
Example 2:
Example 2.1: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt (potassium 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-4-methylbenzoic acid (20 g, 0.093 mol, 1 eq.) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.2 g, 2.5 eq.) were suspended in dioxane (300 mL) and water (10.1 mL). To the mixture was added 1 A7-1 ,2,3-triazole (10.8 mL, 2 eq.) and trans-Λ/,ΛΑ-
dimethylcyclohexane-1 ,2-diamine (1 .32 g, 0.1 eq.). The mixture was heated at reflux for 4 h. IPC showed a conversion of 98.5%. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 75:25. The mixture was concentrated at normal pressure and external temperature of 130 °C. Solvent (100 mL) was removed. To the residue was added dioxane (100 mL) and the mixture was cooled to 45 °C and filtered. The cake was washed with dioxane (80 mL). The solid was dried to obtain 48.8 g of a blue solid. The ratio of N(2) to Λ/(1 ) isomer was 98.7: 1 .3.
Table 4: Characterisation data for 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 1
Example 2.2: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl) benzoic acid
The solid of Example 2.1 was dissolved in water (300 mL) and filtered. To the filtrate were added TBME (200 mL) and 32% aq. HCI (30 mL). The aq. layer was separated and discarded. The organic layer was washed with a mixture of 2N aq. HCI (100 mL) and 32% aq. HCI (10 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 seed crystals (obtained for example using the procedure of reference example 2) were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 1 1 .7 g, 62%. Purity: 100% a/a. tR 0.66 min.
Table 5: Characterisation data for 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Technique Data Summary Remarks
XRPD Crystalline see Fig. 5
Example 2.3: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
4-Methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (5 g, 0.0246 mol) and KHC03 (1 .74 g , 0.7 eq) were suspended in dioxane ( 100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.47 g, 42% . MP: 277 °C (DSC Alupan) 1 H NMR (400 MHz, D20) & 2.32 (s, 3 H), 7.28 (d, J = 7.9 Hz, 1 H), 7.39 (m, 2 H), 7.84 (s, 2 H).
MP: 276.8 °C (DSC shows additionally a broad endothermic event at about 140 °C to 208 °C which may be attributed to endothermic desolvations; melting is immediately followed by exothermic degradation).
XRPD corresponds to crystalline form 1 (see Fig. 4, Example 2.1 ).
Reference Example 3:
Reference Example 3.1: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid sodium salt (sodium 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-5-methylbenzoic acid (20 g, 0.093 mol, 1 eq. ) copper (I) iodide (0.886 g, 0.05 eq.), Na2CC>3 powder (24.6 g, 2.5 eq.) were suspended in dioxane (300 mL) and water (10.1 mL). To the mixture was added 1 /-/-1 ,2,3-triazole ( 10.8 mL, 2 eq.) and 8-hydroxy quinoline ( 1 .35 g, 0.1 eq.). The mixture was heated at reflux for 5 h. IPC showed a conversion of >99%. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 78:22. The mixture was concentrated at normal pressure and external temperature of 135 °C. Solvent (100 mL) was removed. To the residue was added dioxane (100 mL) and the mixture was cooled to 45 °C and filtered. The cake was washed with dioxane (80 mL). The solid was dried to obtain 36.2 g of a yellow solid. The ratio of N(2) to Λ/( 1 ) isomer of was 99: 1 .
Table 6: Characterisation data for 5-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid sodium salt in crystalline form 1
Reference Example 3.2: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid obtaind in Reference Example 3.1 was dissolved in water (300 mL) and filtered. To the filtrate was added TBME (200 mL) and 32% aq. HCI (30 mL) was added. The aq. layer was separated and discarded. The organic layer was washed with 1 N aq. HCI ( 100 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed
under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 seed crystals (obtained for example using the procedure of Reference example 2) were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 12.1 g, 64%. Purity: 100% a/a. tR 0.67 min.
Table 7: Characterisation data for 5-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Reference Example 3.3: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl) benzoic acid sodium salt
5-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid (5 g, 0.0246 mol) and Na2C03 (1 .05 g, 0.4 eq) were suspended in dioxane ( 100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.79 g, 50%. MP: 341 °C (DSC Alupan) 1 H NMR (400 MHz, D20) & 2.32 (s, 3 H), 7.30 (m, 2 H), 7.43 (m, 1 H), 7.83 (s, 2 H).
XRPD corresponds to crystalline form 1 (see Fig. 6, Reference Example 3.1 ).
Reference Example 3.4: 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
2-Bromo-5-methylbenzoic acid (20 g, 0.093 mol, 1 eq. ) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.1 g, 2.5 eq.) were suspended in dioxane (600 mL). To the mixture was added 1 /-/-1 ,2,3-triazole ( 10.8 mL, 2 eq.) and 8-hydroxy quinoline ( 1 .35 g, 0.1 eq.). The mixture was heated at reflux for 4 h. IPC showed a conversion of >94%. The ratio of the desired N(2) to the regioisomeric Λ/( 1 ) isomer was 78:22. The mixture was cooled to 35 °C and filtered. The cake was washed with dioxane (100 mL). The products were dissolved in water and a LC-MS was recorded. The ratio of N(2) to Λ/(1 ) isomer of was 83: 17.
Reference Example 4.1: Methyl (S)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylate
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoic acid (100 g, 0.46 mol) was suspended in DCM (650 mL) and DMF (10 mL) at 20 °C. To this suspension was added oxalyl chloride (51 mL, 0.59 mol) over a period of 30 min. LC-MS showed 60% conversion to acid chloride intermediate. Oxalyl chloride (17.6 mL, 0.45 eq.) was added dropwise. LC-MS showed full conversion to acid chloride intermediate.
Methyl (S)-2-methylpyrrolidine-2-carboxylate hydrochloride (84 g, 0.47 mol) was suspended in DCM (800 mL) in a second flask. The suspension was cooled to 10 °C. Triethylamine (200 mL, 1.41 mol) was added over 15 min. The acid chloride solution was added to the reaction mixture at 10-20 °C over at least 15 min. The reaction mixture was washed with 1 M HCI (500 mL), 1 N NaOH (500 mL) and water (500 mL). The organic layer was concentrated to dryness to give a light-yellow solid as product. Yield: 157 g, 100%, 99% a/a (LC-MS), M+1 =345. 1H NMR (400 MHz, DMSO) δ: 8.06 (s, 2 H), 7.79 (d, J = 8.9 Hz, 1 H), 7.21 (dd, J1 = 2.9 Hz, J2 = 8.9 Hz, 1 H), 6.85 (d, J = 1.9 Hz, 1 H), 3.89 (s, 3 H), 3.66 (s, 3 H), 3.29 (m, 1 H), 3.03 (m, 1 H), 2.08 (m, 1 H), 1.82 (m, 3 H), 1.50 (s, 3 H).
Reference Example 4.2: (S)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylic acid
Methyl (S)-1-(5-methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylate (157 g, 0.46 mol) was dissolved in MeOH (750 mL) at 20 °C. To this solution was added 16% NaOH (300 mL). The resulting solution was heated up to 80 °C and stirred for 60 min. Solvent was distilled off under reduced pressure (850 mL). The residue was taken up in DCM (1500 mL) and water (450 ml) at 20 °C. 32% HCI (200 mL) was added. Layers were separated and the organic layer was washed with water (450 mL). The organic layer was concentrated to the minimum stirring volume under reduced pressure. Toluene (750 mL) was added and solvent was further distilled under vacuum (150 mL distilled). The mixture was cooled to 20 °C and stirred for 15 min. The suspension was filtered at 20 °C. The cake was rinsed with toluene (150 mL) and then dried under reduced pressure at 50 °C to give a white solid as product. Yield: 128 g, 85%, 94% a/a (LC-MS), M+1 =331. Melting point: 178 °C (DSC). 1H NMR (400 MHz, DMSO) δ: 12.3 (s, 1 H), 8.04 (s, 2 H), 7.79 (d, 1 H), 7.20 (dd, J1 = 2.8 Hz, J2 = 8.9 Hz, 1 H), 6.84 (m, 1 H), 3.88 (s, 3 H), 3.29 (m, 1 H), 2.99 (m, 1 H), 2.1 1 (m, 1 H), 1.81 (m, 3 H), 1.47 (s, 3 H).
Reference Example 4.3: (S)-N-(2-amino-4-chloro-3-methylphenyl)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2 methylpyrrolidine-2-carboxamide
(S)-1-(5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylic acid (128 g, 0.39 mol) was suspended in DCM (850 mL) and DMF (6 mL) at 20 °C. To this suspension was added oxalyl chloride (39 mL, 0.45 mol) over a period of 30 min. 4-Chloro-3-methylbenzene-1 ,2-diamine hydrochloride (75 g, 0.39 mol) was suspended in DCM (1300 mL) in a second flask. The suspension was cooled down to 10 °C. Triethylamine (180 mL, 1.27 mol) was added. The acid chloride solution was added to the reaction mixture at 10-20 °C over at least 15 min. Water (650 mL) was added to the reaction mixture. Layers were separated and the organic phase was concentrated under reduced pressure (1900 mL distilled out). TBME (1000 mL) was added and solvent was further distilled under vacuum (400 mL distilled). The mixture was finally cooled down to 20 °C and stirred for 15 min. The resulting suspension was filtered off at 20 °C. The cake was rinsed with TBME (250 mL) and then dried under reduced pressure at 50 °C to give a white solid as product. Yield: 145 g, 80%, 97% a/a (LC-MS), M+1=469. Melting point: 185 °C (DSC). 1H NMR (400 MHz, DMSO) δ: 9.10-9.14 (m, 1 H), 7.88-8.12 (m, 2 H), 7.81-7.82 (m, 1 H), 7.38-7.44 (m, 1 H), 7.21 (dd, J1 = 2.7 Hz, J2 = 8.9 Hz, 1 H), 6.84 (d, J = 7.8 Hz, 1 H), 6.64 (d, J = 8.3 Hz, 1 H), 5.01 (brs, 2 H), 3.88 (s, 3 H), 3.61-3.73 (m, 1 H), 3.14-3.26 (m, 1 H), 2.25-2.30 (m, 1 H), 2.13 (s, 3 H), 1.97 (m, 3 H), 1.47-1.61 (m, 3 H).
Reference Example 4.4: (S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl) (5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone hydrochloride
(S)-/V-(2-amino-4-chloro-3-methylphenyl)-1-(5-methoxy-2-(2H-1 ,2,3-triazol-2-yl) benzoyl)-2 methylpyrrolidine-2-carboxamide (145 g, 0.31 mol) was dissolved in isopropanol (870 mL) at 20 °C. To this solution was added carefully 5-6 N HCI in isopropanol (260 mL) over 10 min. the reaction mixture was then heated up to 90 °C and stirred for 4 hours. Water (28 mL) was added and the reaction mixture was stirred for an additional one hour. The reaction mixture was cooled to 20 °C. A light brown suspension was obtained which was filtered. The cake was rinsed with isopropanol (220 mL). The solid was finally dried under reduced pressure at 60 °C to give a beige solid. Yield: 133 g, 88%, 100% a/a (LC-MS), M+1 =451. Melting point: 277 °C (DSC). Ή NMR (400 MHz, DMSO) δ: 8.06 (s, 2 H), 7.76 (d, J = 8.9 Hz, 1 H), 7.63 (d, J = 8.8 Hz, 2 H), 7.55 (m, 1 H), 7.16 (dd, J1 = 2.7 Hz, J2 = 8.9 Hz, 1 H), 3.98 (m, 1 H), 3.90 (s, 3 H), 3.33 (m, 2H), 3.32 (m, 1 H), 2.74 (s, 3 H), 2.55 (m, 1 H), 2.23 (m, 1 H), 2.10 (m, 2 H), 1.95 (s, 3 H).
“Daridorexant”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03545191 for “Study to Assess the Efficacy and Safety of ACT-541468 in Adult and Elderly Subjects With Insomnia Disorder” at ClinicalTrials.gov
Clinical trial number NCT03575104 for “Study to Assess the Efficacy and Safety of ACT-541468 in Adult and Elderly Subjects Suffering From Difficulties to Sleep” at ClinicalTrials.gov
Clinical trial number NCT03679884 for “Study to Assess the Long Term Safety and Tolerability of ACT-541468 in Adult and Elderly Subjects Suffering From Difficulties to Sleep” at ClinicalTrials.gov
///////////////Daridorexant, Quviviq, FDA 2022, APPROVALS 2022, INSOMNIA, ACT541468A, ACT 541468A. ACT-541468A, ACT541468, FDA 2022, APPROVALS 2022
Monoclonal antibody Treatment and prevention of SARS-CoV-2 infection
Formula
C6488H10034N1746O2038S50
CAS
2420564-02-7
Mol weight
146704.817
2196
AZD-8895
AZD8895
COV2-2196
Tixagevimab
Tixagevimab [INN]
UNII-F0LZ415Z3B
WHO 11776
OriginatorVanderbilt University
DeveloperAstraZeneca; INSERM; National Institute of Allergy and Infectious Diseases
ClassAntivirals; Monoclonal antibodies
Mechanism of ActionVirus internalisation inhibitors
RegisteredCOVID 2019 infections
24 Dec 2021Pharmacodynamics data from a preclinical trial in COVID-2019 infections released by AstraZeneca
16 Dec 2021Pharmacodynamics data from a preclinical trial in COVID-2019 infections released by AstraZeneca
10 Dec 2021Registered for COVID-2019 infections (In the elderly, Prevention, In adults) in USA (IM) – Emergency Use Authorization
Tixagevimab/cilgavimab is a combination of two human monoclonal antibodies, tixagevimab (AZD8895) and cilgavimab (AZD1061) targeted against the surface spike protein of SARS-CoV-2[4][5] used to prevent COVID-19. It is being developed by British-Swedish multinational pharmaceutical and biotechnology company AstraZeneca.[6][7] It is co-packaged and given as two separate consecutive intramuscular injections (one injection per monoclonal antibody, given in immediate succession).[2]
In 2020, researchers at Vanderbilt University Medical Center discovered particularly potent monoclonal antibodies, isolated from COVID-19 patients infected with a SARS-CoV-2 circulating at that time. Initially designated COV2-2196 and COV2-2130, antibody engineering was used to transfer their SARS-CoV-2 binding specificity to IgG scaffolds that would last longer in the body, and these engineered antibodies were named AZD8895 and AZD1061, respectively (and the combination was called AZD7442).[8]
To evaluate the antibodies’ potential as monoclonal antibody based prophylaxis (prevention), the ‘Provent’ clinical trial enrolled 5,000 high risk but not yet infected individuals and monitored them for 15 months.[9][10] The trial reported that those receiving the cocktail showed a 77% reduction in symptomatic COVID-19 and that there were no severe cases or deaths. AstraZeneca also found that the antibody cocktail “neutralizes recent emergent SARS-CoV-2 viral variants, including the Delta variant“.[7]
In contrast to pre-exposure prophylaxis, the Storm Chaser study of already-exposed people (post-exposure prophylaxis) did not meet its primary endpoint, which was prevention of symptomatic COVID-19 in people already exposed. AZD7442 was administered to 1,000 volunteers who had recently been exposed to COVID.[9]
Also in October 2021, AstraZeneca requested Emergency Use Authorization for tixagevimab/cilgavimab to prevent COVID-19 from the U.S. Food and Drug Administration (FDA).[12][13]
Emergency use authorization
On 14 November 2021, Bahrain granted emergency use authorization.[14]
On 8 December 2021, the U.S. Food and Drug Administration (FDA) granted emergency use authorization of this combination to prevent COVID-19 (before exposure) in people with weakened immunity or who cannot be fully vaccinated due to a history of severe reaction to coronavirus vaccines.[15] The FDA issued an emergency use authorization (EUA) for AstraZeneca’s Evusheld (tixagevimab co-packaged with cilgavimab and administered together) for the pre-exposure prophylaxis (prevention) of COVID-19 in certain people aged 12 years of age and older weighing at least 40 kilograms (88 lb).[2] The product is only authorized for those individuals who are not currently infected with the SARS-CoV-2 virus and who have not recently been exposed to an individual infected with SARS-CoV-2.[2]
“Tixagevimab”. Drug Information Portal. U.S. National Library of Medicine.
“Cilgavimab”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT04625972 for “Phase III Double-blind, Placebo-controlled Study of AZD7442 for Post-exposure Prophylaxis of COVID-19 in Adults (STORM CHASER)” at ClinicalTrials.gov
Clinical trial number NCT04625725 for “Phase III Double-blind, Placebo-controlled Study of AZD7442 for Pre-exposure Prophylaxis of COVID-19 in Adult. (PROVENT)” at ClinicalTrials.gov
Tixagevimab (teal, right) and cilgavimab (purple, left) binding the spike protein RBD. From PDB: 7L7E.
It was approved for medical use in the United States in December 2019,[6] in Japan in March 2020,[8] in the European Union in January 2021,[3][4] and in Australia in October 2021.[1]
Trastuzumab deruxtecan (DS-8201a) is a HER2-targeting antibody-drug conjugate or ADC), structurally composed of a humanized anti-human HER2 (anti-hHER2) antibody, an enzymatically cleavable peptide-linker, and a proprietary topoisomerase I inhibitor payload (exatecan derivative or DX-8951 / DXd).
Trastuzumab deruxtecan active substance, also referred to as DS-8201a, results from the conjugation of the following intermediates: – Trastuzumab monoclonal antibody (MAAL-9001); – A drug-linker (MAAA-1162a) comprised of a Topoisomerase I inhibitor derivative of exatecan (MAAA1181a) and a tetrapeptide based cleavable linker (MFAH). MAAL-9001 is covalently conjugated to approximately 8 molecules of MAAA-1162a. The linker is designed to be stable in plasma to reduce systemic exposure to the released MAAA-1181a drug. After cell internalisation, the released MAAA-1181a drug leads to apoptosis of the target tumour cells via the inhibition of topoisomerase I. The released MAAA-1181a drug is cell-membrane permeable, giving the ability to penetrate and act in surrounding cells. The effect of the ADC derives primarily from the released MAAA-1181a drug and to a lesser extent to the antibody-dependent cellular cytotoxic (ADCC) effector function of the conjugated antibody. The quality of MAAL-9001 antibody, MAAA-1162a drug-linker and the conjugated antibody is described in separate sections. The structures of DS-8201a, MAAA-1162a, MAAA-1181a, and MAAL-9001 are provided in Figure 1.
Full information for the active substance intermediate MAAA-1162a (C52H56FN9O13, MW 1034.05) was provided in the dossier. MAAA-1162a is composed of DX-8951·MsOH (drug intermediate) and MFAH (linker intermediate with maleimide functionality). The maleimide moiety reacts with the antibody (MAAL-9001) in the conjugation reaction to yield trastuzumab deruxtecan (DS-8201a). MAAA-1162a contains 3 stereogenic centres. General information was provided for solid state form, melting point, moisture sorption, UV-Vis absorption, optical rotation and solubility.
Fam-trastuzumab deruxtecan-nxki is an ADC that is comprised of an anti-HER2 antibody and the potent topoisomerase inhibitor exatecan [206]. These two entities are connected via a liner consisting of a maleimide conjugation handle that includes a protease-cleavable Gly-Gly-Phe-Gly (GGFG) tetrapeptide linker. This conjugation handle could release deruxtecan after internalization of the conjugate by the cancer cells that recognize the antibody. Fam-trastuzumabderuxtecan-nxki was developed by Daiichi Sankyo and AstraZeneca, and granted approval by the FDA in December 2019 [207]. There are approximately eight payload molecules/antibodies. Fam-trastuzumab deruxtecan-nxki has been approved for the treatment of adult patients with HER2 positive breast cancer that is unresectable or metastatic [208]. The synthesis of GGFG linker is described in Scheme 38 [209,210]. First, commercial tert-butyl 2-aminoacetate 274 was treated with Fmoc-L-Phe-OH 275 in the presence of HOBt and DIC, giving the corresponding product 276. Further deprotection of Fmoc group and amide formation gave the intermediate 278, which then underwent removal of Fmoc group to give GGFG linker 279. Lastly, treatment of 279 with the activated ester 280 provided 281.
Preparation of the payload exatecan derivative is described in Scheme 39 [211]. Aluminum-catalyzed Friedel-Crafts acylation of o-fluorotoluene 282 with succinic andydride 283 gave intermediate 284 in 90% yield. Next, hydrogenation reduction ofthe carbonyl group of 284, followed by reaction with SOCl2 in MeOH, furnished 285, which then underwent nitration with H2SO4 and KNO3 to give compound 286 in 48% overall yield. Hydrolysis of 286 followed by treatment with polyphosphoric acid (PPA) gave the cyclization product 287 in only 27% yield. The transformation of 287 into 288 was realized following the four-step sequence: carbonyl reduction with NaBH4, acid-mediated elimination reaction, PtO2-catalyzed hydrogenation reduction, and acetylation with Ac2O. Regioselective benzylic oxidation of 288 in acetone with KMnO4 gave 289 in 65% yield, further functionalization with butyl nitrile and Zn-mediated acylation gave compound 290 in 66% yield over 2 steps. Treatment of 290 with aqueous HCl provided hydrolysis product 291 in 50% yield, which then coupled with ethyl trifluoroacetate to provide intermediate 292. Polycyclic compound 294 was prepared from 292 and 293 through a [4+2] cycloaddition reaction in refluxing toluene. The key intermediate 294 next underwent acidic hydrolysis and chiral resolution to provide the chiral product 295. Further condensation reaction with 296 in the presence of T3P and Et3N in DCM and TFA-promoted removal of the Boc group formed 297. The synthesis of fam-trastuzumab deruxtecan-nxki is described in Scheme 40 [212]. The linker 281 was coupled to 297 in the presence of T3P and Et3N to give the linker-payload 298. Through transformation of the disulfide bonds into free sulfhydryl groups for linkage (DTT in pH 8.0 buffer), followed by re-oxidation of the remaining disulfide bonds with cysteine, the linker-payload 298 was conjugated to the anti-HER2 mAb to give fam-trastuzumab deruxtecan-nxki (XXIX) based on the amount of protein with approximately eight linker/payloads per antibody.
[206] T.N. Iwata, K. Sugihara, T. Wada, T. Agatsuma, [Fam-] trastuzumab deruxtecan (DS-8201a)-induced antitumor immunity is facilitated by the anti-CTLA-4 antibody in a mouse model, PLoS One 14 (2019) 0222280.
[207] R. Voelker, Another targeted therapy for ERBB2-positive breast cancer, JAMA 323 (2020) 408.
[208] S. Modi, C. Saura, T. Yamashita, Y.H. Park, S.B. Kim, K. Tamura, F. Andre, H. Iwata, Y. Ito, J. Tsurutani, J. Sohn, N. Denduluri, C. Perrin, K. Aogi, E. Tokunaga, S.A. Im, K.S. Lee, S.A. Hurvitz, J. Cortes, C. Lee, S. Chen, L. Zhang, J. Shahidi, A. Yver, I. Krop, Trastuzumab deruxtecan in previously treated HER2-positive breast cancer, N. Engl. J. Med. 382 (2020) 610-621.
[209] C.L. Law, K. Klussman, A.F. Wahl, P. Senter, S. Doronina, B. Toki, Treatment of immunological disorders using anti-CD30 antibodies, 2003.WO2003043583.
[210] S. Doronina, P.D. Senter, B.E. Toki, Pentapeptide compounds and uses related thereto, 2002. WO2002088172. [211] H. Terasawa, A. Ejima, S. Ohsuki, K. Uoto, Hexa-cyclic compound, 1998. US5834476.
[212] G.M. Dubowchik, R.A. Firestone, L. Padilla, D. Willner, S.J. Hofstead, K. Mosure, J.O. Knipe, S.J. Lasch, P.A. Trail, Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity, Bioconjugate. Chem 13 (2002) 855-869.
Trastuzumab deruxtecan-nxki is indicated for the treatment of adults with unresectable (unable to be removed with surgery) or metastatic (when cancer cells spread to other parts of the body) HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting and for adults with locally advanced or metastatic HER2-positive gastric or gastroesophageal junction adenocarcinoma who have received a prior trastuzumab-based regimen.[6][7]
Side effects and label warnings
The most common side effects are nausea, fatigue, vomiting, alopecia (hair loss), constipation, decreased appetite, anemia (hemoglobin in blood is below the reference range), decreased neutrophil count (white blood cells that help lead your body’s immune system response to fight infection), diarrhea, leukopenia (other white blood cells that help the immune system), cough and decreased platelet count (component of blood whose function is to react to bleeding from blood vessel injury by clumping, thereby initiating a blood clot).[6]
The prescribing information for fam-trastuzumab deruxtecan-nxki includes a boxed warning to advise health care professionals and patients about the risk of interstitial lung disease (a group of lung conditions that causes scarring of lung tissues) and embryo-fetal toxicity.[6] Interstitial lung disease and pneumonitis, including cases resulting in death, have been reported with fam-trastuzumab deruxtecan-nxki.[6]
The FDA approved fam-trastuzumab deruxtecan-nxki based on the results of one clinical trial enrolling 184 female patients with HER2-positive, unresectable and/or metastatic breast cancer who had received two or more prior anti-HER2 therapies in the metastatic setting.[6] These patients were heavily pretreated in the metastatic setting, receiving between two and 17 therapies prior to receiving fam-trastuzumab deruxtecan-nxki.[6] Patients in the clinical trial received fam-trastuzumab deruxtecan-nxki every three weeks and tumor imaging was obtained every six weeks.[6] The overall response rate was 60.3%, which reflects the percentage of patients who had a certain amount of tumor shrinkage with a median duration of response of 14.8 months.[6]
On 10 December 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a conditional marketing authorization for the medicinal product Enhertu, intended for the treatment of metastatic HER2-positive breast cancer.[10][11] Enhertu was reviewed under EMA’s accelerated assessment program. The applicant for this medicinal product is Daiichi Sankyo Europe GmbH. Trastuzumab deruxtecan was approved for medical use in the European Union in January 2021.[3][4]
In January 2021, the U.S. Food and Drug Administration (FDA) granted accelerated approval to fam-trastuzumab deruxtecan-nxki for the treatment of adults with locally advanced or metastatic HER2-positive gastric or gastroesophageal (GEJ) adenocarcinoma who have received a prior trastuzumab-based regimen.[7][12]
Efficacy was evaluated in a multicenter, open-label, randomized trial (DESTINY-Gastric01, NCT03329690) in participants with HER2-positive locally advanced or metastatic gastric or GEJ adenocarcinoma who had progressed on at least two prior regimens, including trastuzumab, a fluoropyrimidine- and a platinum-containing chemotherapy.[7] A total of 188 participants were randomized (2:1) to receive fam-trastuzumab deruxtecan-nxki 6.4 mg/kg intravenously every three weeks or physician’s choice of either irinotecan or paclitaxel monotherapy.[7]
References
^ Jump up to:abc“Enhertu”. Therapeutic Goods Administration (TGA). 18 October 2021. Retrieved 22 October 2021.
Clinical trial number NCT03329690 for “DS-8201a in Human Epidermal Growth Factor Receptor 2 (HER2)-Expressing Gastric Cancer [DESTINY-Gastric01]” at ClinicalTrials.gov
////////////Fam-trastuzumab deruxtecan-nxki ,, FDA 2019, APROVALS 2019, DS-8201a
CD-271, Differin, Differine106685-40-9[RN] 2-Naphthalenecarboxylic acid, 6-(4-methoxy-3-tricyclo[3.3.1.13,7]dec-1-ylphenyl)- 6-[3-(Adamantan-1-yl)-4-methoxyphenyl]-2-naphthoic acid 6-[4-methoxy-3-(tricyclo[3.3.1.13,7]dec-1-yl)phenyl]naphthalene-2-carboxylic acid AdapaleneCAS Registry Number: 106685-40-9 CAS Name: 6-(4-Methoxy-3-tricyclo[3.3.1.13,7]dec-1-ylphenyl)-2-naphthalenecarboxylic acid Additional Names: 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid Manufacturers’ Codes: CD-271 Trademarks: Differin (Galderma) Molecular Formula: C28H28O3 Molecular Weight: 412.52 Percent Composition: C 81.52%, H 6.84%, O 11.64% Literature References: Retinoid selective for retinoic acid receptor (RAR) subtypes b and g. Prepn: B. Shroot et al.,EP199636; eidem,US4717720 (1986, 1988 both to Cent. Int. Recher. Dermatol.); and structure-activity study: B. Charpentier et al.,J. Med. Chem.38, 4993 (1995). Pilot-scale synthesis: Z. Liu, J. Xiang, Org. Process Res. Dev.10, 285 (2006). HPLC determn in plasma and tissue: R. Ruhl, H. Nau, Chromatographia45, 269 (1997). Clinical pharmacology: C. E. M. Griffiths et al.,J. Invest. Dermatol.101, 325 (1993). Clinical trial in acne: A. Shalita et al.,J. Am. Acad. Dermatol.34, 482 (1996). Reviews of pharmacology and clinical potential: B. A. Bernard, Skin Pharmacol.6, Suppl. 1, 61-69 (1993); R. N. Brogden, K. L. Goa, Drugs53, 511-519 (1997); of clinical use in acne vulgaris: J. Waugh et al.,Drugs64, 1465-1478 (2004). Properties: White crystals from THF and ethyl acetate, mp 319-322°. pK 4.2. Stable to light. Melting point: mp 319-322° pKa: pK 4.2 Therap-Cat: Antiacne. Keywords: Antiacne.
Adapalene is a third-generation topical retinoid primarily used in the treatment of mild-moderate acne, and is also used off-label to treat keratosis pilaris as well as other skin conditions.[1] Studies have found adapalene is as effective as other retinoids, while causing less irritation.[2] It also has several advantages over other retinoids. The adapalene molecule is more stable compared to tretinoin and tazarotene, which leads to less concern for photodegradation.[2] It is also chemically more stable compared to the other two retinoids, allowing it to be used in combination with benzoyl peroxide.[2] Due to its effects on keratinocyte proliferation and differentiation, adapalene is superior to tretinoin for the treatment of comedonal acne and is often used as a first-line agent. [3]
Adapalene is a third-generation topical retinoid with anti-comedogenic, comedolytic, and anti-inflammatory properties used to treat acne vulgaris in adolescents and adults.
SYN
AU 9047961; EP 0199636; US 4717720; US 5098895; US 5183889
J Med Chem 1995,38(26),4993
Friedel-Crafts condensation of 4-bromophenol (I) with 1-adamantanol (II) in the presence of H2SO4 yielded the adamantyl phenol (III). Subsequent alkylation of the sodium phenoxide of (III) with iodomethane produced the methyl ether (IV). The Grignard reagent (V), prepared from aryl bromide (IV), was converted to the organozincate derivative, and then subjected to a nickel-catalyzed cross-coupling with methyl 6-bromo-2-naphthoate (VI) to furnish adduct (VII). The target carboxylic acid was finally obtained by saponification of the methyl ester (VII).
SYN
CA 2021550; EP 0409740; FR 2649976; JP 1991063246; US 5073361; US 5149631
The bromination of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid methyl ester (I) with Br2 in dichloromethane gives the dibromo derivative (II), which is hydrogenated with tritium gas over Pd/C in THF containing TEA to yield the bis tritiated ester (III). Finally, ester (III) is hydrolyzed with NaOH in refluxing methanol to afford the target tritiated naphthoic acid.
SYN
doi:10.1071/CH9732303c US4717720
SYN
Adapalene (CAS NO.: 106685-40-9), with its systematic name of 2-Naphthalenecarboxylic acid, 6-(4-methoxy-3-tricyclo(3.3.1.1(sup 3,7))dec-1-ylphenyl)-, could be produced through many synthetic methods.
Following is one of the synthesis routes: Firstly, Friedel-Crafts condensation of 4-bromophenol (I) with 1-adamantanol (II) in the presence of H2SO4 yields the adamantyl phenol (III). Next, subsequent alkylation of the sodium phenoxide of (III) with iodomethane produces the methyl ether (IV). The Grignard reagent (V), prepared from aryl bromide (IV), is converted to the organozincate derivative, and then subjects to a nickel-catalyzed cross-coupling with methyl 6-bromo-2-naphthoate (VI) to furnish adduct (VII). Finally, the target carboxylic acid is obtained by saponification of the methyl ester (VII).
Synthesis Reference
Graziano Castaldi, Pietro Allegrini, Gabriele Razzetti, Mauro Ercoli, “Process for the preparation of adapalene.” U.S. Patent US20060229465, issued October 12, 2006.
Adapalene has been approved by the FDA as a cream, a gel, a solution and pledgets for the topical treatment of acne vulgaris and is marketed under the tradename of DIFFERIN®.U.S. Pat. No. 4,717,720 (“the ‘720 patent”) discloses benzonaphthalene derivatives, including adapalene. The ‘720 patent describes a process for preparing adapalene (i.e., according to example 9c followed by example 10) that involves two reaction steps.The first step for preparing adapalene according to the ‘720 patent involves the preparation of the methyl ester of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid. According to example 9c of the ‘720 patent, 2-(1-adamantyl)-4-bromoanisole (also known as 1-(5-bromo-2-methoxyphenyl)adamantane) is converted to its organomagnesium derivative and then into its organozinc derivative. The organozinc derivative is next coupled to methyl 6-bromo-2-naphthoate by adding a catalytic amount of NiCl2/DPPE complex (also known as [bis(diphenylphosphino) ethane]dichloronickel(II)). Upon completion of the reaction, the mixture is poured into water, extracted with dichloromethane, and then dried. The product is next isolated by column chromatography by eluting with a mixture of heptane (70%) and dichloromethane (30%). The resulting product is then recrystallized in ethyl acetate (yield: 78%).The second step for preparing adapalene according to the ‘720 patent involves hydrolyzing the product of step 1 (above). According to example 10 of the ‘720 patent, the ester obtained in Example 9c can be treated with a solution of soda in methanol followed by heating at reflux for 48 hours. The solvents are then evaporated and the resulting residue is taken up in water and acidified with concentrated HCl to neutralize the resulting adapalene sodium salt. The resulting solid is next filtered and dried under vacuum over phosphoric anhydride and then recrystallized in a mixture of tetrahydrofuran and ethyl acetate to yield adapalene (yield: 81%).The process of preparing adapalene according to the ‘720 patent is both difficult and uneconomical to conduct on an industrial scale. Regarding step 1, the use of dichloromethane is both toxic and hazardous for the environment. Additionally, purification of the intermediate product by column chromatography, followed by recrystallization, in order to obtain a crystalline product of acceptable purity is both expensive and laborious. Moreover, the step 1 process produces as a biaryllic C—C bond, and the catalytic coupling is noticeably exothermic. Regarding step 2, the synthesis of adapalene and/or its sodium salt requires a long reaction time (i.e., 48 hours) at methanol reflux and further requires a high ratio of solvent (volume) to product (mass).Additionally, according to the prior art, the manufacture of adapalene is not satisfactory for industrial implementation because the presence of high amounts of undesired by-products makes it necessary to use uneconomical purification procedures to isolate the product according to quality specifications. One significant undesired by-product produced during the Grignard reaction of step 1 in the synthesis of adapalene is 3,3′-diadamantyl-4,4′-dimethoxybiphenyl, which has not been previously described in the literature and which is represented by Compound VI (below):
The level of the by-product in a sample of adapalene, adapalene methyl ester and/or an adapalene salt can be determined using standard analytical techniques known to those of ordinary skill in the art. For example, the level can be determined by HPLC. A specific method for determining the level of this impurity is provided herein.Since the solubility of the dimeric by-product is very low in most solvents, the design of an economical industrial process that yields pure adapalene without the use of expensive chromatographic methods requires the selection of the proper solvents and conditions to inhibit formation of the by-product during the manufacturing process.Additionally, adapalene has been described as being white (see, e.g., Merck Index, 13th ed., p. 29). It has been observed that adapalene has a tendency to yellow under certain synthetic conditions or due to the quality of the starting materials used in its preparation. In this regard, color must be attributed to the presence of some specific impurities that may or may not be detectable by conventional methods such as HPLC.
ExampleStep 2: Preparation of 6-[3-(1-adamantyl)-4-methoxy phenyl]-2-naphthoic acid-potassium Salt (i.e., Adapalene Potassium Salt)In a 2 L, five necked cylindrical reaction vessel equipped with reflux condenser, distillation kit, heat-transfer jacket, anchor impeller and purged with nitrogen, were added 48.38 g (dry equivalent amount) of methyl 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoate (1.134×10−1 mol), wet with methanol, 2.73 g of tetrabutylammonium bromide (8.47×10−3 mol), 18.39 g of potassium hydroxide (85% alkali content, freshly titrated. 2.79×10−1 mol) and 581 mL of toluene. The mixture was heated to reflux temperature, and the methanol/water was removed by distillation. The distilled mixture was replaced by pure toluene and the mixture was stirred at reflux for approximately three hours (including the time required for the distillation). The solution was then cooled to approximately 20-25° C., filtered and the resulting solid was washed with toluene.The solid was next suspended in 187 mL of tetrahydrofuran and stirred for approximately 30 minutes. Then, 375 mL of toluene was added, and the mixture was heated to reflux and maintained at that temperature for approximately 1 hour. The solution was then cooled to approximately 20-25° C., filtered, and the resulting solid washed with toluene. The toluene-wet product was then suspended in 256 mL of methanol, heated to reflux for approximately 30 minutes and cooled to 50-60° C. After cooling, 409 mL of water was added dropwise. The mixture was then again heated to reflux for approximately 15 additional minutes, cooled to room temperature and filtered. The resulting solid was washed with water to yield 50.69 g (wet) of adapalene potassium salt (1.12×10−1 mol, dry equivalent amount calculated from loss on drying; yield: 99.18%). Analytical data: HPLC Purity (HPLC at 272 nm): 99.86%; Impurity (i.e., 3,3′-diadamantyl-4,4′-dimethoxybiphenyl) area percent (HPLC at 272 nm): not detected; 1H-NMR (300 MHz, CD3OD): δ 1.83 (broad s, 6H), 2.08 (broad s, 3H), 2.21 (broad s, 6H), 3.88 (s, 3H), 7.04 (d, 1H, J=8.4 Hz), 7.56 (overlapped, 1H, J=2.4, 9.6 Hz), 7.57 (overlapped s, 1H), 7.74 (dd, 1H, J=8.7, 1.8 Hz), 7.87 (d, 1H, J=9.0 Hz), 7.97 (d, 1H, J=8.7 Hz), 8.00 (broad d, 1H, J=0.9 Hz), 8.06 (dd, 1H, 8.4, J=1.8 Hz), 8.47 (broad d, 1H, J=0.9 Hz); 13C-NMR (75.4 MHz, CD3OD): δ 30.6, 38.3, 41.8, 55.5, 113.3, 125.3, 126.4, 126.6, 127.8, 128.3, 130.0, 130.4, 133.0, 134.2, 136.1, 136.3, 139.7, 141.1, 159.9, 175.4.
ExampleStep 3: Preparation of 6-[3-(1-adamantyl)-4-methoxy phenyl]-2-naphthoic Acid (i.e., Adapalene)In 500 mL of methanol was added 49.59 g (1.10×10−1 mol, dry equivalent amount) of the wet solid obtained in Example/Step 2, and the mixture was heated to reflux for 30 minutes and cooled to approximately 40° C. Next, 33.17 g of concentrated HCl was slowly added over approximately 1 hour with gentle stirring in order to ensure homogeneity, followed by the slow addition of 248 mL of water. The resulting mixture was stirred for approximately 30 additional minutes at approximately 40° C. and then cooled to room temperature, filtered and washed with methanol. The wet solid was then suspended with 1020 mL of tetrahydrofuran and heated to reflux for approximately 10 minutes or until complete dissolution. The solution was then cooled to approximately 35° C., the solid particles were removed by filtration, and the filter was washed with tetrahydrofuran.The collected mother liquors were heated to reflux, and 654 g of tetrahydrofuran was removed by distillation. The mixture was then cooled to approximately 55-60° C. Thereafter, 650 mL of methanol was added over approximately 10 minutes, and the mixture heated to reflux for approximately 30 minutes, cooled, and filtered. The resulting solid was filtered with methanol and dried at 80° C. in a vacuum oven to yield 40.54 g of adapalene (9.83×10−2 mol; yield: 89.29% (from adapalene potassium salt); 88.56% (from adapalene methyl ester); and 78.67% (from methyl 6-bromo-2-naphthoate)). Analytical data: HPLC Purity (HPLC at 272 nm): 100.00%; Assay: 99.99%; Residue on Ignition: 0.02%; IR: matches reference.Table 1 (below) lists the peak assignments of the X-ray powder diffractogram of the adapalene obtained and are illustrated in FIG. 1.
TABLE 1
peak
peak_position
peak_intensity
background
1
9.94547
175.32198
42.94638
2
13.18338
239.32156
48.88440
3
14.87487
234.32591
47.91444
4
15.28319
573.40082
53.73505
5
16.37472
1207.21631
69.64595
6
16.54000
882.00000
68.42529
7
17.39657
110.88804
58.39248
8
17.93203
114.02068
55.36037
9
19.44575
285.34473
113.52401
10
19.94692
569.60516
153.63921
11
22.43198
2846.14307
110.81189
12
24.02238
140.20882
85.37505
13
25.04586
925.64282
121.97974
14
25.41035
240.42351
102.81077
15
26.68556
362.45480
68.05973
16
27.71646
141.77916
72.53469
17
40.51307
133.00453
43.44914
18
46.52728
130.31587
50.16773
ExampleStep 4: Preparation of 3,3′-diadamantyl-4,4′-dimethoxybiphenylTo a 100 mL rounded bottom reaction vessel equipped with a magnetic stirrer, thermometer, reflux condenser, pressure compensated addition funnel, were added 0.15 g of 1-(5-bromo-2-methoxyphenyl)adamantane, 0.47 g of magnesium turnings and 7 mL of tetrahydrofuran. The mixture was heated to approximately 35° C., and 0.13 mL of 1,2-dibromoethane were added to the mixture. Reaction exothermy self-heated the mixture. Next, a solution of 4.85 g of 1-(5-bromo-2-methoxyphenyl)adamantane and 28 mL of tetrahydrofuran was added to the mixture dropwise. During this addition, the temperature of the mixture dropped from reflux temperature to approximately 45° C. The reaction was then stirred for approximately 45 additional minutes at approximately 45° C. and was permitted to cool to approximately 22° C. Next, 2.3 g of ZnCl2 was added to the mixture, resulting in an exothermic reaction that raised the temperature of the mixture to approximately 38° C. The mixture was then permitted to cool to approximately 22° C. and was stirred for approximately 1 hour at this temperature.Next, 0.03 g of Pd(OAc)2 and 3.5 g of 1-(5-bromo-2-methoxyphenyl) adamantane were added to the mixture, followed by 25 mL of tetrahydrofuran in order to improve agitation, and the mixture was heated at reflux for approximately 24 hours. The resulting mixture was then evaporated to dryness and poured into 103 mL of 0.015 N HCl. Next, 150 mL of dichloromethane and 100 mL of water were added to yield a mixture consisting of a solid, an aqueous layer and an organic layer. The mixture was then filtered to separate the solid, the aqueous layer was discarded, and the organic layer was washed with 200 mL of water and decanted again. This process was repeated twice on the filtered solid. The three collected organic layers were evaporated to dryness, washed in methanol, and dried to yield 2.1 g of 3,3′-diadamantyl-4,4′-dimethoxybiphenyl (yield: 39.9%). Analytical data: Melting point: 288.1-289.1° C.; Elemental analysis: C 83.63%, H 8.73%; 1H-NMR (300 MHz, CDCl3): δ 1.78 (broad s, 12H), 2.08 (broad s, 6H), 2.15 (broad s, 12H), 3.86 (s, 6H), 6.92 (dm, 2H, J=8.1 Hz), 7.34 (dd, 2H, J=2.4, 8.1 Hz), 7.39 (d, 2H, J=2.4 Hz); 13C-NMR (75.4 MHz, CDCl3): δ 29.2, 37.1, 37.2, 40.6, 55.1, 111.9, 125.0, 125.5, 134.0, 138.5, 157.8; MS (EI, 70 eV): m/z=484 (6), 483 (36), 412 (1,100), 410 (5), 347 (8), 135 (22), 107 (7), 93 (14), 79 (17), 67 (9), 55 (6); IR (Selected absorption bands): 2992, 2964, 2898, 2850, 1603 cm−1.
The compound 6-[3-(l – Adamantyl) – 4 – methoxy phenyl] – 2 – naphthoic acid of Formula – I known as Adapalene is used in dermatology, particularly in the treatment of acne vulgaris and psoriasis.
Formula – 1Adapalene was first time disclosed in the US patent No. 4,717,720 (herein after referred as ‘720) describe the preparation of compound of Formula – I using Negishi cross Coupling. In this reaction, 2-(l-adamantyl)-4-bromoanisole is converted to its organomagnesium compound followed by conversion to organozinc compound using zinc chloride and reacted with 6-bromo-2-methylnaphthoate employing transition metal as reaction catalyst such as palladium or nickel or one of its complexes with various phosphines. The reaction sequence is as shown in scheme – 1 below:
Scheme – 1 Another US patent No. 5,015,758 describe the process for preparation of 6[3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoate a penultimate step for preparation of Adapalene using Friedel – Crafts alkylation by reacting 1 – acetoxy adamantane with methyl – 6 – (4 – hydroxyphenyl) – 2 – naphthoate in presence of cone. Sulfuric acid in solvent n – heptane.Another improved process was published in the journal, Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. The process involves the preparation of intermediates followed by Negishi cross Coupling, where in 2-(l-adamantyl)-4-bromophenol was prepared using 1 – adamentol and 4- bromo phenol in presence of 98% sulphuric acid and acetic acid, which on methylation with dimethyl sulfate and potassium carbonate in dry acetone yields 2-(l -adamantyl)-4-bromoanisole. The compound is reacted with magnesium to form Grignard reagent and then coupled with 6-bromo-2-methylnaphthoate in presence of novel Pd – Zn double metal catalyst to yield ester, which on saponification followed by treatment with acid yields Adapalene.The recent published application WO 2006/108717 describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – 1. The application describes the preparation of 3-adamantyl-4-methoxyphenyl boronic acid from 2-(l-adamantyl)-4- bromoanisole using n-Butyl Lithium and triisopropyl borate in solvent tetrahydrofuran. Finally 3-adamantyl-4-methoxyphenyl boronic acid is reacted with 6-bromo-2-naphthoic acid involving Suzuki coupling in presence of Palladium acetate catalyst, a ligand 2 – (dicyclohexyl – phosphino) biphenyl, an inorganic base in solvent to get the compound adapalene.Some of the drawbacks of the prior art processes include:- The reported process in US patent 4717720, using Negishi cross coupling involves Grignard reaction. This requires anhydrous condition and a possibility of runaway reaction during Grignard reagent formation. Also the reaction involves the addition of fused ZnC12 and the preparation of the catalyst NiC12 (DPPE) complex, which needs to be freshly prepared increases the reaction step and has to be thoroughly dried before its use for coupling. Further the coupling reaction, results in the formation of dimer impurities during the organozinc compound reaction, with 2-(I -adamantyl)-4-bromoanisole and 6-bromo-2-methylnaphthoate respectively, which are difficult to remove. All these operations make the entire synthesis extremely sensitive and difficult to handle.Some of the above drawbacks were addressed by the authors in the article published in Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. But the use of Pd catalyst with the ligand like PdCl2 (PPh3)2 for the direct conversion of Grignard reagent employing ZnCYl in catalytic amount has its own limitations. The use of Grignard reagent, palladium catalyst with ligand and hygroscopic ZnCl2 demerits this process for industrial application.The recent published application WO 2006/108717; describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – I. The use of organo boronic acids for the Suzuki reaction has some limitations because of the indeterminate stoichiometry associated with the use of boronic acid, and its difficulty in purification and the byproducts formed during the reaction.Therefore there remains a need for an improved process for preparing adapalene that eliminates or substantially reduces the impurities, decreases the number of steps, and employs a more robust process which is convenient and cost efficient.
Examples:Example 1: Preparation of 3 – Adamantyl – 4 – methoxy phenyl potassium trifluoroborate:In a 2.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 100.0 gm of 2-(l- adamantyl) 4-bromo anisole was charged in 600 ml tetrahydrofuran. The reaction mixture was cooled to -55 ± 50C and 302 ml of 1.6 M n – butyl Lithium was slowly added and stirred. 87 ml of tri isopropyl borate was then charged and stirring was continued for 30 minutes at -55 ± 5°C. Cooling was removed and the temperature raised slowly to 25 – 300C. 1.0 L of 1.2N hydrochloric acid was then charged and reaction mass was stirred for 30 minutes and separated the organic layer. The organic layer was charged in 1.0 L round bottom flask and freshly prepared aqueous solution of potassium hydrogen difluoride (230 gm, in 700 ml water) was added at 25 – 300C and stirring was maintained till white precipitate is obtained. The mixture was continued under stirring and cooled to 0 – 50C. The product, 3 – adamantyl – 4 – methoxyphenyl potassium trifluoroborate obtained was filtered, washed with 100 ml of ethyl acetate. The product was dried at 60 – 65°C till constant weight. Yield: 90.5 gm (83%), Purity: 99.0 % by HPLC.Example 2: Preparation of 6 – [3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid:In a 1.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 50.0 gm of 3 – Adamantyl – 4 – methoxyphenyl potassium trifluoroborate, 23 gm of 6- bromo -2-methyl napthoate in 300 ml tetrahydrofuran (THF) was charged. Stirred for 15 min and charged 3.0 gm of 5% Pd / C was and aqueous potassium hydroxide solution (50.0 gm in 300 ml water). Stirring was continued and the temperature was raised to reflux. The reaction mass was maintained for 10 hours at reflux and after the completion of the reaction, 200 ml of tetrahydrofuran: water (1 : 1) mixture was added and then filtered through hyflow bed at 45-500C. The hyflow bed was washed with tetrahydrofuran: water (1 : 1) mixture at 45-500C. 500 ml water was charged and the reaction mass was stirred. The aqueous layer was acidified with 1.2N hydrochloric acid. The precipitated mass was filtered, washed with water till neutral pH. The solid product obtained was dried at 70 – 75°C till constant weight to get 6 – [3-(l- adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid.The dried product was taken in 300 ml of tetrahydrofuran and stirred. The temperature was raised to reflux and was maintained for 30 minutes. The heating was stopped and cooled the reaction mass to 25 – 300C. 500 ml of n – heptane was charged to the reaction mass and stirred for 30 minutes. The reaction mass was then chilled to 0 – 5°C and maintained stirring at 0 – 5°C temperature for 2.0 hours. The precipitated solid was filtered and washed with n – heptane. The pure crystalline 6 – [3-(l- adamantyl) – 4 methoxyphenyl] – 2 – naphthoic acid thus obtained was then dried till constant weight. Yield = 40 – 42 gms (68 – 72 %)
Strategies that were adopted during the process development of adapalene to achieve a cost-effective commercial-scale synthesis are described herein. These included (1) the use of AcOH/H2SO4 to afford 2-(1-adamantyl)-4-bromophenol in quantitative yield; (2) the dimethyl sulfate methylation to enhance the yield of methylation to 95%; (3) direct conversion of the Grignard reagent into methyl 6-(3-(1-adamantyl)-4-methoxyphenyl)-2-naphthoate by the catalysis of both PdCl2(PPh3)2 and ZnCl2 in high yield; (4) the use of EDTA-disodium salt dihydrate to ensure the heavy metal’s content within acceptable limits; (5) the use of toluene to simplify the original chromatographic purification to recrystallization. The pilot-scale synthesis of adapalene is described in detail in the Experimental Section.
6-(3-(1-Adamantyl)-4-methoxyphenyl)-2-naphthoic Acid (Adapalene, 1). Compound 7 (213 g, 0.5 mol) was treated with 2 N NaOH solution (8 L) in methanol under reflux for 8 h. After evaporation of methanol (7 L) and addition of water (1.5 L), the mixture was acidified until pH 1 with 6 N HCl and filtrated through Celite. The residue was washed with water (3 × 5 L), and recrystallized twice in THF (194 g/2 L/time) to give pure (99% HPLC) 1 (177 g, 85%), mp 320-322 °C.1 H NMR (400 MHz, DMSO-d6) δ 1.77 (6 H,s, H on 1-adamantyl), 2.07 (3 H, s, H on 1-adamantyl), 2.14 (6 H, s, H on 1-adamantyl), 3.87 (3 H, s, H on ArOCH3), 7.12 (1 H, d, J ) 8.4 Hz, 5-phenyl H), 7.58 (1 H, d, J ) 2.0 Hz, 2-phenyl H), 7.65 (1 H, dd, J ) 8.4 Hz, J ) 2.0 Hz, 6-phenyl H), 7.89 (1 H, d, J ) 8.8 Hz, 7-naphthyl H), 7.98 (1 H, d, J ) 8.8 Hz, 4-naphthyl H), 8.08 (1 H, d, J ) 8.8 Hz, 8-naphthyl H), 8.15 (1 H, d, J ) 8.8 Hz, 3-naphthyl H), 8.22 (1 H, s, 5-naphthyl H), 8.60 (1 H, s, 1-naphthyl H), 13.05 (1 H, s, -COOH); 13C NMR (100 MHz, DMSO-d6) δ 28.32, 36.47, 40.09, 55.28, 112.68, 123.99, 124.99, 125.38, 125.68, 125.85, 127.55, 128.25, 129.72, 130.13, 130.83, 131.46, 135.38, 138.00, 140.13, 158.53, 167.34.
Adapalene, namely 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid, having the following chemical formula:
is disclosed in U.S. Pat. No. 4,717,720 and used in dermatology, in particular for the treatment of acne vulgaris and psoriasis.According to U.S. Pat. No. 4,717,720 the synthesis is carried out by a coupling reaction between a magnesium, lithium or zinc derivative of a compound of formula (A) and a compound of formula (B), wherein X and Y are Cl, Br, F or I; R is hydrogen or alkyl; and Ad is 1-adamantyl
in an anhydrous solvent, in the presence of a metal transition or a complex thereof as a catalyst.A number of alternative synthetic approaches have been suggested in order to reduce the preparation costs. Surprisingly, particularly advantageous proved the alternative synthesis of the invention, which makes use of easily-available, low-cost 6-hydroxy-2-naphthoic acid alkyl esters as intermediates, and provides good yields.EXAMPLE 1Synthesis of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid methyl ester [adapalene methyl ester]A round-bottom flask is loaded with nickel (II) chloride (0.158 g; 1.2 mmol) and THF (20 ml), and tris(hydroxypropyl)phosphine (1.53 g; 7.3 mmol) is added to the mixture, which is refluxed for an hour, then cooled to a temperature of 50° C. and added in succession with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), potassium phosphate (10.38 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (7-g; 24.4 mmol), water (0.88 g; 48.8 mmol) and THF (50 ml). The mixture is heated under reflux for 24 hours, then cooled to a temperature ranging from 50 to 55° C. and added with water, adjusting pH to a value below 7 with acetic acid. After cooling to a temperature of 15° C., the resulting product is filtered, thereby obtaining crystalline adapalene methyl ester (8.5 g; 20.08 mmol) in 82% yield.1H NMR: (300 MHz, DMSO), δ 8.6 (s, 1H), δ 8.3-7.8 (m, 6H), δ 7.7-7.5 (m, 2H), δ 7.1 (d, 1H), δ 3.9 (s, 3H), δ 3.85 (s, 3H), δ 2 (m, 9H), δ 1.7 (m, 6H).EXAMPLE 2Synthesis of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid sodium salt [adapalene sodium salt]A round-bottom flask is loaded with adapalene methyl ester (7 g; 16.41 mmol), THF (42 ml), water (7 ml) and a 50% w/w sodium hydroxide aqueous solution (1.44 g; 18.05 mmol). The mixture is refluxed for 6 hours, then added with water (133 ml) and THF is distilled off to a residual content of approx. 5% w/w, heated to a temperature of about 80° C. until complete dissolution of the solid, then cooled to 15° C. The crystallized product is filtered and dried under vacuum in a static dryer at a temperature of 50° C., thereby obtaining adapalene sodium salt (6.7 g; 15.42 mmol) in 94% yield.EXAMPLE 3Synthesis of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid [adapalene]A round-bottom flask is loaded with adapalene sodium salt (6.7 g; 15.42 mmol), THF (40 ml) and water (7 ml) and the mixture is refluxed until complete dissolution of the solid. The resulting solution is dropped into a 3% w/w acetic acid aqueous solution, keeping the temperature above 60-70° C., to precipitate adapalene acid (6.3 g; 15.27 mmol), which is filtered and dried under vacuum at a temperature of 50-60° C. The yield is 95%.EXAMPLE 4Synthesis of adapalene methyl esterA round-bottom flask is loaded with nickel (II) chloride (0.158 g; 1.2 mmol) and THF (20 ml), and tris(hydroxypropyl)phosphine (1.53 g; 7.3 mmol) is added. The mixture is refluxed for an hour, then cooled to a temperature of 50° C. and added in succession with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), potassium phosphate (10.38 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (9.1 g; 31.8 mmol), water (10.53 g; 585.3 mmol) and THF (50 ml). The mixture is refluxed for 24 hours, then cooled to a temperature ranging from 50 to 55° C., added with water, and adjusted to pH lower than 7 with acetic acid. After cooling to 15° C., the resulting product is filtered, thereby obtaining adapalene methyl ester (9 g; 21.2 mmol) in 86% yield.EXAMPLE 5Synthesis of adapalene methyl esterA round-bottom flask is loaded with nickel (II) chloride (0.158 g; 1.2 mmol) and THF (15 ml), and tris(hydroxypropyl)phosphine (1.53 g; 7.3 mmol) is added. The mixture is refluxed for an hour, then cooled to a temperature of 50° C. and added in succession with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), potassium carbonate (6.75 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (9.1 g; 31.8 mmol), water (8.11 g; 450.5 mmol) and THF (30 ml). The mixture is refluxed for 24 hours, then cooled to a temperature ranging from 50 to 55° C., added with water, and adjusted to pH lower than 7 with acetic acid. After cooling to 15° C., the resulting product is filtered, thereby obtaining adapalene methyl ester (9.37 g; 21.96 mmol) in 90% yield.EXAMPLE 6Synthesis of adapalene methyl esterA round-bottom flask is loaded with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), THF (70 ml), potassium phosphate (10.38 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (7 g; 24.4 mmol), nickel chloride complexed with tri(cyclohexyl)phosphine (0.83 g; 1.2 mmol) and tri(cyclohexyl)phosphine (1.37 g; 4.88 mmol). The mixture is refluxed for 24 hours, then cooled to a temperature ranging from 50 to 55° C. and added with water, then cooled to 15° C. The resulting product is filtered, thereby obtaining adapalene methyl ester (8.1 g; 19.0 mmol) in 78% yield.
The compound 6-[3-(l – Adamantyl) – 4 – methoxy phenyl] – 2 – naphthoic acid of Formula – I known as Adapalene is used in dermatology, particularly in the treatment of acne vulgaris and psoriasis.
Formula – 1Adapalene was first time disclosed in the US patent No. 4,717,720 (herein after referred as ‘720) describe the preparation of compound of Formula – I using Negishi cross Coupling. In this reaction, 2-(l-adamantyl)-4-bromoanisole is converted to its organomagnesium compound followed by conversion to organozinc compound using zinc chloride and reacted with 6-bromo-2-methylnaphthoate employing transition metal as reaction catalyst such as palladium or nickel or one of its complexes with various phosphines. The reaction sequence is as shown in scheme – 1 below:
Scheme – 1 Another US patent No. 5,015,758 describe the process for preparation of 6[3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoate a penultimate step for preparation of Adapalene using Friedel – Crafts alkylation by reacting 1 – acetoxy adamantane with methyl – 6 – (4 – hydroxyphenyl) – 2 – naphthoate in presence of cone. Sulfuric acid in solvent n – heptane.Another improved process was published in the journal, Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. The process involves the preparation of intermediates followed by Negishi cross Coupling, where in 2-(l-adamantyl)-4-bromophenol was prepared using 1 – adamentol and 4- bromo phenol in presence of 98% sulphuric acid and acetic acid, which on methylation with dimethyl sulfate and potassium carbonate in dry acetone yields 2-(l -adamantyl)-4-bromoanisole. The compound is reacted with magnesium to form Grignard reagent and then coupled with 6-bromo-2-methylnaphthoate in presence of novel Pd – Zn double metal catalyst to yield ester, which on saponification followed by treatment with acid yields Adapalene.The recent published application WO 2006/108717 describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – 1. The application describes the preparation of 3-adamantyl-4-methoxyphenyl boronic acid from 2-(l-adamantyl)-4- bromoanisole using n-Butyl Lithium and triisopropyl borate in solvent tetrahydrofuran. Finally 3-adamantyl-4-methoxyphenyl boronic acid is reacted with 6-bromo-2-naphthoic acid involving Suzuki coupling in presence of Palladium acetate catalyst, a ligand 2 – (dicyclohexyl – phosphino) biphenyl, an inorganic base in solvent to get the compound adapalene.Some of the drawbacks of the prior art processes include:- The reported process in US patent 4717720, using Negishi cross coupling involves Grignard reaction. This requires anhydrous condition and a possibility of runaway reaction during Grignard reagent formation. Also the reaction involves the addition of fused ZnC12 and the preparation of the catalyst NiC12 (DPPE) complex, which needs to be freshly prepared increases the reaction step and has to be thoroughly dried before its use for coupling. Further the coupling reaction, results in the formation of dimer impurities during the organozinc compound reaction, with 2-(I -adamantyl)-4-bromoanisole and 6-bromo-2-methylnaphthoate respectively, which are difficult to remove. All these operations make the entire synthesis extremely sensitive and difficult to handle.Some of the above drawbacks were addressed by the authors in the article published in Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. But the use of Pd catalyst with the ligand like PdCl2 (PPh3)2 for the direct conversion of Grignard reagent employing ZnCYl in catalytic amount has its own limitations. The use of Grignard reagent, palladium catalyst with ligand and hygroscopic ZnCl2 demerits this process for industrial application.The recent published application WO 2006/108717; describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – I. The use of organo boronic acids for the Suzuki reaction has some limitations because of the indeterminate stoichiometry associated with the use of boronic acid, and its difficulty in purification and the byproducts formed during the reaction.Therefore there remains a need for an improved process for preparing adapalene that eliminates or substantially reduces the impurities, decreases the number of steps, and employs a more robust process which is convenient and cost efficient.The present inventors have come out with a novel process which ameliorates the problems in the prior art with a one – pot process for the preparation of adapalene by employing Suzuki – Miyaura coupling involving the use of novel reactant 3-adamantyl-4- methoxyphenyl potassium trifiuoroborate.The novel compound 3 – Adamantyl – 4 – methoxy phenyl potassium trifiuoroborate, exhibit superb behavior in the Suzuki-Miyaura reaction and provides a powerful method for the preparation of 6 – [3-(I – Adamantyl) – 4 – methoxy phenyl] – 2 – naphthoic acid, the compound of Formula – I.
Formula – 1Potassium organotrifluoroborates are air and moisture-stable crystalline solids which can be stored for extended periods of time making it more industrial friendly to use on large scale production.The other advantage of the present invention is in the use of methyl ester of 6 – Bromo – 2 -naphthoic acid and isolating adapalane directly from the reaction instead of its methyl ester, the above process becomes more robust and eliminates the saponification step as reported in prior art. Also the use of readily and cheaply available Pd catalyst on carbon over the conventional and costlier Pd-catalyst with ligands offers further advantage to the current process.Examples:Example 1: Preparation of 3 – Adamantyl – 4 – methoxy phenyl potassium trifluoroborate:In a 2.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 100.0 gm of 2-(l- adamantyl) 4-bromo anisole was charged in 600 ml tetrahydrofuran. The reaction mixture was cooled to -55 ± 50C and 302 ml of 1.6 M n – butyl Lithium was slowly added and stirred. 87 ml of tri isopropyl borate was then charged and stirring was continued for 30 minutes at -55 ± 5°C. Cooling was removed and the temperature raised slowly to 25 – 300C. 1.0 L of 1.2N hydrochloric acid was then charged and reaction mass was stirred for 30 minutes and separated the organic layer. The organic layer was charged in 1.0 L round bottom flask and freshly prepared aqueous solution of potassium hydrogen difluoride (230 gm, in 700 ml water) was added at 25 – 300C and stirring was maintained till white precipitate is obtained. The mixture was continued under stirring and cooled to 0 – 50C. The product, 3 – adamantyl – 4 – methoxyphenyl potassium trifluoroborate obtained was filtered, washed with 100 ml of ethyl acetate. The product was dried at 60 – 65°C till constant weight. Yield: 90.5 gm (83%), Purity: 99.0 % by HPLC.Example 2: Preparation of 6 – [3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid:In a 1.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 50.0 gm of 3 – Adamantyl – 4 – methoxyphenyl potassium trifluoroborate, 23 gm of 6- bromo -2-methyl napthoate in 300 ml tetrahydrofuran (THF) was charged. Stirred for 15 min and charged 3.0 gm of 5% Pd / C was and aqueous potassium hydroxide solution (50.0 gm in 300 ml water). Stirring was continued and the temperature was raised to reflux. The reaction mass was maintained for 10 hours at reflux and after the completion of the reaction, 200 ml of tetrahydrofuran: water (1 : 1) mixture was added and then filtered through hyflow bed at 45-500C. The hyflow bed was washed with tetrahydrofuran: water (1 : 1) mixture at 45-500C. 500 ml water was charged and the reaction mass was stirred. The aqueous layer was acidified with 1.2N hydrochloric acid. The precipitated mass was filtered, washed with water till neutral pH. The solid product obtained was dried at 70 – 75°C till constant weight to get 6 – [3-(l- adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid.The dried product was taken in 300 ml of tetrahydrofuran and stirred. The temperature was raised to reflux and was maintained for 30 minutes. The heating was stopped and cooled the reaction mass to 25 – 300C. 500 ml of n – heptane was charged to the reaction mass and stirred for 30 minutes. The reaction mass was then chilled to 0 – 5°C and maintained stirring at 0 – 5°C temperature for 2.0 hours. The precipitated solid was filtered and washed with n – heptane. The pure crystalline 6 – [3-(l- adamantyl) – 4 methoxyphenyl] – 2 – naphthoic acid thus obtained was then dried till constant weight. Yield = 40 – 42 gms (68 – 72 %)
/////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Per the recommendations of the Global Alliance on Improving Outcomes of Acne, retinoids such as adapalene are considered first-line therapy in acne treatment and are to be used either independently or in conjunction with benzoyl peroxide and/or an antimicrobial agent, like clindamycin, for maximum efficacy.[4][5] Furthermore, adapalene, like other retinoids, increases the efficacy and penetration of other topical acne medications that are used in conjunction with topical retinoids as well as hastens the improvement of the post-inflammatory hyperpigmentation caused by acne.[4] In the long term, it can be used as maintenance therapy.[4]
Adapalene is known to cause mild adverse effects such as photosensitivity, irritation, redness, dryness, itching, and burning.[2] It is common (between 1% and 10% of users)[7] to experience a brief sensation of warmth or stinging, as well as dry skin, peeling and redness during the first 2–4 weeks of using the medication.[4][8] These effects are considered mild and generally decrease over time.[4][8] Any serious allergic reaction is rare.[8] Furthermore, of the three topical retinoids, adapalene is often regarded as the most tolerable.[6]
In pregnancy
Use of topical adapalene in pregnancy has not been well studied, but has a theoretical risk of retinoid embryopathy.[9] Thus far, there is no evidence that the cream causes problems in the baby if used during pregnancy. Use is at the consumer’s own risk.[10]
According to the Drugs and Lactation Database, topical adapalene has poor systemic absorption and results in low blood levels (less than 0.025 mcg/L) despite long term use, suggesting that there is low risk of harm for a nursing infant.[11] However, it is recommended that the topical medication should not be applied to the nipple or any other area that may come into direct contact with the infant’s skin.[11]
Interactions
Adapalene has been shown to enhance the efficacy of topical clindamycin, although adverse effects are also increased.[12][13] Application of adapalene gel to the skin 3–5 minutes before application of clindamycin enhances penetration of clindamycin into the skin, which may enhance the overall efficacy of the treatment as compared to clindamycin alone.[14]
Pharmacology
Unlike the retinoid tretinoin (Retin-A), adapalene has also been shown to retain its efficacy when applied at the same time as benzoyl peroxide due to its more stable chemical structure.[15] Furthermore, photodegradation of the molecule is less of a concern in comparison to tretinoin and tazarotene.[6]
Pharmacokinetics
Absorption of adapalene through the skin is low. A study with six acne patients treated once daily for five days with two grams of adapalene cream applied to 1,000 cm2 (160 sq in) of skin found no quantifiable amounts, or less than 0.35 ng/mL of the drug, in the patients’ blood plasma.[16] Controlled trials of chronic users of adapalene have found drug levels in the patients’ plasma to be 0.25 ng/mL.[9]
Pharmacodynamics
Adapalene is highly lipophilic. When applied topically, it readily penetrates hair follicles and absorption occurs 5 minutes after topical application.[2] After penetration into the follicle, adapalene binds to nuclear retinoic acid receptors (namely retinoic acid receptor beta and gamma).[5][9] These complexes then bind to the retinoid X receptor which induces gene transcription by binding to specific DNA sites, thus modulating downstream keratinocyte proliferation and differentiation.[2][9] This results in normalization of keratinocyte differentiation, allowing for decreased microcomedone formation, decreased clogging of pores, and increased exfoliation by increasing cell turnover.[6][9][17] Adapalene is also regarded as an anti-inflammatory agent, as it suppresses the inflammatory response stimulated by the presence of Cutibacterium acnes,[6] and inhibits both lipoxygenase activity and the oxidative metabolism of arachidonic acid into prostaglandins.[9]
Adapalene selectively targets retinoic acid receptor beta and retinoic acid receptor gamma when applied to epithelial cells such as those found in the skin.[18] Its agonism of the gamma subtype is largely responsible for adapalene’s observed effects. In fact, when adapalene is applied in conjunction with a retinoic acid receptor gamma antagonist, adapalene loses clinical efficacy.[19]
Retinization is a common temporary phenomenon reported by patients when initiating use of retinols.[20] Within the initial period of treatment, skin can become red, irritated, dry and may burn or itch from retinol application; however, this tends to resolve within four weeks with once a day use.[20]
^ Wolf JE, Kaplan D, Kraus SJ, Loven KH, Rist T, Swinyer LJ, Baker MD, Liu YS, Czernielewski J (September 2003). “Efficacy and tolerability of combined topical treatment of acne vulgaris with adapalene and clindamycin: a multicenter, randomized, investigator-blinded study”. Journal of the American Academy of Dermatology. 49 (3 Suppl): S211-7. doi:10.1067/S0190-9622(03)01152-6. PMID12963897.
^ Martin B, Meunier C, Montels D, Watts O (October 1998). “Chemical stability of adapalene and tretinoin when combined with benzoyl peroxide in presence and in absence of visible light and ultraviolet radiation”. The British Journal of Dermatology. 139 Suppl 52: 8–11. doi:10.1046/j.1365-2133.1998.1390s2008.x. PMID9990414. S2CID43287596.
^US Patent 4717720A, Shroot B, Eustache J, Bernardon J-M, “Benzonaphthalene derivatives and compositions”, published 1988-01-05, issued 1988-01-05, assigned to Galderma Research and Development SNC
read https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/071413s019lbl.pdfFluphenazineCAS Registry Number: 69-23-8 CAS Name: 4-[3-[2-(Trifluoromethyl)-10H-phenothiazin-10-yl]propyl]-1-piperazineethanol Additional Names: 1-(2-hydroxyethyl)-4-[3-(trifluoromethyl-10-phenothiazinyl)propyl]piperazine; 10-[3¢-[4¢¢-(b-hydroxyethyl)-1¢¢-piperazinyl]propyl]-3-trifluoromethylphenothiazine; 2-(trifluoromethyl)-10-[3-[1-(b-hydroxyethyl)-4-piperazinyl]propyl]phenothiazine Manufacturers’ Codes: S-94; SQ-4918 Molecular Formula: C22H26F3N3OS, Molecular Weight: 437.52 Percent Composition: C 60.39%, H 5.99%, F 13.03%, N 9.60%, O 3.66%, S 7.33% Literature References: Prepn: H. L. Yale, F. Sowinski, J. Am. Chem. Soc.82, 2039 (1960); GB829246; G. E. Ullyot, US3058979 (1960, 1962 both to SKF); GB833474 (1960 to Scherico), C.A.54, 21143e (1960); E. L. Anderson et al.,Arzneim.-Forsch.12, 937 (1962); H. L. Yale, R. C. Merrill, US3194733 (1965 to Olin Mathieson). Metabolism: J. Dreyfuss, A. J. Cohen, J. Pharm. Sci.60, 826 (1971). Comprehensive description of the enanthate ester: K. Florey, Anal. Profiles Drug Subs.2, 245-262 (1973); of the dihydrochloride: idem,ibid. 263-294; of the decanoate ester: G. Clarke, ibid.9, 275-294 (1980). Properties: Dark brown viscous oil, bp0.5 268-274°; bp0.3 250-252°. Boiling point: bp0.5 268-274°; bp0.3 250-252° Derivative Type: Dihydrochloride CAS Registry Number: 146-56-5 Trademarks: Anatensol (BMS); Dapotum (BMS); Lyogen (Promonta Lundbeck); Moditen (Sanofi Winthrop); Omca (BMS); Pacinol (Schering); Permitil (Schering); Prolixin (Apothecon); Siqualone (BMS); Tensofin (BMS); Valamina (Schering) Molecular Formula: C22H26F3N3OS.2HCl, Molecular Weight: 510.44 Percent Composition: C 51.77%, H 5.53%, F 11.17%, N 8.23%, O 3.13%, S 6.28%, Cl 13.89% Properties: Crystals from abs ethanol, mp 235-237°. Also reported as mp 224.5-226°. Melting point: mp 235-237°; Also reported as mp 224.5-226°
Derivative Type: Decanoate CAS Registry Number: 5002-47-1 Manufacturers’ Codes: SQ-10733; QD-10733 Trademarks: Modecate (Sanofi Winthrop) Molecular Formula: C32H44F3N3O2S, Molecular Weight: 591.77 Percent Composition: C 64.95%, H 7.49%, F 9.63%, N 7.10%, O 5.41%, S 5.42% Properties: Pale yellow-orange, viscous liquid. Slowly crystallizes at room temp. mp 30-32°. Very sol in chloroform, ether, cyclohexane, methanol, ethanol. Insol in water. Melting point: mp 30-32° Derivative Type: Enanthate CAS Registry Number: 2746-81-8 Manufacturers’ Codes: SQ-16144 Molecular Formula: C29H38F3N3O2S, Molecular Weight: 549.69Percent Composition: C 63.36%, H 6.97%, F 10.37%, N 7.64%, O 5.82%, S 5.83% Properties: Pale yellow to yellow-orange viscous liquid or oily solid. Therap-Cat: Antipsychotic. Keywords: Antipsychotic; Phenothiazines.
Fluphenazine is a phenothiazine used to treat patients requiring long-term neuroleptic therapy.
A phenothiazine used in the treatment of psychoses. Its properties and uses are generally similar to those of chlorpromazine.
Fluphenazine is a typical antipsychotic of the phenothiazine class.[1] Its mechanism of action is not entirely clear but believed to be related to its ability to block dopamine receptors.[1] In up to 40% of those on long term phenothiazines, liver function tests become mildly abnormal.[5]
Fluphenazine, 4-[3-[2-(trifluoromethyl)phenothiazin-10-yl]propyl]-1-piperazineethanol (6.1.8), is synthesized by any of the methods described above [21–27]. Alkylation of 2-trifluoromethylphenothiazine using 4-formyl-1-piperazineylpropylchlo-ride in the presence of sodium amide synthesizes 2-trifluoromethyl-10-[3-(4-formyl-1-piperazinyl)propyl]phenothizine (6.1.6). Further alkaline hydrolysis removes the N-formyl group, giving 2-trifluoromethyl-10-[3-(1-piperazinyl)propyl]phenothiazine (6.1.7). This is alkylated by 2-bromethanol-1 acetate, which upon further acidic hydrolysis removes the protecting acetyl group, yielding fluphenazine (6.1.8) [27,28].
Fluphenazine is an extremely strong antipsychotic drug. A stimulatory effect accompanies the neuroleptic effect. It is used in psychiatry for treating various forms of schizophrenia and other mental illnesses. The most common synonyms are fluorphenazine, moditen, dapotum, motival, permitil, and others.SYN
Manufacturing Process
A suspension of 69.0 grams of 2-trifluoromethylphenothiazine in 1 liter of toluene with 10.9 grams of sodium amide is heated at reflux with high speed stirring for 15 minutes. A solution of 54.1 grams of 1-formyl-4-(3’chloropropyl)-piperazine, [prepared by formylating 1-(3′-hydroxypropyl)piperazine by refluxing in an excess of methyl formate, purifying the 1-formyl4-(3′-hydroxypropyl)-piperazine by vacuum distillation, reacting this compound with an excess of thionyl chloride at reflux and isolating the desired 1-formyl-4(3′-chloropropyl)-piperazine by neutralization with sodium carbonate solution followed by distillation] in 200 ml of toluene is added. The reflux period is continued for 4 hours. The cooled reaction mixture is treated with 200 ml of water. The organic layer is extracted twice with dilute hydrochloric acid. The acid extracts are made basic with ammonia and extracted with benzene. The volatiles are taken off in vacuo at the steam bath to leave a dark brown oil which is 10-[3′-(N-formylpiperazinyl)-propyl]-2trifluoromethylphenothiazine. It can be distilled at 260°C at 10 microns, or used directly without distillation if desired. A solution of 103.5 grams of 10-[3′-(N-formylpiperazinyl)-propyl]-2trifluoromethylphenothiazine in 400 ml of ethanol and 218 ml of water containing 26 ml of 40% sodium hydroxide solution is heated at reflux for 2 hours. The alcohol is taken off in vacuo on the steam bath. The residue is swirled with benzene and water. The dried benzene layer is evaporated in vacuo. The residue is vacuum distilled to give a viscous, yellow oil, 10(3’piperazinylpropyl)-2-trifluoromethylphenothiazine, distilling at 210° to235°C at 0.5 to 0.6 mm. A suspension of 14.0 grams of 10-(3′-piperazinylpropyl)-2trifluoromethylphenothiazine, 6.4 grams of β-bromoethyl acetate and 2.6 grams of potassium carbonate in 100 ml of toluene is stirred at reflux for 16 hours. Water (50 ml) is added to the cooled mixture. The organic layer is extracted into dilute hydrochloric acid. After neutralizing the extracts and taking the separated base up in benzene, a viscous, yellow residue is obtained by evaporating the organic solvent in vacuo. This oil is chromatographed on alumina. The purified fraction of 7.7 grams of 10-[3′-(Nacetoxyethylpiperazinyl)-propyl] -2-trifluoromethylphenothiazine is taken up in ethyl acetate and mixed with 25 ml of alcoholic hydrogen chloride. Concentration in vacuo separates white crystals of the dihydrochloride salt, MP 225° to 227°C. A solution of 1.0 gram of 10-[3′-(N-acetoxyethylpiperazinyl)-propyl]-2trifluoromethylphenothiazine in 25 ml of 1 N hydrochloric acid is heated at reflux briefly. Neutralization with dilute sodium carbonate solution and extraction with benzene gives the oily base, 10-[3′-(N-βhydroxyethylpiperazinyl)-propyl]-2-trifluoromethylphenothiazine. The base is reacted with an excess of an alcoholic hydrogen chloride solution. Trituration with ether separates crystals of the dihydrochloride salt, MP 224° to 226°C, (from US Patent 3,058,979).
Chemical Synthesis
Fluphenazine, 4-[3-[2-(trifluoromethyl)phenothiazin-10-yl]propyl]-1- piperazineethanol (6.1.8), is synthesized by any of the methods described above [21–27]. Alkylation of 2-trifluoromethylphenothiazine using 4-formyl-1-piperazineylpropylchloride in the presence of sodium amide synthesizes 2-trifluoromethyl-10-[3-(4-formyl- 1-piperazinyl)propyl]phenothizine (6.1.6). Further alkaline hydrolysis removes the N-formyl group, giving 2-trifluoromethyl-10-[3-(1-piperazinyl)propyl]phenothiazine (6.1.7). This is alkylated by 2-bromethanol-1 acetate, which upon further acidic hydrolysis removes the protecting acetyl group, yielding fluphenazine (6.1.8) [27,28].
Embodiment 1(1) preparation of 2-trifluoromethyl thiodiphenylamine: by 100g(0.356mol) Tecramine adds in reaction flask, be heated to 180-190 DEG C, open and stir, treat that it melts in backward reaction flask completely and add 10g(0.178mol) iron powder, stirring reaction about 2 hours at 180-190 DEG C of temperature, after reaction terminates, reaction solution is cooled to pour in beaker by reaction solution while hot when 100 DEG C, and iron powder stays (used water flushing) bottom reaction flask.Reaction solution is added underpressure distillation in clean reaction flask, collects 134-135 DEG C of (3mmHg) cut, obtain weak yellow liquid 3-trifluoromethyl pentanoic and be about 67.5g, yield about 80%.By 3-trifluoromethyl pentanoic 60g(0.253mol), sublimed sulphur 8g(0.253mol) add in reaction flask, whipped state is warming up to about 130 DEG C, after the complete melting of sulphur, in reaction flask, add 3g elemental iodine, continue to be warming up to 185-190 DEG C, react about 1 hour at this temperature.There is hydrogen sulfide to release in reaction process, note tail gas absorption.After reaction terminates, reaction solution is cooled to about 100 DEG C, adds 200g toluene in reaction flask, about raised temperature to 100 DEG C, in reaction flask, add 100g water, stir layering while hot after 5 minutes, water layer discarded, toluene layer returns reaction flask, and whipped state borehole cooling, to 15-18 DEG C, filters, filtrate retains (to be recycled apply mechanically 3-trifluoromethyl pentanoic), filter cake adopts 60 DEG C, vacuum to dry 10 hours, and obtain 29g intermediate 2-trifluoromethyl thiodiphenylamine, yield is about 85%(and calculates by sulphur)(2) preparation of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine: by 79g(0.5mol) 1,3-bromo-chloropropane, 320g toluene add in reaction flask, 130g(1.0mol is dripped under control 32-35 DEG C condition) 1-(2-hydroxyethyl) piperazine, time for adding about 2 hours.After dropwising, 32-35 DEG C of stirring reaction 10 hours, after reaction terminates, passes into hydrogen chloride gas to reaction system, regulate PH=8, solids removed by filtration, filtrate decompression distillation and concentration removing toluene solvant and unreacted complete 1,3-bromo-chloropropane, obtains viscous liquid product 95g, yield about 92%.(3) fluorine puts forth energy to be the preparation of nearly alkali: by 2-trifluoromethyl thiodiphenylamine 28g(0.105mol), toluene 140g, granular sodium hydroxide 28g(0.7mol) drop in reaction flask, whipped state is warming up to reflux state (110-112 DEG C), drip (the mixing solutions solution of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine and 50g toluene, the dropping process lasts about 1.5 hours of 26g (0.126mol) at reflux.After dropwising, reflux state reaction about 8 hours, whole reflux course notices that system moisture removes by timely water trap.After reaction terminates, be cooled to room temperature, solids removed by filtration insolubles, 150g purifying moisture three washing organic phases.Add the 10% concentration aqueous hydrochloric acid of 100g to organic phase, stir static layering after 10 minutes, discard upper toluene organic phase, retain lower floor’s aqueous phase, wash aqueous phase at twice with 150g toluene.In aqueous phase, add toluene 140g, drip the sodium hydroxide solution of 20% of 62g under whipped state, in process, hierarchy of control temperature is no more than 45 DEG C, after dropwising, stir 20 minutes, static layering, discard lower floor’s aqueous phase, retain upper organic phase, organic phase 15g anhydrous sodium sulfate drying, underpressure distillation removing toluene solvant, residue carries out underpressure distillation, collect 230 DEG C of (0.5mmHg) cuts, obtain 33g fluorine and put forth energy to be nearly alkali, yield 72%.(4) preparation of fluophenazine hydrochloride: 32g alkali is dissolved in 128g dehydrated alcohol, stirring is dissolved backward system completely and is led to hydrogen chloride gas, process temperature is no more than 20 DEG C, logical hydrogen chloride gas is stopped as PH=2, stir after 30 minutes and filter, filter cake 50g absolute ethanol washing, product puts into vacuum drying oven, dry after 10 hours for 45 DEG C and obtain fluophenazine hydrochloride 36g, yield about 95%.embodiment 2.(1) preparation of 2-trifluoromethyl thiodiphenylamine: by 500g(1.78mol) Tecramine adds in reaction flask, be heated to 180-190 DEG C, open and stir, treat that it melts in backward reaction flask completely and add 50g(0.89mol) iron powder, stirring reaction about 2 hours at 180-190 DEG C of temperature, after reaction terminates, reaction solution is cooled to pour in beaker by reaction solution while hot when 100 DEG C, and iron powder stays (used water flushing) bottom reaction flask.Reaction solution is added underpressure distillation in clean reaction flask, collects 134-135 DEG C of (3mmHg) cut, obtain weak yellow liquid 3-trifluoromethyl pentanoic and be about 346g, yield about 82%.By 3-trifluoromethyl pentanoic 300g(1.265mol), sublimed sulphur 40g(1.265mol) add in reaction flask, whipped state is warming up to about 130 DEG C, after the complete melting of sulphur, in reaction flask, add 15g elemental iodine, continue to be warming up to 185-190 DEG C, react about 1 hour at this temperature.There is hydrogen sulfide to release in reaction process, note tail gas absorption.After reaction terminates, reaction solution is cooled to about 100 DEG C, adds 1000g toluene in reaction flask, about raised temperature to 100 DEG C, in reaction flask, add 1000g water, stir layering while hot after 5 minutes, water layer discarded, toluene layer returns reaction flask, and whipped state borehole cooling, to 15-18 DEG C, filters, filtrate retains (to be recycled apply mechanically 3-trifluoromethyl pentanoic), filter cake adopts 60 DEG C, vacuum to dry 10 hours, and obtain 147g intermediate 2-trifluoromethyl thiodiphenylamine, yield is about 86%(and calculates by sulphur)(2) preparation of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine: by 395g(2.5mol) 1,3-bromo-chloropropane, 1600g toluene add in reaction flask, 650g(5.0mol is dripped under control 32-35 DEG C condition) 1-(2-hydroxyethyl) piperazine, time for adding about 2 hours.After dropwising, 32-35 DEG C of stirring reaction 10 hours, after reaction terminates, passes into hydrogen chloride gas to reaction system, regulate PH=8, solids removed by filtration, filtrate decompression distillation and concentration removing toluene solvant and unreacted complete 1,3-bromo-chloropropane, obtains viscous liquid product 470g, yield about 91%.(3) fluorine puts forth energy to be the preparation of nearly alkali: by 2-trifluoromethyl thiodiphenylamine 140g(0.525mol), toluene 700g, granular sodium hydroxide 140g(3.5mol) drop in reaction flask, whipped state is warming up to reflux state (110-112 DEG C), drip (the mixing solutions solution of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine and 300g toluene, the dropping process lasts about 1.5 hours of 130g (0.63mol) at reflux.After dropwising, reflux state reaction about 8 hours, whole reflux course notices that system moisture removes by timely water trap.After reaction terminates, be cooled to room temperature, solids removed by filtration insolubles, 750g purifying moisture three washing organic phases.Add the 10% concentration aqueous hydrochloric acid of 500g to organic phase, stir static layering after 10 minutes, discard upper toluene organic phase, retain lower floor’s aqueous phase, wash aqueous phase at twice with 750g toluene.In aqueous phase, add toluene 720g, drip the sodium hydroxide solution of 20% of 310g under whipped state, in process, hierarchy of control temperature is no more than 45 DEG C, after dropwising, stir 20 minutes, static layering, discard lower floor’s aqueous phase, retain upper organic phase, organic phase 75g anhydrous sodium sulfate drying, underpressure distillation removing toluene solvant, residue carries out underpressure distillation, collect 230 DEG C of (0.5mmHg) cuts, obtain 168g fluorine and put forth energy to be nearly alkali, yield 73%.(4) preparation of fluophenazine hydrochloride: 160g alkali is dissolved in 640g dehydrated alcohol, stirring is dissolved backward system completely and is led to hydrogen chloride gas, process temperature is no more than 20 DEG C, logical hydrogen chloride gas is stopped as PH=2, stir after 30 minutes and filter, filter cake 300g absolute ethanol washing, product puts into vacuum drying oven, dry after 10 hours for 45 DEG C and obtain fluophenazine hydrochloride 182g, yield about 96%.embodiment 3.(1) preparation of 2-trifluoromethyl thiodiphenylamine: by 1000g(3.56mol) Tecramine adds in reaction flask, be heated to 180-190 DEG C, open and stir, treat that it melts in backward reaction flask completely and add 100g(1.78mol) iron powder, stirring reaction about 2 hours at 180-190 DEG C of temperature, after reaction terminates, reaction solution is cooled to pour in beaker by reaction solution while hot when 100 DEG C, and iron powder stays (used water flushing) bottom reaction flask.Reaction solution is added underpressure distillation in clean reaction flask, collects 134-135 DEG C of (3mmHg) cut, obtain weak yellow liquid 3-trifluoromethyl pentanoic and be about 1029g, yield about 82%.By 3-trifluoromethyl pentanoic 600g(2.53mol), sublimed sulphur 80g(2.53mol) add in reaction flask, whipped state is warming up to about 130 DEG C, after the complete melting of sulphur, in reaction flask, add 30g elemental iodine, continue to be warming up to 185-190 DEG C, react about 1 hour at this temperature.There is hydrogen sulfide to release in reaction process, note tail gas absorption.After reaction terminates, reaction solution is cooled to about 100 DEG C, adds 2000g toluene in reaction flask, about raised temperature to 100 DEG C, in reaction flask, add 1000g water, stir layering while hot after 5 minutes, water layer discarded, toluene layer returns reaction flask, and whipped state borehole cooling, to 15-18 DEG C, filters, filtrate retains (to be recycled apply mechanically 3-trifluoromethyl pentanoic), filter cake adopts 60 DEG C, vacuum to dry 10 hours, and obtain 294g intermediate 2-trifluoromethyl thiodiphenylamine, yield is about 86%(and calculates by sulphur)(2) preparation of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine: by 790g(5mol) 1,3-bromo-chloropropane, 3200g toluene add in reaction flask, 1300g(10mol is dripped under control 32-35 DEG C condition) 1-(2-hydroxyethyl) piperazine, time for adding about 2 hours.After dropwising, 32-35 DEG C of stirring reaction 10 hours, after reaction terminates, passes into hydrogen chloride gas to reaction system, regulate PH=8, solids removed by filtration, filtrate decompression distillation and concentration removing toluene solvant and unreacted complete 1,3-bromo-chloropropane, obtains viscous liquid product 940g, yield about 91%.(3) fluorine puts forth energy to be the preparation of nearly alkali: by 2-trifluoromethyl thiodiphenylamine 280g(1.05mol), toluene 1400g, granular sodium hydroxide 280g(7mol) drop in reaction flask, whipped state is warming up to reflux state (110-112 DEG C), drip (the mixing solutions solution of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine and 500g toluene, the dropping process lasts about 1.5 hours of 260g (1.26mol) at reflux.After dropwising, reflux state reaction about 8 hours, whole reflux course notices that system moisture removes by timely water trap.After reaction terminates, be cooled to room temperature, solids removed by filtration insolubles, 1500g purifying moisture three washing organic phases.Add the 10% concentration aqueous hydrochloric acid of 1000g to organic phase, stir static layering after 10 minutes, discard upper toluene organic phase, retain lower floor’s aqueous phase, wash aqueous phase at twice with 1500g toluene.In aqueous phase, add toluene 1400g, drip the sodium hydroxide solution of 20% of 620g under whipped state, in process, hierarchy of control temperature is no more than 45 DEG C, after dropwising, stir 20 minutes, static layering, discard lower floor’s aqueous phase, retain upper organic phase, organic phase 150g anhydrous sodium sulfate drying, underpressure distillation removing toluene solvant, residue carries out underpressure distillation, collect 230 DEG C of (0.5mmHg) cuts, obtain 344g fluorine and put forth energy to be nearly alkali, yield 75%.(4) preparation of fluophenazine hydrochloride: 320g alkali is dissolved in 1280g dehydrated alcohol, stirring is dissolved backward system completely and is led to hydrogen chloride gas, process temperature is no more than 20 DEG C, logical hydrogen chloride gas is stopped as PH=2, stir after 30 minutes and filter, filter cake 500g absolute ethanol washing, product puts into vacuum drying oven, dry after 10 hours for 45 DEG C and obtain fluophenazine hydrochloride 364g, yield about 96%.
A 2018 Cochrane review found that fluphenazine was an imperfect treatment and other inexpensive drugs less associated with side effects may be an equally effective choice for people with schizophrenia.[9]
Side effects
Discontinuation
The British National Formulary recommends a gradual withdrawal when discontinuing antipsychotics to avoid acute withdrawal syndrome or rapid relapse.[10] Symptoms of withdrawal commonly include nausea, vomiting, and loss of appetite.[11] Other symptoms may include restlessness, increased sweating, and trouble sleeping.[11] Less commonly there may be a feeling of the world spinning, numbness, or muscle pains.[11] Symptoms generally resolve after a short period of time.[11]
There is tentative evidence that discontinuation of antipsychotics can result in psychosis.[12] It may also result in reoccurrence of the condition that is being treated.[13] Rarely tardive dyskinesia can occur when the medication is stopped.[11]
Fluphenazine acts primarily by blocking post-synaptic D2 receptors in the basal ganglia, cortical and limbic system. It also blocks alpha-1 adrenergic receptors, muscarinic-1 receptors, and histamine-1 receptors.[14][15]
Values are Ki (nM). The smaller the value, the more strongly the drug binds to the site. All data are for human cloned proteins, except 5-HT3 (rat), D4 (human/rat), H3 (guinea pig), and NMDA/PCP (rat).[16]
In horses, it is sometimes given by injection as an anxiety-relieving medication, though there are many negative common side effects and it is forbidden by many equestrian competition organizations.[27]
^ Tardy M, Huhn M, Engel RR, Leucht S (August 2014). “Fluphenazine versus low-potency first-generation antipsychotic drugs for schizophrenia”. The Cochrane Database of Systematic Reviews. 8 (8): CD009230. doi:10.1002/14651858.CD009230.pub2. PMID25087165.
^ Jump up to:abWorld Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^ Joint Formulary Committee, BMJ, ed. (March 2009). “4.2.1”. British National Formulary (57 ed.). United Kingdom: Royal Pharmaceutical Society of Great Britain. p. 192. ISBN978-0-85369-845-6. Withdrawal of antipsychotic drugs after long-term therapy should always be gradual and closely monitored to avoid the risk of acute withdrawal syndromes or rapid relapse.
^ Parent M, Toussaint C, Gilson H (1983). “Long-term treatment of chronic psychotics with bromperidol decanoate: clinical and pharmacokinetic evaluation”. Current Therapeutic Research. 34 (1): 1–6.
^ Jump up to:ab Jørgensen A, Overø KF (1980). “Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. III. Serum levels”. Acta Psychiatrica Scandinavica. Supplementum. 279: 41–54. doi:10.1111/j.1600-0447.1980.tb07082.x. PMID6931472.
^ Jump up to:ab Reynolds JE (1993). “Anxiolytic sedatives, hypnotics and neuroleptics.”. Martindale: The Extra Pharmacopoeia (30th ed.). London: Pharmaceutical Press. pp. 364–623.
^ Ereshefsky L, Saklad SR, Jann MW, Davis CM, Richards A, Seidel DR (May 1984). “Future of depot neuroleptic therapy: pharmacokinetic and pharmacodynamic approaches”. The Journal of Clinical Psychiatry. 45 (5 Pt 2): 50–9. PMID6143748.
^ Young D, Ereshefsky L, Saklad SR, Jann MW, Garcia N (1984). Explaining the pharmacokinetics of fluphenazine through computer simulations. (Abstract.). 19th Annual Midyear Clinical Meeting of the American Society of Hospital Pharmacists. Dallas, Texas.
^ Janssen PA, Niemegeers CJ, Schellekens KH, Lenaerts FM, Verbruggen FJ, van Nueten JM, et al. (November 1970). “The pharmacology of fluspirilene (R 6218), a potent, long-acting and injectable neuroleptic drug”. Arzneimittel-Forschung. 20 (11): 1689–98. PMID4992598.
^ Beresford R, Ward A (January 1987). “Haloperidol decanoate. A preliminary review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in psychosis”. Drugs. 33 (1): 31–49. doi:10.2165/00003495-198733010-00002. PMID3545764.
^ Reyntigens AJ, Heykants JJ, Woestenborghs RJ, Gelders YG, Aerts TJ (1982). “Pharmacokinetics of haloperidol decanoate. A 2-year follow-up”. International Pharmacopsychiatry. 17 (4): 238–46. doi:10.1159/000468580. PMID7185768.
^ Larsson M, Axelsson R, Forsman A (1984). “On the pharmacokinetics of perphenazine: a clinical study of perphenazine enanthate and decanoate”. Current Therapeutic Research. 36 (6): 1071–88.
A robust, green, and sustainable manufacturing process has been developed for the synthesis of gefapixant citrate, a P2X3 receptor antagonist that is under investigation for the treatment of refractory and unexplained chronic cough. The newly developed commercial process features low process mass intensity (PMI), short synthetic sequence, high overall yield, minimal environmental impact, and significantly reduced API costs. The key innovations are the implementation of a highly efficient two-step methoxyphenol synthesis, an innovative pyrimidine synthesis in flow, a simplified sulfonamide synthesis, and a novel salt metathesis approach to consistently deliver the correct active pharmaceutical ingredient (API) salt form in high purity.
SYN
Organic Process Research & Development (2020), 24(11), 2478-2490.
Gefapixant citrate (MK-7264) is a P2X3 antagonist for the treatment of chronic cough. The second generation manufacturing route developed for the Step 3A/3B formylation–cyclization reaction to generate the key intermediate diaminopyrimidine (1) (AF-072) required a significant excess of ethyl formate (EF), potassium tert-butoxide (KOt-Bu), and guanidine•HCl (G•HCl) when both steps were run as batch processes. It was imperative to develop an alternative process that required less of each reagent and generated less carbon monoxide byproducts, as the annual production of the final active pharmaceutical ingredient (API) is expected to be over 50 MT. In addition, the second generation process was misaligned with our company’s strategy of having the best science in place at the first regulatory filing. The final flow–batch process described herein, which features a flow-based formylation combined with a batch cyclization, has been performed on a 500 kg scale and now requires 35% less EF (leading to a 70% reduction in waste carbon monoxide), 38% less KOt-Bu, and 50% less G•HCl. These improvements, along with a twofold increase in concentration, have resulted in a 54% reduction in the step process mass intensity (step-PMI) from the second generation two-step batch–batch process (PMI of 17.16) to the flow–batch process (PMI of 7.86), without sacrificing reaction performance.
SYN
H. REN*, K. M. MALONEY* ET AL. (MERCK & CO., INC., RAHWAY USA) Development of a Green and Sustainable Manufacturing Process for Gefapixant Citrate (MK-7264) Part 1: Introduction and Process Overview Org. Process Res. Dev. 2020, 24, 2445–2452, DOI: 10.1021/acs.oprd.0c00248.
A scalable two-pot sulfonamidation through the process has been developed for the synthesis of gefapixant citrate, a P2X3 receptor antagonist that is under investigation for the treatment of refractory and unexplained chronic cough. Direct conversion of the diaryl ether precursor to a sulfonyl chloride intermediate using chlorosulfonic acid, followed by treatment with aqueous ammonia hydroxide, provided the desired sulfonamide in high yield. A pH-swing crystallization allowed for the formation of a transient acetonitrile solvate that enables the rejection of two impurities. After drying, the desired anhydrous free base form was isolated in high yield and purity.
Gefapixant is the approved generic name for a compound also known as MK-7264, and prior to that AF-219 and RO-4926219. It is the first-in-class clinically developed antagonist for the P2X3 subtype of trimeric ionotropic purinergic receptors, a family of ATP-gated excitatory ion channels, showing nanomolar potency for the human P2X3 homotrimeric channel and essentially no activity at related channels devoid of P2X3 subunits. As the first P2X3 antagonist to have progressed into clinical studies it has now progressed to the point of successful completion of Phase 3 investigations for the treatment of cough, and the NDA application is under review with US FDA for treatment of refractory chronic cough or unexplained chronic cough. The molecule was discovered in the laboratories of Roche Pharmaceuticals in Palo Alto, California, but clinical development then continued with the formation of Afferent Pharmaceuticals for the purpose of identifying the optimal therapeutic indication for this novel mechanism and establishing a clinical plan for development in the optimal patient populations selected. Geoff Burnstock was a close collaborator and advisor to the P2X3 program for close to two decades of discovery and development. Progression of gefapixant through later stage clinical studies has been conducted by the research laboratories of Merck & Co., Inc., Kenilworth, NJ, USA (MRL; following acquisition of Afferent in 2016), who may commercialize the product once authorization has been granted by regulatory authorities.
SCHEME AExample 1: 5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonamideThe synthetic procedure used in this Example is outlined in Scheme B.
not isolated
SCHEME BStep 1 2-Isopropyl-4-methoxy-phenolTo a cooled solution of l-(2-hydroxy-5-methoxy-phenyl)-ethanone (10.0 kg) in 79.0 kg of THF was gradually added 46.4 kg of 3M solution of MeMgCl in THF at a rate such that the reaction mixture temperature did not exceed 25°C. Following addition of the MeMgCl solution, the reaction mixture was stirred at ambient temperature for 18 hours, at which point HPLC (high pressure liquid chromatography) analysis showed more than 98% conversion of l-(2-hydroxy-5-methoxy-phenyl)-ethanone to 2- (1 -hydroxy- 1- methyl-ethyl)-4-methoxy-phenol (not shown in Scheme D). To the stirred solution was then added 10% palladium on carbon (1.02 kg, 50% water wet) suspended in 3.5 kg of THF. The reaction mixture was cooled and placed under a hydrogen atmosphere at 0.34 atmosphere pressure, and concentrated HCl (19.5 kg) was added while maintaining the reaction temperature at 25°C. The resultant mixture was stirred at ambient temperature for 18 hours, then treated with 44.4 kg water and filtered through a bed of Celite to remove suspended catalyst. The filter cake was rinsed with EtOAc and the combined filtrate was separated. The organic phase was washed with water, then concentrated by distillation to provide an oil. This oil was dissolved in 2-butanone (20.4 kg) and the crude solution was employed directly in the next step. A 161.8 g aliquot of the solution was concentrated under vacuum to provide 49.5 g of 2-isopropyl-4-methoxyphenol as an oil, projecting to 10.4 kg crude contained product in the bulk 2-butanone solution. 1H NMR (DMSO) delta: 1.14 (d, 6H, J = 6.9 Hz), 3.18 (septet, IH, J = 6.9 Hz), 3.65 (s, 3H), 6.56, (dd, IH, J = 8.6 Hz, 3.1 Hz), 6.67 (d, IH, J = 3.1 Hz), 6.69 (d, IH, 8.6 Hz).Step 2 (2-Isopropyl-4-methoxy-phenoxy)-acetonitrileA stirred slurry of toluene-4-sulfonic acid cyanomethyl ester (13.0 kg), potassium carbonate (13.0 kg) and 2-isopropyl-4-methoxyphenol (9.57 kg) in 79.7 kg of 2-butanone was heated to 55-600C for 4 days, then heated to reflux for 18 hours. The resultant slurry was cooled and filtered to remove solids. The filtrate was concentrated under reduced pressure and the residue was redissolved in toluene. The toluene solution was extracted with IN KOH, and the organic phase was concentrated by distillation to give 20.6 g of a 1:1 (by weight) solution of (2-isopropyl-4-methoxy-phenoxy)-acetonitrile in toluene, which was used directly in the next step. A aliquot (96.7 g) of this solution was concentrated to dryness to give 50.9 g of crude (2-isopropyl-4-methoxy-phenoxy)- acetonitrile, projecting to a yield of 10.9 kg in the bulk solution: MS (M+H) = 206; 1H NMR (CDCl3) delta: 1.25 (d, J = 6.9 Hz), 3.31 (septet, IH, J = 6.9 Hz), 3.82 (s, 3H), 4.76 (s, 2H), 6.73 (dd. IH, J = 8.8 Hz, 3.1 Hz), 6.87 (d, IH, J = 3.1 Hz), 6.91 (d, IH, J = 8.8 Hz).Step 3 5-(2-Isopropyl-4-methoxy-phenoxy)-pyrimidine-2,4-diamine An approximately 1:1 (by weight) solution of 10.6 kg of (2-isopropyl-4-methoxy-phen- oxy) -acetonitrile in toluene was concentrated under reduced pressure and the residue was treated with 10.8 kg of tert-butoxybis(dimethylamino)methane (Brederick’s Reagent). The resulting mixture was dissolved in 20.2 kg of DMF and the solution was heated to 1100C for 2 hours, at which point HPLC analysis showed essentially complete conversion to 3,3-bis-dimethylamino-2-(2-isopropyl-4-methoxy-phenoxy)-propionitrile (not isolated, 1H NMR (CDCl3) delta: 1.21 (d, 3H, J = 7.2 Hz), 1.23 (d, 3H, J = 7.1 Hz), 2.46 (s, 6H), 2.48 (s, 6H), 3.43 (d, IH, J = 5.0 Hz), 3.31 (septet, IH, J = 6.9 Hz), 3.79 (s, 3H), 4.93 (d, IH, J = 5.0 Hz), 6.70 (dd, IH, J = 8.8 Hz, 3.0 Hz), 6.82 (d, IH, J = 3.0 Hz), 6.98 (d, IH, J = 8.8 Hz). The DMF solution was cooled and transferred onto 14.7 kg of aniline hydrochloride. The resulting mixture was heated to 1200C for 22 hours, at which point HPLC analysis showed greater than 97% conversion to 2-(2-isopropyl-4-methoxy-phenoxy)-3- phenylamino-acrylonitrile (not isolated, 1H nmr (CDCl3) delta: 1.31 (d, 6H, J = 6.9 Hz), 3.39 (septet, IH, J = 6.9 Hz), 3.82 (s, 3H), 6.61 (d (br), IH, J = 12.7 Hz), 6.73 (dd, IH, J = 8.9 Hz, 3.1 Hz), 6.88 (d, IH, J = 3.0 Hz), 6.93 (m, 2H), 6.97 (d, IH, J = 8.9 Hz), 7.05 (m, IH), 7.17 (d, IH, J = 12.6 Hz), 7.35 (m. 2H)).The mixture was cooled, diluted with 21.5 kg toluene, then with 72.2 L of water. The organic layer was separated, washed with water, and concentrated by distillation. The concentrate was transferred into 23.8 kg DMF, and the DMF solution was transferred onto 6.01 kg of guanidine carbonate. The resulting mixture was heated to 1200C for 3 days, at which point HPLC analysis showed greater than 95% conversion of 2-(2- isopropyl-4-methoxy-phenoxy)-3-phenylamino-acrylonitrile into 5-(2-Isopropyl-4- methoxy-phenoxy)-pyrimidine-2,4-diamine. The reaction mixture was cooled, diluted with 7.8 kg of EtOAc, then reheated to 600C. Water (75.1 L) was added and the resultant mixture was allowed to cool to ambient temperature. The precipitated solid was collected by filtration, rinsed with isopropanol and dried under vacuum at 50 degrees to give 9.62 kg of 5-(2-isopropyl-4-methoxy- phenoxy)-pyrimidine-2,4-diamine: m.p. 170-171 degrees C; MS (M+H) = 275; H nmr (chloroform) delta: 1.25 (d, 6H, J = 6.9 Hz), 3.30 (septet, IH, J = 6.9 Hz), 3.79 (s, 3H), 4.68 (br, 2H), 4.96 (br, 2H), 6.64 (dd, IH, J = 8.9 Hz, 3.0 Hz), 6.73, d, J = 8.9 Hz), 6.85 (d, IH, J = 3 Hz), 7.47 (s, IH).Step 4 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfon- amide, sulfolane solvate Chlorosulfonic acid (13.82 kg) was added to a slurry of 5-(2-isopropyl-4-methoxy-phen- oxy)-pyrimidine-2,4-diamine (10.07 kg) in sulfolane (50.0 kg) at a rate to maintain an internal pot temperature below 65°C. The reaction mixture was aged at 60-650C for 12 hours, at which point HPCL showed that all 5-(2-isopropyl-4-methoxy-phenoxy)- pyrimidine-2,4-diamine starting material had been converted to 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonic acid. MS (M+H) = 355. Phosphorus oxychloride (3.41 kg) was then added to the reaction mixture at 600C. The reaction mixture was heated to 75°C and aged for 12 hours, at which point HPLC showed that approximately 99% of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonic acid had been converted to 5-(2,4-diamino-pyrimidin-5-yloxy)-4-iso- propyl-2-methoxy-benzenesulfonyl chloride. MS (M+H) = 373. The solution of 5-(2,4- diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride was then cooled to around 2°C).To a cooled (ca. 2°C) solution of ammonia (7N) in MeOH (74.1 kg) was added the cooled sulfolane solution of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonyl chloride (a homogeneous syrup) at a rate such that the internal temperature did not exceed 23°C. The resultant slurry was stirred for 18 hours at ambient temperature, then filtered on a coarse porosity frit filter. The collected solids were rinsed with MeOH (15.9 kg), then dried under reduced pressure at 700C to a constant weight of 23.90 kg. HPLC showed 97.5% conversion of 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride to 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide sulfolane solvate. H nmr (DMSOd6) delta: 1.26 (d, 6H, J = 6.9 Hz), 2.07 (sym. m, 8H), 2.99 (sym. m, 8H), 3.41 (septet, IH, J = 6.9 Hz), 3.89 (s, 3H), 6.03 (s (br), 2H), 6.58 (s (br), 2H), 7.00 (s, IH), 7.04 (s (br), 2H), 7.08 (s, IH), 7.35 (s, IH). Step 5 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzene- sulfonamideA slurry of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfon- amide sulfolane solvate (23.86 kg) in a mixture of ethanol (74.3 kg) and 0.44 N HCl (109.4 kg) was heated to reflux to provide a homogeneous solution of the monohydrochloride salt of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonamide. This solution was filterd while hot, then treated with concentrated ammonium hydroxide (3.4 L) to liberate the free base of 5-(2,4-diamino-pyrimidin-5- yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide. The resultant mixture was cooled slowly to 200C and the crystalline product isolated by filtration. The filter cake was washed with water (20.1 kg) and dried under reduced pressure at 700C to a constant weight of 8.17 kg (57.7% yield based on di-solvate of sulfolane).MP = 281-282 0C.1H nmr (DMSOd6) delta: 1.27 (d, 6H, J = 6.9 Hz), 3.41 (septet, IH, J = 6.9 Hz), 3.89 (s, 3H), 5.87 (s (br), 2H), 6.40 (s (br), 2H), 6.98 (s, IH), 7.01 (s (br), 2H), 7.07 (s, IH), 7.36 (s, IH). PATENT US 20080207655https://patents.google.com/patent/US20080207655 PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016004358
[211] A mixture of pyrimidine (0.400 g, 1.5 mmol) in 2 ml chlorosulfonic acid was allowed to stir 20 min. The mixture was poured over ice. The precipitate was filtered, washed by cold H2O and dried under vacuum to afford 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride (0.515 g, 95%) as a white solid; [MH]+= 373.
PATENTDisclosed herein is a novel process for preparing Compound A, a phenoxy diaminopyrimidine compound of the following formula, or a pharmaceutically acceptable salt thereof:
Compound A.Also disclosed herein are various salts and solvates of Compound A.
Scheme 1
Step 1. Preparation of 4-Bromo-2-isopropylphenol DABCO Co-crystalStep 1. Preparation of 4-Bromo-2-isopropylphenol DABCO Co-crystalThe following 4-bromo-2-isopropylphenol hemi-DABCO co-crystal is obtained in greater than 99% purity and at about 85-92% yield by the following process:
To a solution of 2-isopropyl phenol (75.0 g, 550 mmol) in acetonitrile (225 mL) was added MSA (0.520 g, 5.41 mmol). The mixture was cooled to -10 °C and NBS (98.01 g, 550 mmol) was added in portions while maintaining the internal temperature below 10 °C. The reaction was aged for 30 min to 1 h and then warmed to 20 °C, diluted with water (450 mL), and extracted with toluene (225 mL). The organic layer was sequentially washed with 9 wt% phosphoric acid (150 mL) and 5 wt% NaCl (150 mL). The organic layers were concentrated to roughly 150 mL and filtered into a clean reactor. The mixture was heated to 30-40 °C and n- heptane (28.5 mL) was added followed by DABCO (30.89 g, 275 mmol). The mixture was seeded (a seed can be synthesized from a previous batch of this procedure preformed without seeding) with 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (75 mg, 0.277 mmol), diluted with 52.5 mL of n-heptane, and stirred for 1 h. The slurry was cooled to 20 °C over 1 h and 370 mL of n-heptane is added over 2 h. The slurry was cooled to 5 °C over 2 h, aged for 2 h, filtered, and washed with n-heptane (2 x 75 mL). The solid was dried at 20-25 °C under vacuum to yield 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (134.8 g, 90 %) as a solid. 1H NMR (400 MHz, DMSO-76) d 7.20 (d, J= 2.5 Hz, 1H), 7.13 (dd, J= 8.5, 2.6 Hz, 2H), 6.73 (d, J = 8.5 Hz, 2H), 3.16 (hept, J= 6.9 Hz, 2H), 2.60 (s, 12H), 1.14 (d, J= 6.9 Hz, 12H).The crystallization of step 1 generates 4-bromo-2-isopropylphenol hemi-DABCO co-crystal, bromophenol mono-DABCO co-crystal, or a mixture of bromophenol hemi-DABCO co-crystal and bromophenol mono-DABCO co-crystal. An XRPD pattern of bromophenol hemi- DABCO co-crystal is shown in Figure 1.
The bromo-phenol mono-DABCO co-crystal can be generated in the following procedure:
bromophenol DABCO co-crystalTo a vial with a stir bar was charged DABCO (1.7 g, 15 mmol), phenol (2.5 g, 15 mmol), and 2 mL of n-heptane. The resulting slurry was stirred at 23 °C overnight. The slurry was then filtered and the resulting wet cake was washed with 2 mL of 5 °C n-heptane. The cake was dried under vacuum with nitrogen sweep to afford 4-bromo-2-isopropylphenol mono- DABCO co-crystal (2.9 g, 70% yield) as a solid. 1H NMR (500 MHz, DMSO-76) d 9.65 (s, 1H), 7.20 (s, 1H), 7.14 (d, J= 8.5 Hz, 1H), 6.74 (d, J= 8.5 Hz, 1H), 3.17 (hept, J= 6.8 Hz, 1H), 2.61(s, 12H), 1.15 (d, 7 = 6.9 Hz, 6H).An XRPD pattern of bromophenol mono-DABCO co-crystal is shown in Figure 2.Step 2a. Preparation of 2-Isopropyl-4-Methoxyphenol
The 2-isopropyl-4-Methoxyphenol shown below is obtained at about 92% yield by the following process:
bromophenol DABCO co-crystal methoxy phenolTo a solution of 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (120 g, 442 mmol) in 25 wt% sodium methoxide in methanol (430 g) was added 60 mL of DMF. The solution was pressure purged with nitrogen, copper (I) bromide (3.23 g, 22.5 mmol) was added to the mixture, and the reaction was heated to reflux for 12-16 h. The reaction is cooled to 0-5 °C and quenched with 6M HC1 until the pH of the solution is less than 5. The slurry is diluted with 492 mL of toluene and 720 mL of water to provide a homogeneous solution with a rag between the layers. The aqueous layer is cut to waste. The organic layer is filtered to remove the rag and washed with 240 mL of water to provide 2-isopropyl-4-methoxylphenol (491 g, 13.3 wt%, 89% assay yield) as a solution in toluene. 1H NMR (500 MHz, DMSO-76) d 8.73 (s, 1H), 6.68 (d, J = 8.6 Hz, 1H), 6.66 (d, 7= 3.0 Hz, 1H), 6.55 (dd, 7= 8.6, 3.1 Hz, 1H), 3.65 (s, 3H), 3.17 (hept, j = 6.9 Hz, 1H), 1.14 (d, 7= 6.9 Hz, 6H).Step 2b. Preparation of 2-Isopropyl-4-Methoxyphenol
Alternatively, the methoxy phenol is obtained by the following process:
To a high-pressure vessel were charged 400 mL of anhydrous toluene, Re2(CO)io (3.16 g, 4.84 mmol) and mequinol (100 g, 806 mmol) at RT. The vessel was then degassed with propylene, and charged with propylene (85.0 g, 2.02 mol). The vessel was sealed and heated to 170 °C. Internal pressure was measured near 250 psi. The reaction was stirred at this condition for 72 h. The vessel was then allowed to cool down to 23 °C. The internal pressure was carefully released to 1 atmospheric pressure, and the toluene solution was assayed as 91% and used directly in the next step or isolated as a solid.Step 2a/2b results in anhydrous 2-isopropyl-4-methoxyphenol form 1. An XRPD pattern of the methoxy phenol form 1 is shown in Figure 3.In another embodiment, the product is isolated as a DMAP co-crystal:
To a vial with a stir bar was charged DMAP (3.67 g, 30.1 mmol), 2.5 ml of toluene, and 2-isopropyl-4-methoxylphenol (5.00 g, 30.1 mmol). The reaction mixture was stirred at RT for 5 min, and a homogeneous solution was formed. The reaction mixture was then cooled to 5 °C. Ten mL of n-heptane was slowly charged over 20 min. The resulting slurry was stirred at 5 °C overnight. The slurry was filtered and the resulting wet cake was washed with 3 mL of 5 °C n-heptane. The cake was dried under vacuum with a nitrogen sweep to provide 2- isopropyl-4-methoxylphenol DMAP co-crystal (7.01 g, 81%) as a solid. 1H NMR (500 MHz, DMSO-76) d 8.78 (s, 1H), 8.10 (d, J= 6.1 Hz, 2H), 6.71 – 6.65 (m, 2H), 6.57 (dd, J= 11.3, 6.0 Hz, 3H), 3.66 (s, 3H), 3.17 (hept, J= 6.8 Hz, 1H), 2.95 (s, 6H), 1.14 (d, J= 6.9 Hz, 6H).The crystallization generates anhydrous 2-isopropyl -4-methoxyphenol DMAP co crystal. An XRPD pattern of the 2-isopropyl-4-methoxyphenol DMAP co-crystal is shown in Figure 4.Step 3a. Preparation of the Cvanoether. 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile
The cyanoether is obtained at about 95 % yield by the following process:
A 12-15 wt% solution of 2-isopropyl-4-methoxylphenol (314.3 g, 12 wt%, 226.8 mmol) was concentrated to greater than 50 wt% 2-isopropyl-4-methoxyphenol in toluene under vacuum at 40-50°C. To the solution was added 189 mL of NMP, and the mixture was cooled to 5 °C. Sodium hydroxide (27.2 g, 50 wt% in water, 340 mmol) and chloroacetonitrile (36 g, 340 mmol) were added sequentially to the mixture while maintaining the internal temperature below 10 °C. The reaction was aged for 2 h and then diluted with 150 mL of toluene and 226 mL of water while maintaining the temperature below 10 °C. The mixture was warmed to 20-25 °C, the layers were separated, and the organic layer was washed with 75 mL of 20 wt% NaCl (aq.). The organic layer was and filtered to provide 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile (56.8 g, 74.6 wt%) as a solution in toluene. The filter was washed with NMP to provide additional 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile (27.1 g, 5.0 wt%) as a solution in NMP. The combined yield was about 94 %. 1H NMR (500 MHz, DMSO-i¾) d 7.05 (d, J= 8.8 Hz, 1H), 6.81 (d, 7= 3.0 Hz, 1H), 6.78 (dd, j= 8.8, 3.1 Hz, 1H), 5.11 (s, 2H), 3.73 (s, 3H), 3.20 (hept, j = 6.9 Hz, 1H), 1.17 (d, 7= 6.9 Hz, 6H).Step 3b. Preparation of the Cvanoether. 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile
Alternatively, the cyanoether shown below is obtained at about 92% yield by the following process:
A solution of 2-isopropyl-4-methoxyphenol in toluene (491 g, 13.3 wt%, 393 mmol) was concentrated and solvent switched to acetonitrile under vacuum at 40-50 °C.Potassium carbonate (164.5 g, 1190 mmol) and tetrabutylammonium hydrogensulfate (1.5 g, 4.42 mmol) were added to a separate vessel, and the vessel was pressure purged with nitrogen gas.The solution of phenol in acetonitrile and chloroacetonitrile was added sequentially to the reaction vessel. The vessel was heated to 40 °C and aged for 4 h. The mixture was allowed to cool to 25 °C, and was diluted with 326 mL water. The layers were separated, and the organic layer was washed with 130 mL of 10 wt% NaCl. A solvent switch to toluene was performed under vacuum, and the organic layer was filtered through two 16D Cuno #5 cartridges. The organic layer was concentrated to provide 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile in toluene (128.2 g, 58 wt%, 92% yield).Step 4 Preparation of the Dia inopyrimidine 5-(2-isopropyl-4-methoxyphenoxy)pyrimidine-2.4-di amineThe diaminopyrimidine is obtained at about 90 % yield by the following process:
A solution of potassium tert-butoxide (44.8 g, 0399 mmol) in NMP (180 mL) was cooled to -10 °C. A solution of 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile, the cyanoether, (59.3 g, 61.4 wt%, 177 mmol) in toluene and ethyl formate (26.3 g, 355 mmol) was charged to the base solution while maintaining the internal temperature between -12 °C and -8 °C. After a 3 h age, guanidine hydrochloride (136 g, 1420 mmol) was added to the mixture and the reaction was heated to 115 °C for 6 h. The mixture was allowed to cool to 90 °C, diluted with 200 mL of water, and aged until the reaction mixture was homogeneous (about 30-45 min). After all solids dissolved, vacuum (400 mm Hg) was applied to the reactor to remove toluene. Vacuum was disconnected and the solution was allowed to cool to 85°C. 5-(2-Isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine seed (49.8 mg) (a seed can be synthesized by a route described in U.S. Patent 7,741,484) was charged, the solution was aged for 2 h, 200 mL of water was added, and the batch was allowed to cool to 20 °C over 6 h. The slurry was aged for 10 h at 20 °C, filtered, washed with 2: 1 water :NMP (3 x 100 mL) and water (3 x 100 mL), and dried under vacuum at 50 °C to provide the title compound (42.2 g, 88%) as a solid. 1H NMR (500 MHz, DMSO-r¾) d 7.23 (s, 1H), 6.83 (d, J= 3.0 Hz, 1H), 6.70 (dd, J= 8.9, 3.0 Hz, 1H), 6.63 (d, j= 8.8 Hz, 1H), 6.32 (s, 2H), 5.75 (s, 2H), 3.71 (s, 3H), 3.28 (hept, j= 6.9 Hz, 1H), 1.20 (d, j = 6.9 Hz, 6H); 13C NMR (126 MHz, DMSO-r¾) d 159.7, 157.2, 155.1, 148.4, 144.2, 139.0, 130.4,116.9, 112.5, 111.3, 55.4, 26.57, 22.83.The crystallization of step 4 generates an anhydrous 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1. An XRPD pattern of the 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1 is shown in Figure 5.In one embodiment, 5-(2-isopropyl-4-methoxyphenoxy)pyrimidine-2, 4-diamineNMP solvate 1 is obtained by adding excess amount of 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1 into NMP in a closed vessel to form a suspension. The suspension is stirred at RT until the completion of form transition. The crystals of 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine NMP solvate 1 can be collected by filtration and measured immediately by XRPD to prevent desolvation. An XRPD pattern of the 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine NMP solvate 1 is shown in Figure 6.Step 5. Preparation of Compound A Free BaseCompound A free base is obtained at about 91% yield by a process comprising the steps:
To a suspension of 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine, the diaminopyrimidine, (47.0 g, 171 mmol) in 141 mL of acetonitrile at -10 °C was added chlorosulfonic acid (63.1 mL, 942 mmol) while maintaining the internal temperature below 25 °C. The solution was aged for 1 h at 25 °C and then heated to 45 °C for 12 h. The solution was allowed to cool to 20 °C and added to a solution of 235 mL ammonium hydroxide and 71 mL of acetonitrile at -10 °C while maintaining the internal temperature below 15 °C. The slurry was aged at l0°C for 1 h, heated to 25 °C, and aged for 1 h. The slurry was diluted with 564 mL of water and 188 mL of 50 wt% sodium hydroxide to provide a homogeneous solution that was heated to 35 °C for 2 h. The solution was allowed to cool to 22 °C and the pH of the solution was adjusted to 12.9 with a 2M solution of citric acid. The solution was seeded with Compound A free base (470 mg, 1.19 mmol) (a seed can be synthesized by a route described in U.S. Patent 7,741,484), aged for 2 h, acidified to pH 10.5-11.3 with a 2M solution of citric acid over 5-10 h, and then aged for 2 h. The slurry was filtered, the resulting cake was washed with 90: 10 water: acetonitrile (2 x 118 mL) and water (2 x 235 mL), and dried at 55 °C under vacuum to provide Compound A free base (50.9 g, 91%) as a solid. 1H NMR (500 MHz, DMSO-i¾) d 7.36 (s, 1H), 7.07 (s, 1H), 7.05 – 6.89 (m, 3H), 6.37 (s, 2H), 5.85 (s, 2H), 3.89 (s, 3H), 3.41 (hept, J = 6.6 Hz, 1H), 1.27 (d, J= 6.8 Hz, 6H).The crystallization of step 5 generates anhydrous Compound A free base form 1. In one embodiment, Compound A free base acetonitrile solvate 1 can be prepared by adding excess amount of Compound A free base form 1 into acetonitrile in a closed vessel to form a suspension. The suspension is stirred at 50 °C until the completion of form transition.The crystals of Compound A free base acetonitrile solvate 1 can be collected by filtration and measured immediately by XRPD to prevent desolvation. An XRPD pattern of Compound A free base acetonitrile solvate 1 is shown in Figure 7.Step 6a. Preparation of Compound A Citrate SaltCompound A citrate salt is obtained by a process comprising the steps:
Compound A free base (30.0 g, 84.9 mmol) and glycolic acid (22.6 g, 297 mmol) were added to methanol (360 mL). The solution was heated to 60 °C, aged for 1 h, and filtered through a 0.6 pm filter into a clean vessel. A solution of citric acid (32.6 g, 170 mmol) in 2- propanol (180 mL) at RT was filtered through a 0.6 pm filter into the methanol solution over 30 min while the temperature of the methanol solution was maintained between 58-62 °C. The solution was seeded with Compound A citrate salt (450 mg, 0.825 mmol) (a seed can be synthesized by a route described in patent application number PCT/US17/66562), aged for 1 h, and diluted with 180 mL of 2-propanol over 3 h while the temperature was maintained between 58-62 °C. The slurry was cooled to 50 °C over 3 h. The slurry was filtered at 50 °C, washed with 1 : 1 methanol :2-propanol (120 mL) and 2-propanol (120 mL) at 50 °C, and dried under vacuum at 35 °C to provide Compound A citrate salt (45.1 g, 97%) as a solid. 1H NMR (400 MHz, DMSO-76) d 10.89 (s, 3H), 7.33 (s, 1H), 7.10 (s, 1H), 7.07 (s, 3H), 7.04 (s, 2H), 6.44 (s, 2H), 3.91 (s, 3H), 3.34 (hept, J= 6.7 Hz, 1H), 2.69 (d, 7= 15.3 Hz, 2H), 2.60 (d, 7= 15.3 Hz, 2H), 1.26 (d, 7= 6.9 Hz, 6H). Step 6b. Alternative preparation of Compound A Citrate SaltAlternatively, Compound A citrate salt is obtained by a process comprising the steps:
To a suspension of Compound A citrate salt (4.5 g, 8.25 mmol) in methanol (72 mL) and 2-propanol (36 mL) at 50 °C were added simultaneously through separate 0.6 pm filters a solution of Compound A free base (30.0 g, 84.9 mmol) and glycolic acid (22.6 g, 297 mmol) in 360 mL of methanol at 50 °C and a solution of citric acid (19.5 g, 101 mmol) in 180 mL of 2- propanol at 25 °C over 8 h while maintaining the seed solution temperature of 60 °C. After the simultaneous addition is complete, citric acid (13.2 g, 68.7 mmol) in 180 mL of 2-propanol was added to the slurry over 8 h while the temperature was maintained at 60 °C. The slurry was allowed to cool to 50 °C and aged for 1 h, filtered at 50 °C, washed with 1 : 1 methanol :2- propanol (2 x 120 mL) and 2-propanol (120 mL), and dried under vacuum at 35 °C to provide Compound A citrate salt (45.1 g, 88%) as a solid.The crystallization of step 6a/6b generates anhydrous Compound A citrate form 1. In another embodiment, Compound A citrate methanol solvate 1 can be prepared via a saturated solution of Compound A citrate form 1 in methanol at 50C. The solution is naturally cooled to ambient temperature or evaporated at ambient temperature until the crystals of Compound A citrate methanol solvate 1 can be acquired. An XRPD pattern of Compound A citrate methanol solvate 1 is shown in Figure 8. PATENT https://patents.google.com/patent/CN111635368B/enPreparation of the Compound Gefapixant of example 11Adding compound 7(16g) and dichloromethane (64mL) into a 250mL three-necked bottle, stirring for dissolving, cooling to below 5 ℃ in an ice bath, dropwise adding a mixed solution of chlorosulfonic acid (21.1g) and dichloromethane (16mL) into the reaction solution, and stirring for 1 hour at the temperature of not higher than 5 ℃; then heating to room temperature and continuing stirring for 10 hours, after the reaction is finished, pouring the reaction liquid into ice water, and quickly separating a water layer; the organic layer was washed once with ice water, dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give a crude product. Dissolving the crude product with 30ml of acetonitrile, and cooling to below 5 ℃; 16ml of ammonia water (25-28%) is dripped into the solution, and after the dripping is finished, the solution is heated to room temperature and stirred for 20 hours. After the reaction is completed, concentrating the reaction solution under reduced pressure to remove acetonitrile, and separating out a white solid; and filtering again, and drying the filter cake at 70 ℃ under reduced pressure for 24h to obtain Gefapixant: white powder (19.50g), yield 94.6%, purity: 97.2 percent.Example 12 purification of the Compound GefapixantAdding a compound Gefapixant (20.77g) into a 500mL reaction bottle, adding 0.44N hydrochloric acid (95.4mL), absolute ethyl alcohol (64.4g) and nitrogen protection, heating to 75 ℃, stirring for dissolving, then carrying out heat preservation and reflux for 1 hour, filtering while hot, after filtering, heating the filtrate again to 60 ℃, dropwise adding ammonia water (25-28 percent and 2.96mL), closing and heating after dropwise adding, slowly cooling to room temperature, and gradually precipitating white solids. And continuously cooling the reaction solution to 20 ℃, keeping the temperature and stirring for 4h, filtering, washing a filter cake with 15ml of water, and performing vacuum drying on the obtained wet product at 60 ℃ for 24h to obtain Gefapixant: white powder (6.58g), yield 53.2%, purity: 99.5 percent.1H NMR(400MHz,DMSO)δ7.37(s,1H),7.08(s,1H),7.02(s,2H),7.00(s,1H),6.43(brs,2H),5.89(s,2H),3.90(s,3H),3.42(m,1H),1.28(d,J=8.0Hz,6H);LC-MS:m/z=354.1[M+H]+。
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
^ Muccino D, Green S (June 2019). “Update on the clinical development of gefapixant, a P2X3 receptor antagonist for the treatment of refractory chronic cough”. Pulmonary Pharmacology & Therapeutics. 56: 75–78. doi:10.1016/j.pupt.2019.03.006. PMID30880151.
^ Marucci G, Dal Ben D, Buccioni M, Martí Navia A, Spinaci A, Volpini R, Lambertucci C (December 2019). “Update on novel purinergic P2X3 and P2X2/3 receptor antagonists and their potential therapeutic applications”. Expert Opinion on Therapeutic Patents. 29 (12): 943–963. doi:10.1080/13543776.2019.1693542. hdl:11581/435751. PMID31726893. S2CID208037373.
^ Ford, Anthony P.; Dillon, Michael P.; Kitt, Michael M.; Gever, Joel R. (November 2021). “The discovery and development of gefapixant”. Autonomic Neuroscience. 235: 102859. doi:10.1016/j.autneu.2021.102859.
17 Dec 2021Vincerx Pharma plans phase II trials for Cancer (IV, Infusion), in the second half of 2022
16 Dec 2021Phase-I clinical trials in Chronic lymphocytic leukaemia (Second-line therapy or greater) in USA (IV)
16 Dec 2021Phase-I clinical trials in Richter’s syndrome (Second-line therapy or greater) (IV) in USA
First-in-human dose escalation study of cyclin-dependent kinase-9 inhibitor VIP152 in patients with advanced malignancies shows early signs of clinical efficacyJennifer R. Diamond, Valentina Boni, Emerson Lim, Grzegorz Nowakowski, Raul Cordoba, Daniel Morillo, Ray Valencia, Isabelle Genvresse, Claudia Merz, Oliver Boix, Melanie M. Frigault, Joy M. Greer, Ahmed M. Hamdy, Xin Huang, Raquel Izumi, Harvey Wong and Victor Moreno DOI: 10.1158/1078-0432.CCR-21-3617
Abstract
Purpose: To report on the first-in-human phase I study of VIP152 (NCT02635672), a potent and highly selective CDK9 inhibitor. Patients and Methods: Adults with solid tumors or aggressive non-Hodgkin lymphoma (NHL) who were refractory to or had exhausted all available therapies received VIP152 monotherapy as a 30-minute intravenous, once weekly infusion, as escalating doses (5, 10, 15, 22.5, or 30 mg in 21-day cycles) until the maximum tolerated dose (MTD) was determined. Results: Thirty-seven patients received {greater than or equal to} 1 VIP152 dose, with 30 mg identified as the MTD based on dose-limiting toxicity of grade 3/4 neutropenia. The most common adverse events were nausea and vomiting (75.7% and 56.8%, respectively), all of grade 1/2 severity. Of the most common events, Grade 3/4 events occurring in > 1 patient were neutropenia (22%), anemia (11%), abdominal pain (8%), increased alkaline phosphatase (8%), and hyponatremia (8%). Day 1 exposure for the MTD exceeded the predicted minimum therapeutic exposure and reproducibly achieved maximal pathway modulation; no accumulation occurred after multiple doses. Seven of 30 patients with solid tumors had stable disease (including 9.5 and 16.8 months in individual patients with pancreatic cancer and salivary gland cancer, respectively), and 2 of 7 patients with high-grade B-cell lymphoma with MYC and BCL2/BCL6 translocations (HGL) achieved durable complete metabolic remission (ongoing at study discontinuation, after 3.7 and 2.3 years of treatment). Conclusion: VIP152 monotherapy, administered intravenously once weekly, demonstrated a favorable safety profile and evidence of clinical benefit in patients with advanced HGL and solid tumors.
CLIP
Preclinical bioconjugation platform designed to overcome limitations of small–molecule and antibody–drug conjugates use to treat cancer
Vincera Pharma, Inc., a biopharmaceutical company aspiring to address the unmet medical needs of patients with cancer through paradigm-shifting therapeutics, today announced the signing of an exclusive license agreement with Bayer AG for the development and commercialization of an early development oncology portfolio. The license will become effective upon the closing of the transaction with LSAC (described below), and Vincera intends to use the funds it will receive upon closing of such transaction to initiate its clinical program.
Under the terms of the license agreement, Vincera will in-license VIP152 (formerly BAY 1251152 & CAS RN.: 1610358-56-9), a clinical-stage, highly selective, positive transcription elongation factor b (PTEFb)/cyclin-dependent kinase 9 (CDK9) inhibitor for the treatment of cancer. Additionally, Vincera will receive assets and license technology for a preclinical bioconjugation platform to address the limitations of small-molecule and antibody-drug conjugates in oncology. The preclinical assets include VIP236, a small molecule drug conjugate (SMDC) targeting advanced and metastatic cancer; as well as VIP943 (formerly BAY-943) and VIP924 (formerly BAY-924), two antibody-drug conjugates (ADC) targeting hematologic tumors; and VIP217, an oral PTEFb/CDK9 inhibitor in discovery. “This license agreement with Bayer creates the foundation of Vincera’s targeted clinical oncology pipeline, with a potentially best-in-class asset, while positioning us for long-term growth across two therapeutic platforms,” said Ahmed Hamdy M.D., Chief Executive Officer of Vincera. “Our lead asset, VIP152, is a small molecule PTEFb/CDK9 inhibitor with very encouraging data from monotherapy Phase 1 studies, including 2 of 7 patients with durable remissions of over 2 years in the very aggressive indication of relapsed/refractory double-hit DLBCL. In addition, preclinical data support our belief that VIP152 is the most selective CDK9 inhibitor in the clinic with on-target depletion of oncogenic MYC and MCL1 mRNA transcripts in patients. These results, combined with the acceptable safety profile seen to date, suggest that VIP152 could be an important new treatment option for patients with MYC- and MCL1-driven malignancies. Importantly, with proof-of-concept clinical data in hand, we are poised to execute on a strategic clinical development plan with the potential for multiple accelerated approvals in the U.S. Expansion of the current Phase 1b study to include these patient populations is expected to begin in 2021.”
Title: 5-FLUORO-N-(PYRIDIN-2-YL)PYRIDIN-2-AMINE DERIVATIVES CONTAINING A SULFOXIMINE GROUP.
Abstract
The present invention relates to 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfoximine group of general formula (I) as described and defined herein, and methods for their preparation, their use for the treatment and/or prophylaxis of disorders, in particular of hyper-proliferative disorders and/or virally induced infectious diseases and/or of cardiovascular diseases. The invention further relates to intermediate compounds useful in the preparation of said compounds of general formula (I).
“CDK9 represents a validated target for malignancies such as CLL where other less selective CDK inhibitors have shown clinical activity in high-risk patients,” says Dr. John C. Byrd, Chair of the Scientific Advisory Board of Vincera. “VIP-152 represents an exciting new therapy for this disease, particularly those with prior resistance to ibrutinib and venetoclax where a true unmet need exists for new treatments.”
Dr. Hamdy continued, “In addition to our planned clinical program, we intend to advance, in parallel, the development of our preclinical bioconjugation platform. We believe our next-generation platform has the potential to generate first-in-class and best-in-class opportunities in oncology, improving the specificity of drug targeting and release through a modular platform with innovative warhead design and linker-payload technologies. We are thrilled that the Bayer license will allow us to pursue the commercial potential of this promising oncology portfolio and look forward to providing updates as we execute across our pipeline in the coming quarters.”
In exchange for this license, Vincera will pay Bayer an upfront license fee and development and commercial sales milestone payments. In further consideration of the rights granted, we will also pay an annual royalty on the commercial sale of licensed products in the single- to low-double-digit percentage range on net commercial sales of licensed products.
On September 29, 2020, Vincera announced that it has entered into a merger agreement with LifeSci Acquisition Corp. (“LSAC”), a publicly-traded blank check company targeting biopharma, medical technology, digital health, and healthcare services sectors. Following the completion of the merger, the combined company is expected to have approximately $60 million in cash to fund its preclinical and clinical pipeline. Additional information about the merger and related transactions, including a copy of the merger agreement, are included in a Current Report on Form 8-K filed by LSAC with the SEC on September 29, 2020, and available at www.sec.gov.
About Vincera Pharma, Inc.
Vincera is a recently formed clinical-stage life sciences company focused on leveraging its extensive development and oncology expertise to advance new therapies intended to address unmet medical needs for the treatment of cancer. Vincera’s executive team has assembled a management team of biopharmaceutical experts with extensive experience in building and operating organizations that develop and deliver innovative medicines to patients. Vincera’s current pipeline is derived from an exclusive license agreement with Bayer and includes (i) a clinical-stage and follow-on small molecule drug program and (ii) a preclinical stage bioconjugation/next-generation antibody-drug conjugate platform. The company intends to develop multiple products through clinical proof-of-concept and potentially through Accelerated Approval in the United States. For more information, please visit www.vincerapharma.com.
Preparation of Intermediate 1.12-Chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine
A batch with 2-chloro-5-fluoro-4-iodopyridine (1000 mg; 3.88 mmol; APAC Pharmaceutical, LLC), (4-fluoro-2-methoxyphenyl)boronic acid (660 mg; 3.88 mmol; Aldrich Chemical Company Inc.) and tetrakis(triphenylphosphin)palladium(0) (449 mg; 0.38 mmol) in 1,2-dimethoxyethane (10.0 mL) and 2 M aqueous solution of potassium carbonate (5.8 mL) was degassed using argon. The batch was stirred under an atmosphere of argon for 4 hours at 100° C. After cooling, the batch was diluted with ethyl acetate and THF and washed with a saturated aqueous solution of sodium chloride. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography (hexane to hexane/ethyl acetate 50%) to give the desired product (947 mg; 3.70 mmol).
A batch containing 2-chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine (400 mg; 1.57 mmol), 4-[(methylsulfanyl)methyl]pyridin-2-amine (483 mg; 3.13 mmol; UkrOrgSynthesis Ltd.), (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (40 mg; 0.07 mmol) and cesium carbonate (765 mg; 2.35 mmol) in dioxane (6.0 mL) was degassed using argon. Tris(dibenzylideneacetone)dipalladium(0) (21 mg; 0.02 mmol) was added under argon and the batch was stirred in a closed pressure tube for 5 hours at 100° C. After cooling, the batch was diluted with an aqueous solution of sodium chloride and extracted with DCM (3×). The combined organic phases were filtered using a Whatman filter and concentrated. The residue was purified by chromatography (hexane to hexane/ethyl acetate 30%) to give the desired product (556 mg; 1.48 mmol).
Under argon, a solution of 2,2,2-trifluoroacetamide (195 mg; 1.73 mmol) in dioxane (0.5 mL) was added dropwise to a solution of sodium tert.-butoxide (111 mg; 1.15 mmol) in dioxane (0.6 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (247 mg; 0.86 mmol) in dioxane (0.6 mL)/THF (1.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine (430 mg; 1.15 mmol) in dioxane (1.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. The mixture was stirred for 60 minutes at 10° C. The batch was diluted with toluene (2.0 mL) under cooling and an aqueous solution of sodium sulfite (145 mg; 1.15 mmol in 2.0 mL water) was added so that the temperature of the mixture remained below 15° C. An aqueous solution of sodium chloride was added and the batch was extracted with ethyl acetate (3×). The combined organic phases were filtered using a Whatman filter and concentrated to give crude 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide, that was used without further purification.
Acetone (6.0 mL) and potassium permanganate (814 mg; 5.15 mmol) were added to the residue and the mixture was stirred at 50° C. for 90 minutes. Additional potassium permanganate (223 mg; 1.42 mmol) was added and stirring was continued at 50° C. for 4 hours. Finally, additional potassium permanganate (305 mg; 1.93 mmol) was added and stirring was continued at 50° C. for 150 minutes. After cooling, the batch was filtered, the residue was washed with acetone and the combined filtrates were concentrated. The residue was dissolved in MeOH (60 mL), potassium carbonate (182 mg; 1.32 mmol) was added and the reaction mixture was stirred for 20 minutes at RT. The batch was diluted with an aqueous solution of sodium chloride and extracted with DCM (3×). The combined organic phases were filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC to give the desired product (50 mg; 0.12 mmol).
[TABLE-US-00003] System:Waters Autopurificationsystem: Pump 254, Sample Manager 2767, CFO, DAD 2996, SQD 3100Column:XBrigde C18 5 μm 100 × 30 mmSolvent:A = H2O + 0.2% NH3 (32%) B = MeCNGradient:0-8 min 15-50% BFlow:50 mL/minTemperature:RTSolution:132 mg/2 mL DMF/MeOH 1:1Injection:2 × 1 mLDetection:DAD scan range 210-400 nm MS ESI+, ESI−, scan range 160-1000 m/zRetention:3.39-3.88 minMS(ES+):m/z = 404
Alternative Procedure for the Preparation of Intermediate 1.25-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 1.3(2-{[5-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methanol
A batch containing 2-chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine (411 mg; 1.61 mmol), (2-aminopyridin-4-yl)methanol (200 mg; 1.61 mmol; ABCR GmbH & CO. KG), (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (418 mg; 0.72 mmol) and cesium carbonate (784 mg; 2.41 mmol) in dioxane (8.0 mL) was degassed using argon. Tris(dibenzylideneacetone)dipalladium(0) (147 mg; 0.16 mmol) was added under an atmosphere of argon and the batch was stirred for 29 hours at 100° C. After cooling, additional (2-aminopyridin-4-yl)methanol (100 mg; 0.81 mmol), (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (118 mg; 0.20 mmol) and tris(dibenzylideneacetone)dipalladium(0) (74 mg; 0.08 mmol) were added and the mixture was stirred for 19 hours at 100° C. After cooling, the batch was diluted with ethyl acetate and washed with an aqueous solution of sodium chloride. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by chromatography (DCM/EtOH 9:1) to give the desired product (389 mg; 1.13 mmol).
Preparation of End Product (Alternative Preparation of Intermediate 1.2)
Thionyl chloride (0.19 ml; 2.55 mmol) was added dropwise to a stirred solution of (2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methanol (350 mg; 1.01 mmol) in DCM (4.0 ml) and NMP (0.4 ml) at 0° C. The mixture was stirred for 7 hours at RT. The batch was diluted with aqueous sodium bicarbonate solution and aqueous sodium chloride solution and extracted with DCM (3×). The combined organic phases were filtered using a Whatman filter and concentrated to give crude N-[4-(chloromethyl)pyridin-2-yl]-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine, that was used without further purification in the next step.
The residue was re-dissolved in EtOH (12.0 ml) and the resulting solution was cooled to 0° C. Sodium methanethiolate (158 mg; 2.26 mmol) was added portionwise to the stirred solution at 0° C. The mixture was stirred for 4 hours at RT before it was diluted with DCM and washed with aqueous sodium chloride solution. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by chromatography (DCM/EtOH 95:5) to give the desired product (301 mg; 0.81 mmol).
Alternative Procedure for the Preparation of Example 1Preparation of Intermediate 1.4(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Under an atmosphere of argon, a solution of 2,2,2-trifluoroacetamide (2.53 g; 22.4 mmol) in THF (10.0 mL) was added dropwise to a solution of sodium tert.-butoxide (1.43 g; 14.9 mmol) in THF (12.0 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (3.20 g; 11.2 mmol) in THF (12.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine (5.57 g; 14.9 mmol; Intermediate 1.2) in dioxane (12.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. The mixture was stirred for 60 minutes at 10° C. The batch was diluted with toluene (40.0 mL) under cooling and an aqueous solution of sodium sulfite (1.88 g; 14.9 mmol in 40.0 mL water) was added so that the temperature of the mixture remained below 15° C. The batch was extracted three times (3×) with ethyl acetate. The combined organic layers were washed with an aqueous solution of sodium chloride, filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (DCM to DCM/EtOH 95:5) to give the desired product (4.71 g; 9.72 mmol).
Alternative Preparation of End Product (Example 1)
An aqueous solution of potassium hydroxide (25%) was added dropwise to a stirred solution of 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (4.64 g; 9.58 mmol) in DMF (350 mL), methanol (100 mL) and water (100 mL) to adjust the pH to 10.5. Oxone® (5.00 g; 8.14 mmol) was added and the mixture was stirred at room temperature for 4.5 hours. During this time, the pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. The mixture was filtered and the filter cake was washed with plenty of DCM. The pH of the filtrate was adjusted to 6-7 using an aqueous solution of hydrogen chloride (15%). The filtrate was washed with an aqueous solution of sodium chloride, followed by an aqueous solution of sodium thiosulfate (10%). During evaporation of solvents using a rotary evaporator, a solid substance precipitated from the solution. The precipitated solid was isolated by suction filtration, washed with DCM and diisopropyl ether, and dried to give the desired product (2.61 g; 6.43 mmol).
Enantiomers of 5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine (3.47 g) was separated into the single enantiomers by preparative chiral HPLC.
Example 4(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 4.15-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
A solution of lithium bis(trimethylsilyl)amide in THF (1M; 20.5 mL; 20.53 mmol; Aldrich Chemical Company Inc.) was added to a mixture of 2-chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine (2.50 g; 9.78 mmol; Intermediate 1.1), tris(dibenzylideneacetone)dipalladium (0) (0.18 g; 0.20 mmol; Aldrich Chemical Company Inc.) and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (0.19 g; 0.39 mmol; Aldrich Chemical Company Inc.) in THF (16.3 mL) under an atmosphere of argon at room temperature. The mixture was stirred at 60° C. for 6 hours. The mixture was cooled to −40° C. and water (10 ml) was added. The mixture was slowly warmed to room temperature under stirring, solid sodium chloride was added and the mixture was extracted with ethyl acetate twice (2×). The combined organic layers were filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 60%) to give the desired product (2.04 g; 8.64 mmol).
Preparation of Intermediate 4.2(2-Chloro-6-methylpyridin-4-yl)methanol
To a stirred solution of 2-chloro-6-methylpyridine-4-carboxylic acid (10.00 g; 55.4 mmol; Maybridge) in THF (100 mL) at 0° C. was added a 1M solution of borane-tetrahydrofuran complex in THF (221.5 mL; 221.5 mmol). The mixture was allowed to react at RT overnight. Then, MeOH (22 mL) was cautiously added to the stirred mixture while cooling with an ice bath. The batch was diluted with ethyl acetate and washed with aqueous sodium hydroxide solution (1N) and saturated aqueous sodium chloride solution. The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (DCM/EtOH 95:5) to give the pure product (7.24 g; 45.9 mmol).
Preparation of Intermediate 4.32-Chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine
To a stirred solution of (2-chloro-6-methylpyridin-4-yl)methanol (7.20 g; 45.7 mmol) in DMF (200 mL) at 0° C. was added dropwise thionyl chloride (8.3 mL; 114.2 mmol). The mixture was allowed to react at 10° C. for 2 hours. Then, the mixture was concentrated to give the crude product 2-chloro-4-(chloromethyl)-6-methylpyridine (17.08 g).
Crude 2-chloro-4-(chloromethyl)-6-methylpyridine (8.04 g).was dissolved in acetone (250 mL) and an aqueous solution of sodium methanethiolate (21%, 18.3 mL, 54.8 mmol; Aldrich Chemical Company Inc.) was added dropwise under stirring. The mixture was stirred at RT for 3 hours before additional aqueous solution of sodium methanethiolate (21%, 15.3 mL, 45.7 mmol; Aldrich Chemical Company Inc.) was added and the mixture was stirred at RT overnight. Finally, additional aqueous solution of sodium methanethiolate (21%, 15.3 mL, 45.7 mmol; Aldrich Chemical Company Inc.) was added and the mixture was stirred at RT for 6 hours. The batch was diluted with ethyl acetate and an aqueous solution of sodium chloride. The mixture was extracted twice with ethyl acetate. The combined organic layers were filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 20%) to give the desired product (7.05 g; 37.6 mmol).
Preparation of Intermediate 4.45-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
A batch containing 5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (852 mg; 3.61 mmol), 2-chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine (677 mg; 3.61 mmol) and cesium carbonate (1410 mg; 4.33 mmol) in dioxane (8.3 mL) was degassed using argon. (9,9-Dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (81 mg; 0.14 mmol) and tris(dibenzylideneacetone)dipalladium(0) (69 mg; 0.08 mmol) were added under an atmosphere of argon and the batch was stirred in a closed pressure tube for 3 hours at 100° C. Additional (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (81 mg; 0.14 mmol) and tris(dibenzylideneacetone)dipalladium(0) (69 mg; 0.08 mmol) were added under an atmosphere of argon and the batch was stirred in the closed pressure tube for additional 20 hours at 100° C.
After cooling, the mixture was diluted with ethyl acetate and washed with an aqueous solution of sodium chloride. The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 50%) to give the desired product (628 mg; 1.62 mmol).
Preparation of Intermediates 4.5 and 4.6(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide and (rac)-N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
Under an atmosphere of argon, a solution of 2,2,2-trifluoroacetamide (125 mg; 1.11 mmol) in THF (1.0 mL) was added dropwise to a solution of sodium tert.-butoxide (71 mg; 0.74 mmol) in THF (1.0 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (158 mg; 0.55 mmol) in THF (1.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine (286 mg; 0.74 mmol) in THF (1.5 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. The mixture was stirred for 60 minutes at 10° C. The batch was diluted with toluene (4.0 mL) under cooling and an aqueous solution of sodium sulfite (93 mg; 0.74 mmol in 7.0 mL water) was added so that the temperature of the mixture remained below 15° C. The batch was extracted three times with ethyl acetate. The combined organic layers were washed with an aqueous solution of sodium chloride, filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 100%) to give the desired product 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (134 mg; 0.27 mmol) and the side product N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}-2,2,2-trifluoroacetamide (110 mg; 0.19 mmol).
An aqueous solution of potassium hydroxide (25%) was added dropwise to a stirred solution of 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (126 mg; 0.25 mmol) in methanol (5.0 mL) and water (1.8 mL) to adjust the pH to 10.5. Oxone® (132 mg; 0.22 mmol) was added and the mixture was stirred at room temperature for 4.5 hours. During this time, the pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. After 4.5 hours, additional Oxone® (33 mg; 0.05 mmol) was added and the mixture was stirred at room temperature for additional 2.5 hours. The pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. The mixture was filtered and the filter cake was washed with plenty of DCM. The filtrate was washed with an aqueous solution of sodium chloride, followed by an aqueous solution of sodium thiosulfate (10%). The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by chromatography (DCM to DCM/ethanol 10%) to give the desired product (38 mg; 0.09 mmol).
Alternative Procedure for the Preparation of Example 4Preparation of Intermediate 4.15-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
2-Chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine (20.00 g; 78.23 mmol), tris(dibenzylideneacetone)dipalladium (0) (1.433 g; 1.563 mmol) and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (1.492 g; 3.129 mmol) in anhydrous THF (200 mL) were degassed three times with argon. After 10 minutes of stirring at RT a solution of lithium bis(trimethylsilyl)amide (156.5 mL; 1.0M; THF) was added and the reaction mixture was degassed three more times with argon. The reaction mixture was stirred 2.5 hours at 60° C.
The reaction mixture was cooled to −20° C. Diluted aqueous hydrochloric acid (1.0M) was added so that the pH was adjusted to approximately 5. The reaction mixture was allowed to reach RT and stirred for 10 minutes at this temperature. Then, the pH was adjusted to 10-11 with aqueous sodium hydroxide solution (5.0M). The reaction mixture was diluted with ethyl acetate and washed twice with half saturated sodium chloride solution. The organic layer was dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (gradient: hexane to ethyl acetate 100%, with 5% dichloromethane during the first 4 column volumes and afterwards 10% dichloromethane) to give the desired compound (12.04 g; 50.97 mmol).
Preparation of Intermediate 4.32-Chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine
An aqueous solution of sodium methanethiolate (21%, 13.15 mL, 39.38 mmol) was added dropwise to a stirred solution of 4-(bromomethyl)-2-chloro-6-methylpyridine hydrochloride (4.60 g; 17.90 mmol; Aldlab Chemicals, LLC; for the free base see CAS 1227588-90-0) in acetone (100 mL) while cooling with a water bath at RT. The mixture was stirred at RT over night. EtOAc was added and the layers were separated. The organic layers were washed with saturated aqueous sodium chloride solution, dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (gradient: hexane to hexane/EtOAc 8:2) to give the desired product (2.60 g, 13.85 mmol).
Preparation of Intermediate 4.45-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
A batch containing 5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (692.2 mg; 2.93 mmol), 2-chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine (500 mg; 2.66 mmol) and cesium carbonate (1302 mg; 4.00 mmol) in dioxane (15 mL) was degassed with argon. (9,9-Dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) (67.8 mg; 0.117 mmol) and tris(dibenzylideneacetone)dipalladium(0) (36.6 mg; 0.04 mmol) were added under an atmosphere of argon and the batch was stirred in a closed pressure tube for 10 hours at 100° C.
Five of these batches were combined and diluted with EtOAc. The organic layer was washed twice with saturated aqueous sodium chloride solution, dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (gradient: hexane to hexane/EtOAc 1:1) affording the desired product (3.75 g; 9.68 mmol).
Preparation of Intermediate 4.5(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Preparation of Intermediate 4.6(rac)-N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
Under an atmosphere of argon, a solution of 2,2,2-trifluoroacetamide (450.7 mg; 3.99 mmol) in anhydrous THF (2.0 mL) was added dropwise to sodium tert.-butoxide (255.5 mg; 2.60 mmol) in anhydrous THF (3.0 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (456.1 mg; 1.60 mmol) in anhydrous THF (3.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine (1030 mg; 2.66 mmol) in anhydrous THF (3.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. The mixture was stirred 1 hour at 10° C. The batch was diluted with toluene (8.0 mL) under cooling and an aqueous solution of sodium sulfite (335 mg; 2.66 mmol in 15.0 mL water) was added under cooling so that the temperature of the mixture remained below 15° C. After 10 minutes the batch was extracted three times with ethyl acetate. The combined organic phases were washed with saturated aqueous sodium chloride solution, dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (gradient: hexane to ethyl acetate 100%) to yield the desired product 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (1202 mg; 2.41 mmol; containing 5,5-dimethylhydantoin) and the side product N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}-2,2,2-trifluoroacetamide (7 mg; 0.012 mmol).
To remove the 5,5-dimethylhydantoin 3.76 g of the product from 4 batches were purified by column chromatography on silica gel (gradient: dichloromethane to dichloromethane/methanol 95:5) to yield the desired product (3.39 g; 6.80 mmol).
3.76 g of racemic 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide were separated by chiral HPLC:
[TABLE-US-00005] System:Agilent: Prep 1200, 2xPrep Pump, DLA, MWD, Prep FCColumn:Chiralpak IA 5 μm 250 × 30 mm Nr.: 010Solvent:hexane/ethanol/diethylamine 50:50:0.1 (v/v/v)Flow:45 mL/minTemperature:RTSolution:3760 mg/30.4 mL DCM/MeOHInjection:38 × 0.8 mLDetection:UV 280 nm Fractionsretention time in minpurity in %yieldSpecific optical rotation Intermediate 4.7 5.3-6.8 min95.5%;1520 mg[α]D20 = +113.4° ee: 100%(3.05 mmol)(1.00, DMSO)Intermediate 4.87.2-10.5 min97.1%;1480 mg[α]D20 = −112.1° ee: 98.7%(2.97 mmol)(1.00, DMSO)
Alternative Preparation of End Product (Example 4)(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (150 mg; 0.301 mmol) was dissolved in methanol (18.0 mL) and water (9.0 mL). At 0-5° C. the pH was adjusted to 9-10 with an aqueous potassium hydroxide solution (15%). At this temperature Oxone® (157.0 mg; 0.256 mmol) was added in several portions and the pH was held at 9-10. The mixture was stirred for 1 hour at 0-5° C. and the pH was held at 9-10.
The reaction mixture was adjusted with 2.0M hydrochloric acid to pH 6-7. Saturated aqueous sodium chloride solution was added and the reaction mixture was extracted three times with dichloromethane. The combined organic phases were washed with an aqueous sodium thiosulfate solution (10%), dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (gradient: dichloromethane to dichloromethane/ethanol 9:1) to afford the desired product (100 mg; 0.239 mmol).
An aqueous solution of potassium hydroxide (25%) was added dropwise to a stirred solution of N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}-2,2,2-trifluoroacetamide (161 mg; 0.28 mmol, Intermediate 4.6) in methanol (15.0 mL) and water (5.0 mL) to adjust the pH to 10.5. Oxone® (146 mg; 0.24 mmol) was added and the mixture was stirred at room temperature for 4 hours. During this time, the pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. After 4 hours, an additional portion of Oxone® (50 mg; 0.08 mmol) was added and the mixture was stirred at room temperature for additional 2.5 hours. The pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. The mixture was filtered and the filter cake was washed with plenty of DCM/MeOH (2:1). The pH of the filtrate was adjusted to pH 6.5 using an aqueous solution of hydrogen chloride (15%), diluted with DCM and washed with an aqueous solution of sodium chloride. The organic layer was finally washed with an aqueous solution of sodium thiosulfate (10%). The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by chromatography (DCM to DCM/ethanol 5%) to give the desired product (44 mg; 0.09 mmol).
Example 6(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 6.1:2-Chloro-6-methoxy-4-[(methylsulfanyl)methyl]pyridine
An aqueous solution of sodium methanethiolate (21%, 1.4 mL, 4.2 mmol; Aldrich Chemical Company Inc.) was added dropwise to a stirred solution of 4-(bromomethyl)-2-chloro-6-methoxypyridine (1000 mg; 4.2 mmol, ZereneX Molecular Limited) in acetone (50 mL) while cooling with a water bath at RT. The mixture was stirred at RT for 3 hours. The batch was diluted with ethyl acetate and an aqueous solution of sodium chloride. The mixture was extracted twice (2×) with ethyl acetate. The combined organic layers were filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 10%) to give the desired product (738 mg; 3.6 mmol).
Preparation of Intermediate 6.25-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
A mixture of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (1281 mg; 5.4 mmol, Intermediate 4.1), 2-chloro-6-methoxy-4-[(methylsulfanyl)methyl]pyridine (724 mg; 3.6 mmol), chloro(2-dicyclohexylphosphino-2′,4′,6′-tri-iso-propyl-1,1′-biphenyl)[2-(2-aminoethyl)phenyl]palladium(II) methyl-tert-butylether adduct (294 mg; 0.36 mmol; ABCR GmbH & CO. KG) and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (170 mg; 0.36 mmol; Aldrich Chemical Company Inc.) and potassium phosphate (3773 mg; 17.77 mmol) in toluene (84 ml) and NMP (10 mL) was stirred under an atmosphere of argon at 130° C. in a closed vessel for 4 hours. After cooling, the batch was diluted with DCM and washed with aqueous sodium chloride solution. The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 35%) to give the pure product (1212 mg; 3.00 mmol).
Preparation of Intermediate 6.3(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methoxypyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Under an atmosphere of argon, a solution of 2,2,2-trifluoroacetamide (252 mg; 2.23 mmol) in THF (2.0 mL) was added dropwise to a solution of sodium tert.-butoxide (143 mg; 1.49 mmol) in THF (2.0 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (255 mg; 0.89 mmol) in THF (2.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine (600 mg; 1.49 mmol) in THF (3.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. The mixture was stirred for 3.5 hours at 10° C. The batch was diluted with toluene (8.0 mL) under cooling and an aqueous solution of sodium sulfite (187 mg; 1.49 mmol in 14.0 mL water) was added so that the temperature of the mixture remained below 15° C. The batch was extracted three times with ethyl acetate. The combined organic layers were washed with an aqueous solution of sodium chloride, filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (DCM to DCM/ethanol 5%) to give the desired product (37 mg; 0.07 mmol).
An aqueous solution of potassium hydroxide (25%) was added dropwise to a stirred solution of 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methoxypyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide (32 mg; 0.06 mmol) in methanol (1.0 mL) and water (0.6 mL) to adjust the pH to 10.5. Oxone® (32 mg; 0.05 mmol) was added and the mixture was stirred at RT for 2.5 hours. During this time, the pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. The mixture was filtered and the filter cake was washed with plenty of DCM. The filtrate was washed with an aqueous solution of sodium chloride, followed by an aqueous solution of sodium thiosulfate (10%). The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC to give the desired product (9 mg; 0.02 mmol).
[TABLE-US-00006] System:Waters Autopurificationsystem: Pump 2545, Sample Manager 2767, CFO, DAD 2996, ELSD 2424, SQD 3001Column:XBrigde C18 5 μm 100 × 30 mmSolvent:A = H2O + 0.1% HCOOH B = MeCNGradient:0-1 min 1% B, 1-8 min 1-99% B, 8-10 min 99% BFlow:50 mL/minTemperature:RTSolution:Max. 250 mg/max. 2.5 mL DMSO or DMFInjection:1 × 2.5 mLDetection:DAD scan range 210-400 nm MS ESI+, ESI−, scan range 160-1000 m/z
Alternative Procedure for the Preparation of Example 6(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
A freshly prepared 1.5 M solution of sodium ethanolate in ethanol (1.5 mL; 2.25 mmol) was added under an atmosphere of argon to a solution of (rac)-ethyl{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methoxypyridin-4-yl)methyl](methyl)oxido-λ 6-sulfanylidene}carbamate (290 mg; 0.57 mmol; Example 15) in ethanol (6.3 mL). The batch was stirred at 60° C. for 4 hours. After cooling the batch was diluted with an aqueous solution of sodium chloride and extracted three times with ethyl acetate. The combined organic layers were filtered using a Whatman filter and concentrated to give the desired product (257 mg; 0.0.59 mmol).
Example 7(rac)-N-{6-Chloro-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
Preparation of Intermediate 7.12-Chloro-6-methoxy-4-[(methylsulfanyl)methyl]pyridine
A mixture of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (2000 mg; 8.47 mmol, Intermediate 4.1), (2,6-dichloropyridin-4-yl)methanol (1507 mg; 8.47 mmol; ABCR GmbH & CO. KG), chloro(2-dicyclohexylphosphino-2′,4′,6′-tri-iso-propyl-1,1′-biphenyl)[2-(2-aminoethyl)phenyl]palladium(II) methyl-tert-butylether adduct (700 mg; 0.85 mmol; ABCR GmbH & CO. KG) and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (404 mg; 0.85 mmol; Aldrich Chemical Company Inc.) and potassium phosphate (8986 mg; 42.33 mmol) in toluene (40 ml) and NMP (4 mL) was stirred under an atmosphere of argon at 110° C. for 135 minutes. After cooling, the batch was diluted with ethyl acetate and washed with aqueous sodium chloride solution. The organic layer was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 50%) to give the pure product (1350 mg; 3.57 mmol).
Preparation of Intermediate 7.2N-{6-Chloro-4-[(methylsulfanyl)methyl]pyridin-2-yl}-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
To a stirred solution of (2-chloro-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methanol (1.47 g; 3.89 mmol) in DMF (43 mL) at 0° C. was added dropwise thionyl chloride (0.71 mL; 9.73 mmol). The mixture was allowed to react at RT for 2 hours. Then, the mixture was concentrated to give crude N-[6-chloro-4-(chloromethyl)pyridin-2-yl]-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (2.85 g).
Crude N-[6-chloro-4-(chloromethyl)pyridin-2-yl]-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (2.85 g) was dissolved in acetone (87 mL) and an aqueous solution of sodium methanethiolate (21%, 5.2 mL, 15.58 mmol; Aldrich Chemical Company Inc.) was added dropwise under stirring. The mixture was stirred at RT for 6 hours. The mixture was diluted with an aqueous solution of sodium chloride and extracted twice with ethyl acetate. The combined organic layers were filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 20%) to give the desired product (1.24 g; 3.04 mmol).
Preparation of Intermediate 7.3(rac)-N-{[(2-chloro-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
Under an atmosphere of argon, a solution of 2,2,2-trifluoroacetamide (312 mg; 2.76 mmol) in THF (2.0 mL) was added dropwise to a solution of sodium tert.-butoxide (176 mg; 1.84 mmol) in THF (2.0 mL), so that the temperature of the mixture remained below 10° C. Subsequently, a freshly prepared solution of 1,3-dibromo-5,5-dimethylhydantoin (394 mg; 1.38 mmol) in THF (3.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 10° C. Then the mixture was stirred for 10 minutes at 10° C. Finally, a solution of N-{6-chloro-4-[(methylsulfanyl)methyl]pyridin-2-yl}-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine (750 mg; 1.84 mmol) in THF (3.0 mL) was added dropwise to the stirred mixture, so that the temperature of the mixture remained below 5° C. The mixture was stirred for 3 hours at 5° C. The batch was diluted with toluene (5.0 mL) under cooling and an aqueous solution of sodium sulfite (232 mg; 1.84 mmol in 5.0 mL water) was added so that the temperature of the mixture remained below 15° C. The batch was extracted three times with ethyl acetate. The combined organic layers were washed with an aqueous solution of sodium chloride, filtered using a Whatman filter and concentrated. The residue was purified by column chromatography on silica gel (hexane to hexane/ethyl acetate 85%) to give the desired product (363 mg; 0.70 mmol).
An aqueous solution of potassium hydroxide (25%) was added dropwise to a stirred solution of N-{[(2-chloro-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}-2,2,2-trifluoroacetamide (495 mg; 0.95 mmol) in methanol (15.0 mL) and water (6.7 mL) to adjust the pH to 10.5. Oxone® (498 mg; 0.81 mmol) was added and the mixture was stirred at RT for 90 minutes. During this time, the pH was kept between 10-11, by dropwise addition of an aqueous solution of potassium hydroxide (25%), if necessary. The mixture was filtered and the filter cake was washed with plenty of DCM and methanol. The pH of the filtrate was adjusted to 6-7 using an aqueous solution of hydrogen chloride (15%). The filtrate was washed with an aqueous solution of sodium chloride, followed by an aqueous solution of sodium thiosulfate (10%). The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by chromatography (DCM to DCM/ethanol 50%) to give the desired product (118 mg; 0.27 mmol).
A batch with 2-chloro-5-fluoro-4-iodopyridine (1000 mg; 3.88 mmol; APAC Pharmaceutical, LLC), (4-fluoro-2-methoxyphenyl)boronic acid (660 mg; 3.88 mmol; Aldrich Chemical Company Inc.) and tetrakis(triphenylphosphin)palladium(0) (449 mg; 0.38 mmol) in 1,2-dimethoxyethane (10.0 mL) and 2 M aqueous solution of potassium carbonate (5.8 mL) was degassed using argon. The batch was stirred under an atmosphere of argon for 4 hours at 100 °C. After cooling, the batch was diluted with ethyl
acetate and THF and washed with a saturated aqueous solution of sodium chloride. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography (hexane to hexane / ethyl acetate 50%) to give the desired product (947 mg; 3.70 mmol).
FDA APPROVED 1/25/2022, Kimmtrak, To treat unresectable or metastatic uveal melanoma
Immunocore Limited
T cell receptor α chain (synthetic human) fusion protein with T cell receptor β chain (synthetic human) fusion protein with immunoglobulin, anti-(human CD3 antigen) (synthetic scFv fragment)
Tebentafusp is a bispecific gp100 peptide-HLA-directed CD3 T cell engager used to treat unresectable or metastatic uveal melanoma.
Tebentafusp is a gp100 peptide-HLA-directed CD3 T cell engager.5 It is a bispecific, fusion protein and first-in-class drug of immune-mobilizing monoclonal T cell receptors against cancer (ImmTACs), a recently developed cancer immunotherapy with a novel mechanism of action. ImmTACs bind to target cancer cells that express a specific antigen of interest and recruit cytotoxic T cells to lyse the cells, such as melanocytes.1,2
Uveal melanoma is a rare ocular tumour with often poor prognosis and limited treatment options. Even after surgical ablation or removal of the ocular tumour, almost 50% of patients with uveal melanoma develop metastatic disease.1 On January 26, 2022, tebentafusp was first approved by the FDA for the treatment of HLA-A*02:01-positive adults with unresectable or metastatic uveal melanoma. This approval marks the first bispecific T cell engager to be approved by the FDA to treat a solid tumour and being the first and only therapy for the treatment of unresectable or metastatic uveal melanoma to be approved by the FDA.5
FDA approves tebentafusp-tebn for unresectable or metastatic uveal melanoma
On January 25, 2022, the Food and Drug Administration approved tebentafusp-tebn (Kimmtrak, Immunocore Limited), a bispecific gp100 peptide-HLA-directed CD3 T cell engager, for HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma.
Efficacy was evaluated in IMCgp100-202 (NCT03070392), a randomized, open-label, multicenter trial of 378 patients with metastatic uveal melanoma. Patients were required to be HLA-A*02:01 genotype positive identified by a central assay. Patients were excluded if prior systemic therapy or localized liver-directed therapy were administered. Prior surgical resection of oligometastatic disease was permitted. Patients with clinically significant cardiac disease or symptomatic, untreated brain metastases were excluded.
Patients were randomized (2:1) to receive tebentafusp-tebn (N=252) or investigator’s choice (N=126) of either pembrolizumab, ipilimumab, or dacarbazine. Tebentafusp-tebn was administered weekly by intravenous infusion at 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15 and every subsequent week until disease progression or unacceptable toxicity. The main efficacy outcome measure was overall survival (OS). An additional efficacy outcome was investigator-assessed progression-free survival (PFS) per RECIST 1.1. Median OS was 21.7 months (95% CI: 18.6, 28.6) for patients treated with tebentafusp-tebn and 16 months (95% CI: 9.7, 18.4) in the investigator’s choice arm (HR=0.51, 95% CI: 0.37, 0.71, p<0.0001) PFS was 3.3 months (95% CI: 3, 5) for those receiving tebentafusp-tebn and 2.9 months (95% CI: 2.8, 3) in the investigator’s choice arm (HR=0.73, 95% CI: 0.58, 0.94, p=0.0139).
The most common adverse reactions (≥30%) were cytokine release syndrome, rash, pyrexia, pruritus, fatigue, nausea, chills, abdominal pain, edema, hypotension, dry skin, headache, and vomiting. The most common laboratory abnormalities (≥50%) were decreased lymphocyte count, increased creatinine, increased glucose, increased aspartate aminotransferase, increased alanine aminotransferase, decreased hemoglobin, and decreased phosphate.
The recommended tebentafusp-tebn dose administered intravenously is:
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), Health Canada, and the United Kingdom’s Medicines and Healthcare product Regulatory Agency (MHRA). The application reviews may be ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
Tebentafusp is indicated for HLA-A*02:01-positive adults with unresectable or metastatic uveal melanoma.[1][2]
History
Efficacy was evaluated in IMCgp100-202 (NCT03070392), a randomized, open-label, multicenter trial of 378 participants with metastatic uveal melanoma.[2] Participants were required to be HLA-A*02:01 genotype positive identified by a central assay.[2] Participants were excluded if prior systemic therapy or localized liver-directed therapy were administered.[2] Prior surgical resection of oligometastatic disease was permitted.[2] Participants with clinically significant cardiac disease or symptomatic, untreated brain metastases were excluded.[2]
Mechanism of ActionHuman growth hormone replacements
Orphan Drug StatusYes – Somatotropin deficiency
RegisteredSomatotropin deficiency
21 Jan 2022Pfizer and OPKO health receives complete response letter from the US FDA for somatrogon in Somatotropin deficiency (In children)
20 Jan 2022Registered for Somatotropin deficiency (In children) in Japan (SC)
01 Dec 2021CHMP issues a positive opinion and recommends approval of somatrogon for Somatotropin deficiency in the European Union
Somatrogon, sold under the brand name Ngenla, is a medication for the treatment of growth hormone deficiency.[1][2] Somatrogon is a glycosylated protein constructed from human growth hormone and a small part of human chorionic gonadotropin which is appended to both the N-terminal and C-terminal.[2]
Somatrogon is a long-acting recombinant human growth hormone used as the long-term treatment of pediatric patients who have growth failure due to growth hormone deficiency.
omatrogon is a long-acting recombinant human growth hormone. Growth hormone is a peptide hormone secreted by the pituitary gland that plays a crucial role in promoting longitudinal growth during childhood and adolescence and regulating metabolic function in adulthood.2 Recombinant growth hormone therapy for growth hormone deficiency and other conditions has been available since 1985, with daily administration being the standard treatment for many years. More recently, longer-acting forms of growth hormone were developed to improve patient adherence and thus, improve the therapeutic efficacy of treatment.1 Somatrogon was produced in Chinese Hamster Ovary (CHO) cells using recombinant DNA technology. It is a chimeric product generated by fusing three copies of the C-terminal peptide (CTP), or 28 carboxy-terminal residues, from the beta chain of human chorionic gonadotropin (hCG) to the N-terminus and C-terminus of human growth hormone.2,6 The glycosylation and the presence of CTPs in the protein sequence prolongs the half-life of somatrogon and allows its once-weekly dosing.6
In October 2021, Health Canada approved somatrogon under the market name NGENLA as the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone caused by growth hormone deficiency, marking Canada as the first country to approve this drug.4 It is available as a once-weekly subcutaneous injection.5
////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Phase 3 study in naive growth hormone deficiency pediatric population was completed.
The study was conducted in over 20 countries. This study enrolled and treated 224 pre-pubertal, treatment-naive children with growth hormone deficiency.
MAA submission in Europe to follow upon completion of open label study demonstrating benefit and compliance with reduced treatment burden
Study expected to be completed in Q3 2020
References
Hershkovitz O, Bar-Ilan A, Guy R, et al. In vitro and in vivo characterization of MOD-4023, a long-acting carboxy-terminal peptide (CTP)-modified human growth hormone. Mol Pharm. 2016; 13:631–639 [PDF]
Strasburger CJ, Vanuga P, Payer J, et al. MOD-4023, a long-acting carboxy-terminal peptide-modified human growth hormone: results of a Phase 2 study in growth hormone-deficient adults. Eur J Endocrinol. 2017;176:283–294 [PDF]
Zelinska N, Iotova V, Skorodok J, et al. Long-acting CTP-modified hGH (MOD-4023): results of a safety and dose-finding study in GHD children. J Clin Endocrinol Metab. 2017;102:1578–1587 [PDF]
Fisher DM, Rosenfeld RG, Jaron-Mendelson M, et al. Pharmacokinetic and pharmacodynamic modeling of MOD-4023, a long-acting human growth hormone, in GHD Children. Horm Res Paediatr. 2017;87:324–332 [PDF]
Kramer W, Jaron-Mendelson M, Koren R, et al. Pharmacokinetics, Pharmacodynamics and Safety of a Long-Acting Human Growth Hormone (MOD-4023) in Healthy Japanese and Caucasian Adults. Clin Pharmacol Drug Dev. 2017 [in press]
Society and culture
Legal status
On 16 December 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Ngenla, intended for the treatment of growth hormone deficiency (GHD) in children and adolescents from 3 years of age.[3] The applicant for this medicinal product is Pfizer Europe MA EEIG.[3]
Somatrogon was approved for medical use in Australia in November 2021.[1]
References
^ Jump up to:abcd“Ngenla”. Therapeutic Goods Administration (TGA). 13 December 2021. Retrieved 28 December 2021.
Faricimab was developed by Roche. Faricimab completed Phase III trials[3] and was approved for use in the United States by the Food and Drug Administration in January 2022.[1][4]
FDA Approves Faricimab to Treat Wet AMD and DME\
FDA Approves Faricimab to Treat Wet AMD and DMEFebruary 1, 2022
The FDA has approved faricimab-svoa (Vabysmo; Genentech) to treat 2 leading causes of vision loss: wet, or neovascular, age-related macular degeneration (AMD) and diabetic macular edema (DME).
After 4 initial monthly doses, faricimab is delivered as injections from 1 to 4 months apart in the first year while the current standard of care for wet AMD and DME requires injections every 1 to 2 months. In wet AMD, patients receive the 4 monthly injections first and then based on outcomes may receive their subsequent treatments every 2, 3, or 4 months. For DME, after the 4 initial monthly injections, treatment is extended or reduced based on outcomes, with a range of 1 to 4 months between doses.
The treatment targets and inhibits pathways involving angiopoietin-2 and vascular endothelial growth factor-A (VEGF-A), which are thought to contribute to vision loss by destabilizing blood vessels.
“Vabysmo represents an important step forward for ophthalmology. It is the first bispecific antibody approved for the eye and a major advance in treating retinal conditions such as wet AMD and diabetic macular edema,” Charles Wykoff, MD, PhD, director of research at Retina Consultants of Texas in Houston and a Vabysmo phase 3 investigator, said in a statement. “With Vabysmo, we now have the opportunity to offer patients a medicine that could improve their vision, potentially lowering treatment burden with fewer injections over time.”
The FDA approved faricimab on the results from 4 phase 3 studies: TENAYA and LUCERNE for wet AMD and YOSEMITE and RHINE for DME. All 4 studies were randomized, multicenter, double-masked, global trials.
TENAYA and LUCERNE were identical: 1329 treatment-naive patients with wet AMD, aged 50 and older, were assigned 1:1 to faricimab up to every 16 weeks or aflibercept every 8 weeks. YOSEMITE and RHINE were also identical: 1891 patients with vision loss due to DME were randomly assigned 1:1:1 to faricimab every 8 weeks, faricimab per personalized treatment interval, or aflibercept every 8 weeks.
For all trials, faricimab was noninferior to aflibercept and the incidence of ocular adverse events was comparable. The researchers determined that the longer time between dosing intervals combined with the visual benefits of faricimab reduced the burden in patients.
The 1-year results from these studies were published January 24 in The Lancet.1,2
“These data published in The Lancet reinforce the potential of faricimab as an important treatment option that may help improve and maintain vision while extending the time between treatments up to 4 months,” Levi Garraway, MD, PhD, chief medical officer and head of Global Product Development, said in a statement. “We remain deeply committed to developing new medicines such as faricimab that may help preserve sight in many people living with serious retinal conditions.”
Now that faricimab is approved, Genentech expects it to become available in the United States within weeks. Meanwhile, the European Medicines Agency is currently evaluating a Marketing Authorization Application for faricimab to treat wet AMD and DME.
There are additional trials—COMINO and BALATON—underway to evaluate the efficacy and safety of faricimab in people with macular edema following retinal vein occlusion. In addition, 2-year results for faricimab in DME will be presented at the Angiogeneisis, Exudation, and Degeneration 2022 meeting in February.
References
1. Heier JS, Khanani AM, Quezada Ruiz C, et al; TENAYA and LUCERNE Investigators. Efficacy, durability, and safety of intravitreal faricimab up to every 16 weeks for neovascular age-related macular degeneration (TENAYA and LUCERNE): two randomised, double-masked, phase 3, non-inferiority trials. Lancet. Published January 24, 2022. doi:10.1016/S0140-6736(22)00010-1
2. Wykoff CC, Abreu F, Adamis AP, et al. Efficacy, durability, and safety of intravitreal faricimab with extended dosing up to every 16 weeks in patients with diabetic macular oedema (YOSEMITE and RHINE): two randomised, double-masked, phase 3 trials. Lancet. Published online January 24, 2022. doi:10.1016/S0140-6736(22)00018-6
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
^ Nicolò M, Ferro Desideri L, Vagge A, Traverso CE (March 2021). “Faricimab: an investigational agent targeting the Tie-2/angiopoietin pathway and VEGF-A for the treatment of retinal diseases”. Expert Opinion on Investigational Drugs. 30 (3): 193–200. doi:10.1080/13543784.2021.1879791. PMID33471572. S2CID231665201.
^World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 80”. WHO Drug Information. 32 (3). hdl:10665/330907.
External links
“Faricimab”. Drug Information Portal. U.S. National Library of Medicine.
////////////Faricimab-svoa, APPROVALS 2022, FDA 2022, RO6867461, RO 6867461, PEPTIDE, MONOCLONAL ANTIBODY, RG 7716, WHO 10563, peptide
5,7-Diiodo-8-quinolinol 5,7-Diiodooxine 5,7-diiodoquinolin-8-ol 83-73-8[RN] 8-Hydroxy-5,7-diiodoquinoline 8-Quinolinol, 5,7-diiodo- дийодогидроксихинолин[Russian][INN] ثنائي إيودوهيدروكسيكينوليين[Arabic][INN] 双碘喹啉[Chinese][INN] 201-497-9[EINECS] 5,7-Diiodo-8-hydroxyquinoline IodoquinolCAS Registry Number: 83-73-8 CAS Name: 5,7-Diiodo-8-quinolinol Additional Names: diiodohydroxyquin; diiodo-oxyquinoline; 5,7-diiodo-8-hydroxyquinoline Manufacturers’ Codes: SS-578 Trademarks: Diodoquin (Searle); Disoquin; Floraquin (Searle); Dyodin; Dinoleine; Searlequin; Diodoxylin; Rafamebin; Ioquin (Abbott); Direxiode (Delalande); Stanquinate; Yodoxin (Searle); Zoaquin; Enterosept; Embequin (M & B) Molecular Formula: C9H5I2NO, Molecular Weight: 396.95 Percent Composition: C 27.23%, H 1.27%, I 63.94%, N 3.53%, O 4.03% Literature References: Prepd by the action of iodine monochloride on 8-hydroxyquinoline: Papesch, Burtner, J. Am. Chem. Soc.58, 1314 (1936); by the action of KIO3 on 8-hydroxyquinoline: Zeifman, C.A.34, 3745. Electrolytic prepn: Brown, Berkowitz, Trans. Electrochem. Soc.75, 385 (1939). See also Claus, DE78880; Passek, DE411050; Matsumura, C.A.21, 1461 (1927); Pirrone, Cherubino, C.A.28, 3073 (1934).Properties: Crystals from xylene. The medicinal grade is a yellowish-brown powder. mp 200-215° (extensive decompn). Almost insol in water. Sparingly sol in alcohol, ether, and acetone; sol in hot pyridine and in hot dioxane. Melting point: mp 200-215° (extensive decompn) Therap-Cat: Antiamebic. Keywords: Antiamebic.
The quinoline derivative diiodohydroxyquinoline (INN), or iodoquinol (USAN), can be used in the treatment of amoebiasis.[1]
Iodoquinol is an amebocide used against Entamoeba histolytica, and it is active against both cyst and trophozoites that are localized in the lumen of the intestine. It is considered the drug of choice for treating asymptomatic or moderate forms of amebiasis. The full mechanism of action is unknown. Iodoquinol is used for diseases caused by moderate intestinal amebiasis.
It acts as an amoebicidal and so used in the treatment of amoebiasis, balantidiasis (an infection caused by protozoa).
Indications
Iodoquinol (diiodohydroxyquin, Yodoxin, Moebiquin) is a halogenated 8-hydroxyquinoline derivative whose precise mechanism of action is not known but is thought to involve an inactivation of essential parasite enzymes. Iodoquinol kills the trophozoite forms of E. histolytica, B. coli, B. hominis, and Dientamoeba fragilis. Iodoquinol is absorbed from the gastrointestinal tract and is excreted in the urine as glucuronide and sulfate conjugates. Most of an orally administered dose is excreted in the feces. Iodoquinol has a plasma half-life of about 12 hours. Iodoquinol is the drug of choice in the treatment of asymptomatic amebiasis and D. fragilis infections. It is also used in combination with other drugs in the treatment of other forms of amebiasis and as an alternative to tetracycline in the treatment of balantidiasis. Adverse reactions are related to the iodine content of the drug; the toxicity is often expressed as skin reactions, thyroid enlargement, and interference with thyroid function studies. Headache and diarrhea also occur. Chronic use of clioquinol, a closely related agent, has been linked to a myelitislike illness and to optic atrophy with permanent loss of vision.
Manufacturing Process
5,7-Diiodo-8-quinolinol widely used as an intestinal antiseptic, especially as an antiamebic agent. It is also used topically in other infections and may cause CNS and eye damage. It is known by very many similar trade names worldwide. 0.01 mol 8-oxychinoline and 0.01 mol salicylic acid were dissolved in 500 ml of water and then 0.05 mol potassium iodide was added. The mixture was heated to temperature 90°-100°C. After that 0.01 mol of KIO3 by little tiles was added. The next tile was added after a disappearence of discharging iodine. Then 10 ml 2 N HCl was added. The solid product was fallen, filtered off, washed with hot water and in 0.25 N NaOH dissolved. The solution was filtered and the clear filtrate precipitated with a very little excess of HCl. The product 5,7-diiodo-8-quinolinol was filtered, washed with hot water and dried. MP: 200°-250°C (with decomposition).
brand name
Quinadome (Bayer); Yodoxin (Glenwood).
Therapeutic Function
Antibacterial
Clinical Use
5,7-Diiodo-8-quinolinol, 5,7-diiodo-8-hydroxyquinoline,or diiodohydroxyquin (Yodoxin, Diodoquin, Diquinol) is ayellowish to tan microcrystalline, light-sensitive substancethat is insoluble in water. It is recommended for acute andchronic intestinal amebiasis but is not effective in extraintestinaldisease. Because a relatively high incidence of topicneuropathy has occurred with its use, iodoquinol should notbe used routinely for traveler’s diarrhea.
Safety Profile
Poison by ingestion and intravenous routes. Human systemic effects by ingestion: eye effects. Mutation data reported. When heated to decomposition it emits very toxic fumes of Iand Nox
Chemical Synthesis
Iodoquinol, 5,7-diiodo-8-quinolinol (37.2.2), is made by iodination of 8-oxyquinoline (37.2.1) using a mixture of potassium iodide/potassium iodate. The initial 8-hydroxyquinolin (37.2.1) is made from 2-aminophenol and glycerol in the presence of sulfuric acid and nitrobenzene (Skraup synthesis).
Purification Methods
It crystallises from xylene and is dried at 70o in a vacuum. [Beilstein 21 II 58.]
Synthesis of 5,7-Diiodo-8-quinolinol from 8-Hydroxyquinoline
SYN
DE 411050 DOI: 10.1021/ja01298a506
CLIP
Iodoquinol, 5,7-diiodo-8-quinolinol (37.2.2), is made by iodination of 8-oxyquinoline (37.2.1) using a mixture of potassium iodide/potassium iodate. The initial 8-hydroxyquinolin (37.2.1) is made from 2-aminophenol and glycerol in the presence of sulfuric acid and nitrobenzene (Skraup synthesis) [39,40]
Iodoquinol is an amebocide used against E. histolytica, and it is active against both cysts and trophozoites that are localized in the lumen of the intestine. It is considered the drug of choice for treating asymptomatic or moderate forms of amebiasis. The mechanism of action is unknown. Iodoquinol is used for diseases caused by moderate intestinal amebiasis. Synonyms of this drug are diquinol, iodoxin, diiodoquin, amebaquin, and others
39. F. Passek, Ger. Pat. 411.050 (1925). 40. V. Papesch, R.R. Burtner, J. Am. Chem. Soc., 58, 1314 (1936).
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
^ Ghaskadbi S, Vaidya VG (March 1989). “In vivo antimutagenic effect of ascorbic acid against mutagenicity of the common antiamebic drug diiodohydroxyquinoline”. Mutat. Res. 222 (3): 219–22. doi:10.1016/0165-1218(89)90137-7. PMID2493578.
^ Aggett, P.J.; Delves, H.T.; Harries, J.T.; Bangham, A.D. (March 1979). “The possible role of Diodoquin as a zinc ionophore in the treatment of acrodermatitis enteropathica”. Biochemical and Biophysical Research Communications. 87 (2): 513–517. doi:10.1016/0006-291X(79)91825-4. PMID375935.
Literature References: Prepn of the hydrochloride: Holm, Acta Chem. Scand.17, 2437 (1963); idem,GB939856 corresp to US3177209 (1963, 1965, both to Kefalas A/S). Crystal structure: J. Lopez de Lerma et al.,Acta Crystallogr.B35, 1739 (1979). Toxicity data: P. V. Petersen et al.,Acta Pharmacol. Toxicol.24, 121 (1966).
The pharmacology of melitracen has not been properly investigated and is largely unknown, but it is likely to act in a similar manner to other TCAs. Indeed, melitracen is reported to have imipramine and amitriptyline-like effects and efficacy against depression and anxiety, though with improved tolerability and a somewhat faster onset of action.[9][10]
Fig. 10 Synthesis of melitracen HCl-(36) by Kiil and co-workers making use of a one-flow system. Adapted with permission from Org. Process Res. Dev., 2018, 22, 228–235. Copyright 2018 American Chemical Society.35
Grignard reactions are commonly used for the construction of carbon–carbon bonds and show exothermic behaviour which can be dangerous in large-scale batch processes. The use of Grignard reagents in flow can be beneficial because of the high control of reaction conditions, facile heat transport and small effective reaction volume.6,34 A recent example was published by Kiil and co-workers, who synthesised melitracen (36) in a one-flow system.35 Kiil hypothesised that the seven unit operations required in batch could be decreased by combining a hydrolysis and dehydration step, and removing a phase separation (Fig. 10).
The investigation commenced with finding a suitable solvent for the Grignard reaction in which starting materials 34, 35 and intermediate products would dissolve. After having identified THF as the most suitable option, the next challenge was to find an acid that could induce both hydrolysis and dehydration in a single step. Hydrochloric acid was able to perform both transformations, however, precipitation was observed. Thus, hydrochloric acid molarities ranging from 1–12 M were tested. However, while even at the lowest molarity precipitation was observed, it also appeared that below 6 M the dehydration reaction did not proceed. Since the precipitation could not be prevented, a molarity of 12 M was eventually used. The individually optimised transformations were then combined in a one-flow continuous system. Most troublesome was that addition of HCl to the reaction mixture led to an exothermic reaction and boiling of the solvent. Therefore, a back-pressure regulator was employed so that melitracen (36) could be successfully synthesised as its HCl-salt in approximately 85% yield.
A Grignard-based batch process, for the preparation of Melitracen HCl, has been redesigned to fit a continuous reactor system. The Grignard addition is carried out at room temperature, with subsequent hydrolysis of the magnesium alkoxide intermediate followed by dehydration of the resulting alcohol. The product undergoes further workup by simple gravimetric phase separation and then crystallization with 2 M HCl in diethyl ether to afford pure Melitracen HCl. All steps in the laboratory setup were concatenated, and the setup was proven capable of producing a significant portion of the commercial quantities of Melitracen HCl. The flow setup profits from a reduced footprint, lower energy consumption, fewer synthetic steps, and reduced raw material usage compared to the batch process.
As illustrated in Scheme 1, four synthetic steps are involved in the manufacturing of Melitracen HCl (6). The four steps are a classic Grignard addition to a ketone, a hydrolysis of a magnesium alkoxide, a dehydration of an alcohol and a salt precipitation to isolate the API. The Grignard addition is between 10,10-dimethylanthrone (10,10-DMA (1)) and 3-(N,N-dimethylamino)propylmagnesium chloride (DMPC-MgCl (2)), resulting in formation of the magnesium alkoxide 3. The magnesium alkoxide 3 is then hydrolyzed to the alcohol 4 and dehydrated to form product 5. The last step is a crystallization of the API as a salt, where HCl is added to obtain the Melitracen HCl (6)
Scheme 1: Syntheses of magnesium alkoxide 3, alcohol 4 and dehydrated product 5 in the manufacturing process of Melitracen HCl 6, from ketone 1 and Grignard reagent 2.
Current Batch Synthesis The current batch synthesis involves individual synthetic steps, as illustrated in Figure 1. DMPC-MgCl 2 is made in-house before it is used, due to its limited storage shelf life, in a toluene-THF solvent mixture. THF is present in trace amounts in order to stabilize the magnesium in the Grignard reagents.45 A solution of 10,10-DMA 1 is prepared in toluene and is slowly transferred to the DMPC-MgCl 2, maintaining a temperature of 50°C. DMPC-MgCl 2 is used in an equivalence of 1.6 compared to 10,10-DMA 1. The formed magnesium alkoxide 3 is hydrolyzed with water and acetic acid (80%). The aqueous phase is discarded and concentrated hydrochloric acid (37%) is used to dehydrate alcohol 4 to form dehydrated product 5. Toluene is replaced with ethanol by a solvent swap. Crystallization of the dehydrated product 5 from the ethanol phase is done with HCl gas to obtain the final Melitracen HCl (6), which is subsequently isolated by filtration.
Precipitation of Melitracen HCl from THF The dehydrated product 5 was crystallized as the final HCl salt in the THF in a batch experiment, in order to remove a solvent swap to ethanol. The crystallization was carried out with 2 M HCl in Et2O, as this was considered more suited for a later flow process and more easily implemented in the laboratory setup. An equivalence of 1.1 HCl was used and the requirement was an achievement of pH<2. The mixture was kept stirred during the crystallization and carried out at ambient temperature. After 10 minutes, fine white solids started to form, followed by a massive precipitation of Melitracen HCl 6. The Melitracen HCl 6 was filtered with a Büchner funnel and washed with THF. The isolated yield was 80% and within the specifications for the in-house analysis methods used in the routine production (CHN, TGA, UV-vis, HPLC, melting point). Figure 3 is a microscope picture of the isolated Melitracen HCl 6. For full-scale production, the HCl gas would still be more desirable for the crystallization and the 2 M HCl in Et2O merely serves as a proof of concept for the laboratory flow setup.
Melitracen (Melitracen), is a kind of tricyclics, entitled 10, the 10- dimethyl -9- γ-two of chemistry Methylamino acrylic -9,10- dihydro-anthraquinone, Clinical practice is its hydrochloride.Melitracen can suppress in presynaptic membrane To the effect of the reuptake of norepinephrine and serotonin, and therefore improve containing for monoamine transmitterses in synaptic cleft Amount.
On the preparation method of melitracen, document report both domestic and external is seldom, existing as described below:
US3177209, GB939856, DK97400, are the compound patents of Lundbeck drugmaker of Denmark, it is mentioned that Synthetic method is that, with 10,10- dimethylanthracene -9- ketone and N, TMSDMA N dimethylamine base propyl group magnesium chloride is generated in the middle of melitracen Body, then by intermediate be dissolved under chloroform, reflux state lead to hydrogen chloride prepare melitracen crude product, then crystallized again with acetone Melitracen is obtained, this method needs to be passed through hydrogen chloride at reflux, there is substantial amounts of smog to produce, and reaction condition is not yet It is easy to control, it there is larger safety factor.
CN103877088A is Lundbeck drugmaker of Denmark in a kind of safe melitracen group disclosed in 2014 Compound, wherein the purity to melitracen in drug regimen proposes more strict requirements, especially to that may make in clinic Cause the impurity (formula I, formula II) of the adverse reactions such as anxiety, irritated and excitement in, even more propose:Formula I<0.1%, formula II< 0.1, I+formula of formula II<0.1% rigors.The melitracen of patent US3177209, GB939856, DK97400 method synthesis Impurity is more, and primary purification can not obtain satisfactory active pharmaceutical ingredient (API).
It is also mentioned that the preparation method of melitracen hydrochloride, this method is with 10,10- diformazans in patent CN103877088A The γ of base-9-dimethylaminopropyl-9- anthrols are raw material, add dichloromethane and hydrochloric acid, are heated to reflux, reaction system alkaline hydrolysis from The free alkali obtained afterwards, is re-dissolved in acetone and leads to hydrogen chloride into salt, obtain melitracen crude product, then isolated and purified with column chromatography Obtain the melitracen of high-purity.The melitracen yield that it is prepared into is low, and purifies and separates process needs column chromatography, it is impossible to meet The need for large-scale production.
Embodiment 1
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
10,10- dimethylanthracene -9- ketone carry out grignard reaction with 3- dimethylaminos-n-propyl chloride in the presence of initiator, obtain To melitracen intermediate, detailed process is as follows:
340g magnesium rods and 17.5L absolute ethers are added in 20L glass reaction kettles, stirring is warming up to 30~35 DEG C, addition 1.75kg 3- dimethylaminos-n-propyl chloride, finish insulated and stirred, add 1g iodine and 2mL 1,2- Bromofume as initiator, 9h is stirred at reflux, magnesium rod disappears completely, reaction system is cooled into 10~20 DEG C, 1.5kg 10,10- dimethyl is slowly added to Anthracene -9- ketone, then it is warming up to 30~35 DEG C, back flow reaction 1 hour;TLC monitoring reactions are complete, and reaction system is cooled into 10~20 DEG C, then add 5.5L water, ether layer is separated, anhydrous sodium sulfate is added and is concentrated under reduced pressure drying, obtain melitracen intermediate 2.03kg, receive The ﹪ of rate 97.2, purity 98.5%.
TLC monitoring methods:Add water and be quenched after sampling, take organic layer point plate;Solvent is petroleum ether:Ethyl acetate=2:1 (volume ratio);The Rf of 10,10- dimethylanthracene -9- ketone is 0.6, and the Rf of melitracen intermediate is 0.1.
(2) melitracen crude product is prepared
2kg melitracens intermediate, 10L chloroforms and 2.4L concentrated hydrochloric acids are put into 20L glass reaction kettles, stirred molten Solution, obtains pale yellow solution, and 60 DEG C of heating stirring reaction 2 hours, TLC monitoring reactions are complete, and separate aqueous layer, organic phase is concentrated under reduced pressure Dry, it is melitracen crude product 2.03kg, yield 95.7%, purity 99.41%, containing Formulas I to obtain white solid:0.20%, formula II:0.13%;Formulas I, II1HNMR spectrograms, melitracen crude product liquid phase spectrogram are shown in accompanying drawing 1,2,3 respectively;
TLC monitoring methods:Organic phase point plate is extracted reaction solution, solvent is dichloromethane:Methanol:Acetic acid=150:10:2 (volume ratio).
Take 2.03kg melitracens crude product (purity 99.41%, Formulas I:0.20%, Formula II:0.13%) 4 times of amount (W/, are added V isopropanol), 20~25 DEG C of stirring 4h (mashing), is filtered, and is dried, is obtained product 2.0kg, yield is 98.5%, and purity is 99.61%, containing Formulas I:0.054%, without Formula II;Melitracen crude product is shown in accompanying drawing 4 through isopropanol mashing sample liquid chromatography(LC figure;
Product after 2kg is beaten is added in 30L glass reaction kettle, adds 16kg isopropanols, and backflow is dissolved, then Cool to 10 DEG C and stir crystallization and stay overnight, suction filtration is dried under reduced pressure, and obtains melitracen 1.89kg, and yield 94.5%, purity 99.98% contains Formulas I:0.0026%, without Formula II;See accompanying drawing 5 through isopropanol recrystallization liquid phase spectrogram.
Embodiment 2
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 1;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 1;
(3) purifying of melitracen crude product
Take 10g melitracens crude product (purity 99.41%, Formulas I:0.20%, Formula II:0.13%) 4 times of amounts (W/V), are added Ethanol, 20~25 DEG C stirring 4h (mashing), filtering, drying, obtain product 9.79g, yield is 97.9%, purity 99.69% contains Formula I 0.047%, containing formula II 0.005%;Melitracen crude product is shown in accompanying drawing 6 through ethanol mashing sample liquid chromatography(LC figure;
Product after 9.0g ethanol is beaten is added in 250mL round-bottomed flask, adds the dissolving of 230mL alcohol refluxs, Then 10 DEG C are cooled to stir crystallization and stay overnight, suction filtration is dried under reduced pressure, obtain melitracen 8.4g, yield 93.3%, purity 99.98%, Containing Formulas I:0.0041%, without Formula II;See accompanying drawing 7 through ethanol recrystallization liquid phase spectrogram.
Embodiment 3
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 1;
(2) melitracen crude product is prepared
100g melitracens intermediate, 500mL chloroforms and 120mL concentrated hydrochloric acids are put into 1L three-necked bottles, stirred molten Solution, obtains pale yellow solution, and 60 DEG C of heating stirring reaction 2 hours, TLC monitoring reactions are complete, and separate aqueous layer, organic phase is concentrated under reduced pressure Dry, it is melitracen crude product 104g, yield 98.3%, purity 99.38%, containing Formulas I to obtain white solid:0.22%, Formula II: 0.15%;Melitracen crude product liquid phase spectrogram is shown in accompanying drawing 8;
TLC monitoring methods:Organic phase point plate is extracted reaction solution, solvent is dichloromethane:Methanol:Acetic acid=150:10:2 (volume ratio).
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the methanol of 4 times of amounts (W/V) is added, 20~25 DEG C of stirring 4h (mashing) obtain product Weight is 18.48g, and yield is 92.4%, and purity is 99.66%, containing Formulas I:0.05%, Formula II:0.008%, see accompanying drawing 9.
Embodiment 4
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the n-butanol of 4 times of amounts (W/V) is added, 20~25 DEG C of stirring 5h (mashing) are produced Thing weight is 19.6g, and yield is 98%, and purity is 99.54%, containing Formulas I:0.05%, Formula II:0.009%, see accompanying drawing 10.
Embodiment 5
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the isopropanol of 4 times of amounts (W/V) is added, 30~35 DEG C of stirring 5h (mashing) are produced Thing weight is 18.06g, and yield is 90.3%.
Embodiment 6
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the isopropanol of 4 times of amounts (W/V) is added, 50 DEG C of stirring 3h (mashing) obtain product weight Measure as 14.2g, yield is 71%.
Embodiment 7
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the isopropanol of 4 times of amounts (W/V) is added, 5-10 DEG C of stirring 5h (mashing) obtains product Weight is 19.7g, and yield is 98.5%, and purity is 99.53%, containing Formulas I:0.054%, Formula II:0.014%, see accompanying drawing 11.
Embodiment 8
With reference to CN103877088A, crystallized using acetone, that is, take 10g melitracens intermediate and 24mL dichloromethane, 6.7mL concentrated hydrochloric acids are heated to reflux 2h and are cooled to room temperature, and pH is to 8-9 for regulation, then are extracted with dichloromethane and product, are concentrated to give free Alkali cpd, acetone is dissolved in by the free alkali compound, concentrated hydrochloric acid is added dropwise to pH=0.1, stirring, cooling separate out solid 7.1g, This solid crystallizes to obtain sample 6.4g with acetone again, and total recovery is 60.9%, and purity is 99.64%, containing Formulas I:0.09%, Formula II: 0.04%.Melitracen is shown in accompanying drawing 12 only with acetone crystallization liquid chromatography(LC figure.
Repeat literature method crystallized only with acetone obtained by product in impurity Formulas I, Formula II impurity summation be 0.13%, The adverse reactions such as anxiety, irritated and excitement may be caused in Clinical practice.
In summary, the effect of mashing is to make melitracen crude product rapid dispersion, and the effect of methanol mashing is similar with ethanol, But it is good without isopropanol effect, but methanol mashing yield is decreased obviously trend;N-butanol mashing needs the extension time to reach To the effect same with ethanol, but be not as good as isopropanol effect, and because the viscosity of n-butanol is slightly larger, melitracen crude product is at it In disperse slightly worse, invention has the granular solids that not readily dissolve after filtering, and the removal effect to other impurities is also poor;Isopropanol Temperature is raised during mashing, yield is decreased obviously, and reduces temperature, yield has no raising, though to the removal effect of impurity Formula II It can so control in the range of conforming to quality requirements, but compared to being decreased obviously in embodiment 1.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
^ Van Moffaert M, Dierick M, De Meulemeester F, Vereecken A (1983). “Treatment of depressive anxiety states associated with psychosomatic symptoms. A double-blind multicentre clinical study: mianserin versus melitracen-flupentixol”. Acta Psychiatrica Belgica. 83 (5): 525–39. PMID6670581.
^ Bin Yaacob H (April 1985). “Flupenthixol and Melitracen in the management of trigeminal neuralgia”. Dental Journal of Malaysia. 8 (2): 37–8. PMID3917005.
Literature References: Antimycotic deriv of imidazole. Prepn: E. Regel et al.,DE2461406; eidem,US4118487 (1976, 1978 both to Bayer). Series of articles on in vitro and in vivo antimycotic efficacy, microscopic studies, pharmacokinetics, efficacy in dermatomycoses and comparison with clotrimazole and miconazole, q.q.v.:Arzneim.-Forsch.33, 517-551, 745-754 (1983). Toxicology: G. Schlüter, ibid. 739.
Properties: Crystals from acetonitrile, mp 142°. Very lipophilic. Sol in alcohols, DMF, DMSO. Soly in water at pH 6: <0.1 mg/100 ml. Stable in aq soln at pH 1-12. LD50 in male mice, rats (mg/kg): 2629, 2854 orally (Schlüter).
Melting point: mp 142°
Toxicity data: LD50 in male mice, rats (mg/kg): 2629, 2854 orally (Schlüter)
(CAS NO.: ), with its systematic name of , 1-(alpha-(4-biphenylyl)benzyl)-, could be produced through many synthetic methods.
Following is one of the synthesis routes: (I) could be reduced with NaBH4 in ethanol to produce 4-phenylbenzhydrol (II), and the yielding product is then condensed with imidazole (III) in the presence of SOCl2 in acetonitrile.
The The present invention relates to a process for the preparation of Bifonazole (1- [biphenyl-4-yl (phenyl) methyl] -1H-imidazole) by reacting 1-biphenyl-4-yl (phenyl) methanol with a chlorinating reagent in cyclohexane and subsequent coupling with imidazole.
[0002]The compound bifonazole (1- [biphenyl-4-yl (phenyl) methyl] -1H-imidazole) is off DE-A 2 461 406 known and corresponds to the formula (I). Due to its antifungal activity, it can be used as an agent for the treatment of fungal diseases.
[0003]Various methods for preparing this compound are known. So describes DE-A 2 461 406 the synthesis (process 1) of bifonazole (Example 1) starting from biphenyl-4-yl (phenyl) methanol by reaction with imidazole and thionyl chloride in acetonitrile with a yield of only 56% of theory. An alternative synthesis described therein (process 2) starting from 4- [chloro (phenyl) methyl] biphenyl, which is prepared from biphenyl-4-yl (phenyl) methanol by reaction with thionyl chloride in toluene, by reaction with trimethylsilylimidazole bifonazole provides only in a yield of 52% of theory.
[0004]ES-A 2 024 363 describes also starting from 4- [chloro (phenyl) methyl] biphenyl, which is prepared from biphenyl-4-yl (phenyl) methanol by reaction with hydrogen chloride in acetonitrile, by reaction with imidazole in acetonitrile using a phase transfer catalyst, the synthesis (method 3) of bifonazole.
[0005]AT-B 396 931 describes the preparation (method 4) of bifonazole by means of reductive amination of biphenyl-4-yl (phenyl) methanone with imidazole and formic acid. However, this requires high reaction temperatures (220 ° C.) and long reaction times. DE-A 3 538 873 describes a comparable process (process 5) with the additional use of p-toluenesulfonic acid, wherein the reaction temperature is 180 ° C.
[0006]This in ES 539 345 described method (method 6) for the preparation of bifonazole involves a Gringard reaction between 4-biphenylmagnesium bromide and benzoylated imidazole. Finally, it is tosylated and reduced to bifonazole.
[0007]ES 549 793 describes the synthesis (method 7) of bifonazole starting from a cyclocondensation between biphenyl-4-yl (phenyl) methylamine, 2-chloro-1-aminoethane and ethyl orthoacetate. The final dehydrogenation is carried out by reaction with 2,3-dichloro-5,6-dicyano-p-benzoquinone in benzene.
[0008]All known processes have various disadvantages which are particularly unfavorable in the preparation of the compound of the formula (I) on an industrial scale. The solvents used in processes 1 and 2 acetonitrile and toluene are of concern to health. Their use should be avoided in the manufacture of active ingredients used in medicines. By using toluene in process 2, chlorination to give 4- [chloro (phenyl) methyl] biphenyl also produces a toluene-specific, undesired by-product which can only be removed incompletely and thus deteriorates the product quality. The yield is unsatisfactory in both processes. A significant disadvantage of method 3 is, in addition to the use of acetonitrile as solvent, the use of a phase transfer catalyst, which is difficult to separate from the product during work-up. Methods 4 and 5 both operate at very high temperatures and are therefore disadvantageous in a technical use due to the energy consumption and the potential hazard. In method 6, the use of the Gringard reagent is disadvantageous, since this must be produced under considerable safety expense and difficult to handle on an industrial scale. Disadvantage in process 7 is the use of the very toxic compounds 2,3-dichloro-5,6-dicyano-p-benzoquinone and benzene. Their use should be avoided especially in the production of active ingredients used in pharmaceuticals
The following scheme illustrates the individual reaction steps.
Embodiment:
Synthesis of bifonazole (1- [Biphenyl-4-yl (phenyl) methyl] -1H-imidazole)
[0038]140 g (0.54 mol) dry (water content <0.3%) biphenyl-4-yl (phenyl) methanol (II) are suspended in 1550 ml of cyclohexane and treated with 90 g (0.76 mol) thionyl chloride at a temperature of 50 to 55 ° C added. The reaction mixture is stirred for 0.5 h at a temperature of 50 to 55 ° C stirred. Subsequently, in the Vacuum (<100 mbar) Distilled off thionyl chloride and cyclohexane. A distillation bottoms containing 4- [chloro (phenyl) methyl] biphenyl remains.
[0039]162 g (2.4 mol) of imidazole are suspended in 1350 ml of acetone and dissolved at 50 ° C. This solution is added to the distillation bottoms from step 1 containing 4- [chloro (phenyl) methyl] biphenyl (III). The reaction mixture is heated at reflux for 3 h. After cooling, the reaction solution is mixed with 2 g of activated carbon and 2 g of bleaching earth at a temperature of 50 to 55 ° C, stirred for 0.5 h and filtered. The filtrate is cooled to about 0 ° C. The title compound crystallizes by addition of seed crystals, is filtered off and washed with a mixture of acetone / water (1: 1). For recrystallization, the product is dissolved in 1250 ml of isopropanol, treated with 0.5 g of activated charcoal and 0.5 g of bleaching earth, heated to reflux and filtered hot. The filtrate is cooled to 10 ° C. The title compound crystallizes out by addition of seed crystals, is filtered off, washed with isopropanol and dried. The yield is 101 g (61.9% of theory). The purity of the product is 98.68% by weight. Melting point: 142 ° C
Comparative method:
[0040]In the comparative method, instead of cyclohexane, toluene is used as solvent in step 1 as in DE-A 2 461 406 described. Step 2 is performed as described above. 1- [biphenyl-4-yl (phenyl) methyl] -1H-imidazole (bifonazole) is obtained in a purity of 97.66% by weight.
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The most common side effect is a burning sensation at the application site. Other reactions, such as itching, eczema or skin dryness, are rare.[3] Bifonazole is a potent aromatase inhibitorin vitro.[4][5]
^ Jump up to:abc Haberfeld H, ed. (2015). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Canesten Bifonazol-Creme.
^ Trösken ER, Fischer K, Völkel W, Lutz WK (February 2006). “Inhibition of human CYP19 by azoles used as antifungal agents and aromatase inhibitors, using a new LC-MS/MS method for the analysis of estradiol product formation”. Toxicology. 219 (1–3): 33–40. doi:10.1016/j.tox.2005.10.020. PMID16330141.
^ Berg D, Regel E, Harenberg HE, Plempel M (1984). “Bifonazole and clotrimazole. Their mode of action and the possible reason for the fungicidal behaviour of bifonazole”. Arzneimittel-Forschung. 34 (2): 139–46. PMID6372801.
Further reading
Lackner TE, Clissold SP (August 1989). “Bifonazole. A review of its antimicrobial activity and therapeutic use in superficial mycoses”. Drugs. 38 (2): 204–25. doi:10.2165/00003495-198938020-00004. PMID2670516.
Literature References: Derivative of artemisinin, q.v. Prepn: Y. Li et al.,K’o Hsueh T’ung Pao24, 667 (1979), C.A.91, 211376u (1979); eidem,Acta Pharm. Sin.16, 429 (1981). Absolute configuration: X.-D. Luo et al.,Helv. Chim. Acta67, 1515 (1984). NMR spectral study: F. S. El-Feraly et al.,Spectrosc. Lett.18, 843 (1985). Inhibition of protein synthesis: H. M. Gu et al.,Biochem. Pharmacol.32, 2463 (1983). Antimalarial activity: S. Thaithong, G. H. Beale, Bull. WHO63, 617 (1985). Series of articles on chemistry, pharmacology and antimalarial efficacy: China Cooperative Research Group on Qinghaosu, J. Tradit. Chin. Med.2, 3-50 (1982). Toxicity data: eidem,ibid. 31. Clinical trial in cerebral malaria in children: M. B. van Hensbroek et al.,N. Engl. J. Med.335, 69 (1996). Review: R. N. Price, Expert Opin. Invest. Drugs9, 1815-1827 (2000).
Properties: Crystals, mp 86-88°. [a]D19.5 +171° (c = 2.59 in CHCl3). LD50 i.m. in mice: 263 mg/kg (China Cooperative Research Group on Qinghaosu).
Melting point: mp 86-88°
Optical Rotation: [a]D19.5 +171° (c = 2.59 in CHCl3)
Toxicity data: LD50 i.m. in mice: 263 mg/kg (China Cooperative Research Group on Qinghaosu)
Therap-Cat: Antimalarial.
Keywords: Antimalarial.
Artemether is an antimalarial agent used in combination with lumefantrine for the treatment of acute uncomplicated malaria caused by Plasmodium falciparum.
Artemether is an antimalarial agent used to treat acute uncomplicated malaria. It is administered in combination with lumefantrine for improved efficacy. This combination therapy exerts its effects against the erythrocytic stages of Plasmodium spp. and may be used to treat infections caused by P. falciparum and unidentified Plasmodium species, including infections acquired in chloroquine-resistant areas.
Artemether is a natural product which effectively kills both malarial parasites P. falciparum and P. vivax. Artemether is usually used in combination with Lumefantrine for the treatment of malaria. Arthemether also kills trematodes of the species Schistosoma, providing protection against schistosomiasis. Sesquiterpene lactones like artemether, artesunate, and artemisinin have potential applications in certain types of cancer and inflammatory conditions.
Artemether causes relatively few side effects.[5] An irregular heartbeat may rarely occur.[5] While there is evidence that use during pregnancy may be harmful in animals, there is no evidence of concern in humans.[5] The World Health Organization (WHO) therefore recommends its use during pregnancy.[5] It is in the artemisinin class of medication.[5]
Haynes RK, Vonwiller SC: Extraction of artemisinin and artemisinic acid: preparation of artemether and new analogues. Trans R Soc Trop Med Hyg. 1994 Jun;88 Suppl 1:S23-6. Pubmed.
Malaria is a serious parasitic disease caused by Plasmodium parasites in the human body. Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malaria and Plasmodium knowlesi are the parasites that live in humans, of which P. vivax and P. falciparum are the most common.
Traditional anti-malarial drugs mainly include quinine, chloroquine, primaquine, and pyrimethamine. In 1972, the antimalarial active ingredient artemisinin extracted from the Compositae plant Artemisia annuaL by Chinese scientists is the most popular antimalarial effect after chloroquine, pyrimethamine, primary amine and sulfonamide. Drugs, especially for the treatment of cerebral malaria and anti-chloroquine malaria.
At present, a large number of artemisinin derivatives have been synthesized and screened for antimalarial activity. Artemether is a compound with excellent curative effect. In addition to the advantages of artemisinin’s quick effect and low toxicity, its solubility in oil is also higher than that of artemisinin. Artemisinin is large, which is especially beneficial for the preparation of preparations. Since artemether has two products, α and β epimers, and the antimalarial activity of artemether is mainly isomer β, so the industrial automation and intelligent production of β-artemether and the improvement of the process are realized. , reducing the impurities produced by the reaction, improving the quality of the product, and improving the purity of the product are the problems that need to be solved in today’s scientific research.
Patent CN104557965B discloses a preparation process of β-artemether, which mainly includes adding dihydroartemisinin and etherification reagent to alcohol to form a reaction system, and then adding acid to the reaction system for reaction. Water or non-alkaline aqueous solution is added to the reaction system to crystallize, namely β-artemether. The preparation process claims to effectively inhibit the production of isomer α-artemether in the reaction, and can make the etherification reaction proceed mildly, with simple post-treatment and high purity; although the purity of the product has been improved, the yield and Purity needs to be further improved.
Patent CN102731523B discloses a method for preparing β-artemether, which mainly includes the reaction of artemisinin under the action of a reducing agent to generate dihydroartemisinin, and the reaction of dihydroartemisinin with p-toluenesulfonic acid to generate β-artemisinin. The crude artemether is crystallized with methanol, ethanol, ethylene glycol or isopropanol, filtered, washed and dried. The method for preparing B-artemether of the invention has mild conditions, is environmentally friendly, is suitable for industrial production, and has a product yield of over 90 percent and a purity of 99.2 percent. The crystallization step of the invention adopts organic reagents, which adversely affects the quality control of subsequent products.
Patent CN103180325B discloses a method for preparing β-artemether, which uses dihydroartemisinin as a raw material and undergoes etherification reaction with trimethyl orthoformate in organic solvents including esters and alkanes to obtain β-artemether. The method of the invention is easy to control in process operation, high in yield, low in cost and high in product quality, and is suitable for industrial production. The method requires vacuum distillation, the obtained crude product needs to be redissolved with methanol, decolorized with activated carbon, etc., new impurities are easily introduced, the operation is not simple enough, and the efficiency is low.
Patent CN107793428A discloses a preparation method of artemether, hydrogenating artemisinin to obtain dihydroartemisinin, adding trimethyl orthoformate, reacting with boron trifluoride ether solution, slowly adding saturated sodium bicarbonate solution dropwise, The system was adjusted to neutrality, the liquids were separated, the aqueous phase was extracted with dichloromethane, the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure to obtain a solid; the obtained solid was dissolved in methanol, and an appropriate amount of activated carbon was added to obtain a solid. Reflux and decolorize, filter, add pure water dropwise to the filtrate, crystallize, wash with water, and dry to obtain artemether. However, this method requires steps such as extraction with an organic reagent dichloromethane and decolorization with activated carbon, which is cumbersome to handle.
Therefore, the following problems generally exist in the process of preparing β-artemether at present:
(1) when preparing β-artemether, the reaction time is longer, the impurities are large, and the purity and yield of the product are not high enough;
(2) The use of organic reagents in the subsequent purification process has a certain impact on the quality control of the product;
(3) The batch production equipment is adopted, the subsequent process steps are many, the degree of industrialization is low, the production efficiency is low, and it does not meet the requirements of GMP.
Example 1
This embodiment includes the following steps:
(1) at room temperature, add methanol 2400L in the 3000L stirred tank (1), then add 600kg of dihydroartemisinin through the solid feed pump, and circulate and disperse evenly;
(2) add etherification agent trimethyl orthoformate and acid catalyst acetyl chloride through three-way automatic feeding mixing reactor again, the volume ratio is 500:100:3, the mixing reactor control temperature is 5 ℃, and the flow rate of control feeding is 5L /min;
(3) in the continuous flow pipeline, enter the second mixer and add 5% sodium bicarbonate solution to neutralize, and the adding speed is 1.0L/min, and is filtered through the fine filter;
(4) Then directly enter the 2000L crystallization reaction kettle 11 with 300L of water added in advance and keep the temperature at 10°C. At the same time, purified water was added to the reaction kettle at a rate of 12L/min, and the crystallization was continued for 1.5h; the jacket of the crystallization kettle was fed with -10°C chilled water for 30min, and the temperature of the system was controlled to 5°C.
(5) centrifugal washing, obtaining crude artemether 704.5kg, drying to obtain artemether fine product 608.6kg, β-artemether purity 99.83%, α-artemether impurity 0.12%, and other single impurities less than 0.1%, The content is 99.8%, the mass yield is 96.1%, and the molar yield is 91.42%.
Example 2
This embodiment includes the following steps:
(1) at room temperature, 2400L of methanol was pumped into the 3000L reactor 1, and then 800kg of dihydroartemisinin was added by the solid feed pump, and the circulation was uniformly dispersed;
(2) add etherifying agent dimethyl phosphate and acid catalyst boron trifluoride ether through the three-way automatic feeding mixing reactor again, the volume ratio is 500:105:3.5, the mixing reactor control temperature is 3 ℃, and the control feeding flow rate is 3L/min;
(3) in the continuous flow pipeline, enter the second mixer and add 3% sodium bicarbonate solution to neutralize, and the speed of addition is 1.8L/min, through the fine filter;
(4) Directly enter the 2000L crystallization reaction kettles 11 and 12 with 300L of water added in advance and the temperature kept at 10°C. At the same time, purified water was added to the reaction kettle at 9 L/min, and the crystallization was continued for 2.5 hours; the jacket of the crystallization kettle was fed with -10 °C chilled water for 30 minutes, and the temperature of the system was controlled to 10 °C
(5) centrifugal washing, obtain crude artemether 939.3kg, oven dry to obtain artemether fine product 809.7kg, β-artemether purity 99.81%, α-artemether impurity 0.11%, other single impurities are less than 0.1%, The content is 99.8%, the mass yield is 96.2%, and the molar yield is 91.6%.
Example 3
This embodiment includes the following steps:
(1) 2400L of methanol was pumped into the 3000L reactor F1 at room temperature, and then 400kg of dihydroartemisinin was added through the solid feed pump, and the circulation was uniformly dispersed;
(2) Add etherification agent dimethyl phosphate and acid catalyst trimethylchlorosilane through the three-way automatic feeding mixing reactor, the volume ratio is 500:95:2.5, the mixing reactor is controlled at a temperature of 8 °C, and the feeding liquid is controlled to be added. The flow rate is 7L/min, and the reaction time is;
(3) in the continuous flow pipeline, enter the second mixer and add 8% sodium bicarbonate solution for neutralization, and the rate of addition is 0.6L/min, passing through the fine filter;
(4) Directly enter into the 2000L crystallization reactor J2 with 300L water added in advance and keeping the temperature at 10°C. At the same time, purified water was added to the reaction kettle at 15 L/min, and the crystallization was continued for 1 hour; the jacket of the crystallization kettle was fed with -10 °C chilled water for 30 minutes, and the temperature of the system was controlled to 0 °C
(5) centrifugal washing, obtain crude artemether 939.3kg, oven dry to obtain artemether fine product 809.7kg, β-artemether purity 99.81%, α-artemether impurity 0.11%, other single impurities are less than 0.1%, The content is 99.8%, the mass yield is 95.5%, and the molar yield is 90.9%.
Comparative Example 1
The difference between this embodiment and Example 1 is that hydrochloric acid is used instead of the acidic catalyst. Finally, 633.6kg of crude artemether was obtained, and 550.3kg of fine artemether was obtained by drying. The purity of β-artemether was 94.20%, and the impurities of α-artemether were 3.66%. %, and the molar yield was 80.6%.
Comparative Example 2
The difference between this embodiment and Example 1 is that the step of adding water in advance in the crystallization kettle is removed. Finally, 645.1kg of crude artemether was obtained, and 562.2kg of fine artemether was obtained by drying. The purity of β-artemether was 99.68%, the impurity of α-artemether was 0.22%, and the average of single and impurity was less than 0.1%. The mass yield was 88.7%. %, and the molar yield was 84.4%.
In Comparative Example 2, the step of adding water in advance in the crystallization was removed, the purity of β-artemether was 99.68%, and the yield was 88.7%. The yield dropped by 7.6%.
The above detailed description is a specific description of one of the feasible embodiments of the present invention, and this embodiment is not intended to limit the patent scope of the present invention. Any equivalent implementation or modification that does not depart from the present invention shall be included in the present invention. within the scope of the technical solution.
SYN1
Synthetic Reference
Continuous synthesis of artemisinin-derived medicines; Gilmore, Kerry; Kopetzki, Daniel; Lee, Ju Weon; Horvath, Zoltan; McQuade, D. Tyler; Seidel-Morgenstern, Andreas; Seeberger, Peter H. Chemical Communications (Cambridge, United Kingdom); Volume 50; Issue 84; Pages 12652-12655; Journal; 2014
SYN2
Synthetic Reference
An Improved Manufacturing Process for the Antimalaria Drug Coartem. Part I; Boehm, Matthias; Fuenfschilling, Peter C.; Krieger, Matthias; Kuesters, Ernst; Struber, Fritz; Organic Process Research & Development; Volume 11; Issue 3; Pages 336-340; Journal; 2007
SYN3
Synthetic Reference
Some transition metal complexes bearing artemisinin derivatives and (N-N-O) tridentate chromium (III) complexes ligated by 2-benzolmidazo-yl-6-acetyl-pyridines for catalytic behaviour towards ethylene; Obaleye, Joshua Ayoola; Amolegbe, Saliu Alao; Adewuyi, Sheriff; Sun, Wenhua; Oshodi, Margaret Damilola; Journal of Chemistry and Chemical Engineering; Volume 4; Issue 12; Pages 23-32; Journal; 2010
SYN4
Synthetic Reference
Method and apparatus for the synthesis of dihydroartemisinin and artemisinin derivatives; Kopetzki, Daniel; McQuade, David Tyler; Seeberger, Peter H.; Gilmore, Kerry; Assignee Max-Planck-Gesellschaft zur Foerderung der Wissenschaften e.V., Germany; 2015; Patent Information; Jan 21, 2015; EP 2826779 A1
An efficient one pot green synthesis of β-artemether/arteether from artemisinin has been developed using a sodium borohydride-cellulose sulfuric acid (CellSA) catalyst system. The green methodology is high yielding and the catalyst has good recyclability.
Fig. 2 Conventional approaches for synthesis of artemether from artemisinin.
Scheme 1 One-pot conversion of β-artemisinin to artemether.
Experimental section
Representative procedure for catalyst preparation
Preparation of cellulose sulfuric acid.To a magnetically stirred mixture of 5.00 g of cellulose (DEAE for column chromatography, Merck) in 20 ml of n-hexane, 1.0 g of chlorosulfonic acid (9 mmol) was added dropwise at 0 °C over 2 h. HCl gas was removed from the reaction vessel immediately. After the addition was complete, the mixture was stirred for 2 h. Then, the mixture was filtered, washed with 30 ml of acetonitrile, and dried at room temperature to obtain 5.47 g cellulose sulfuric acid as a white powder.17
General procedure for the arteether from artemisinin in one-pot
To a solution of artemisinin (200 mg, 0.71 mmol) in ethanol (15 ml) and trimethyl orthoacetate (0.5 ml) was added NaBH4 (67 mg, 1.77 mmol, 2.5 equ.) and cellulose sulfuric acid (0.015 g). Reaction mixture was carried out at −5 to 0 °C for 60 min, and then stirred at room temperature for 1.5 h. Then we added a solution of sodium bicarbonate to quenched the reaction. The slurry was stirred in an below 20 °C for 1 h and allowed to settle for 30 min. Solid crude arteether was collected by filtration, and the cake was washed with of ethanol. The reaction mass was heated to 40 ± 5 °C in water. The reaction mass was seeded with pure β-arteether. Then it was filtered, washed with chilled 50% solution of ethanol in water and dried.
General procedure for the artemether from artemisinin in one-pot
Artemisinin (200 mg, 0.71 mmol) in methanol (15 ml) and trimethylorthoformate (0.5 ml), cellulose sulfuric acid (0.015 g), was carried out at −5 to 0 °C for 60 min, and then stirred at room temperature for 1.5 h. The reaction was monitored by TLC and HPLC to check completion of the reaction. The cellulose sulfuric acid was removed by filtration, the filtrate was concentrated. Then we added a solution of sodium bicarbonate to terminate the reaction. Then, follow above recrystallization method.
Preparation of cellulose sulfuric acid. To a magnetically stirred mixture of 5.00 g of cellulose (DEAE for column chromatography, Merck) in 20 ml of n-hexane, 1.0 g of chlorosulfonic acid (9 mmol) was added dropwise at 0 0 C over 2 h. HCl gas was removed from the reaction vessel immediately. After the addition was complete, the mixture was stirred for 2 h. Then, the mixture was filtered, washed with 30 ml of acetonitrile, and dried at room temperature to obtain 5.47 g cellulose sulfuric acid as a white powder. K General procedure for the arteether from artemisinin in one-pot. To a solution of artemisinin (200 mg, 0.71 mmol) in ethanol (15 mL) and trimethyl orthoacetate (0.5 mL) was added NaBH4 (67 mg, 1.77 mmol, 2.5 equ.) and cellulose sulfuric acid (0.015 g). Reaction mixture was was carried out at -5 to 0°C for 60 min, and then stirred at room temperature for 1.5 h. Then we added a solution of sodium bicarbonate to quenched the reaction. The slurry was stirred in an below 20 0 C for 1 h and allowed to settle for 30 min. Solid crude arteether was collected by filtration, and the cake was washed with of ethanol. The reaction mass was heated to 40± 5 0 C in water. The reaction mass was seeded with pure β–arteether. Then it was filtered, washed with chilled 50% solution of ethanol in water and dried. General procedure for the artemether from artemisinin in one-pot. Artemisinin (200 mg, 0.71 mmol) in methanol (15 ml) and trimethylorthoformate (0.5 ml), cellulose sulfuric acid (0.015 g), was carried out at -5 to 0°C for 60 min, and then stirred at room temperature for 1.5 h. The reaction was monitored by TLC and HPLC to check completion of the reaction. The cellulose sulfuric acid was removed by filtration, the filtrate was concentrated. Then we added a solution of sodium bicarbonate to terminate the reaction. Then, follow above recrystallization method.
Approximately, out of the 4 billion people suffering from malaria, 1-3 million, mostly children die every year worldwide. The rapidly spreading multidrug resistant parasite to standard quinoline based antimalarial drugs such as chloroquine and mefloquine based antimalarial complicate chemotherapy treatment of malaria patients.
Artemether is a methyl ether derivative of dihydroartemisinin. Dihydroartemisinin is derived from arternisinin, a novel sesquiterpene endoperoxide isolated from the plant Artemisia annua. Artemisinin and its derivative artemether, arteether, artelinate and artesunate a novel class of antimalarials derived from Artemisia annua are now proving their promising activity and being used for the treatment; of uncomplicated severe complicated/cerebral and multi drug resistant malaria.
Artemether, developed in France and China has undergone extensive preclinical, animal, toxicological studies as well as clinical studies. Artemether is more potential as compared to artemisinin and an antimalarial drug especially for treating multi drug resistant and complicated strains of Plasmodium falciparum.
Artemether shows rapid shizonticidal action with quicker parasite clearance rate, short half life less side effect and low recrudence rate. Brossi, et al (Brossi, A; Venugopalan, B, Domingueg, G L; Yeh, H. J. C; Flippend-Anderson, J. L.; Buchs, P; Luo, X. D.; Milhous,W and peters, W; J. Med. Chem. 31, 646-649, 1988) reported the preparation of arteether, the ethyl ether derivative of dihydroartemisinin in two steps: First artemisinin was reduced with an excess of sodium borohydride in methanol at 0 to −5 degree C. in 3 hours to dihydroartemisinin in 79% yield. In the second step arteether is prepared by dissolving the dihydroartemisinin in the solvent mixture of benzene and ethanol at 45 degree C. followed by addition of BF3 etherate and refluxing the reaction mixture at 70 degree C. for one hour. After completion of the reaction it was worked up, dried over anhydrous sodium sulphate with removal of the solvent dichloromethane. The reaction yielded arteether along with some impurities. Column chromatography of the reaction mixture over silica gel, 1:20 ratio yielded pure alpha and beta arteether in nearly qualitative yield.
EL-Feraly etal. (E L Feraly, F. S; Al-Yahya M A; Orabi, K. Y; Mc-Phail D R and Me Phail A. T. J.Nat.Prod. 55, 878-883 1992) reported the preparation of arteether by a process in which anhydrodihydroartemisinin, prepared from artemisinin was dissolved in absolute alcohol. The reaction mixture was stirred in the presence of p-toluene sulphonic acid used as a catalyst. On workup it yielded a mixture of beta arteether and C-11 epimer in the ratio of 3:1. In this process only beta arteether, is obtained and separation of C-11 epimer is difficult and preparation of anhydrodihydroartemisinin is a tedious process. The reaction took 22 hours to complete. The lewis acid catalyst used in this reaction is required in large amount (60 mg. acid catalyst by 100 mg. anhydrodihydroartemisinin).
In another method Bhakuni etal (Bhakuni, R. S.; Jain D. C and Sharma R. P. Indian. J. Chemistry, 34B, 529-30, 1995) arteether, artemether and other ether derivatives were prepared from dihydroartemisinin in different alcohol and benzene in the presence of chlorotrimethylsilane catalyst in 2-4 hours at room temperature. After workup of the reaction mixture and removal of the solvent, the impure reaction products were purified over silica gel column to obtained the pure mixture of alpha, beta ethers.
Another method is reported by Lin et al. (Lin, A. J. and Miller, R. E, J.Med Chero. 38,764-770, 1995) In this method the new ether derivatives were prepared by dissolving dihydroarternisinin in anhydrous ether and appropriate alcohol followed by BF3-etherate. The reaction mixture was stirred at room temperature for 24 hours. The yield of the purified products ranged from 40-90%. Purification was achieved by the use of silica gel chromatography.
Another method described by Jain et al (Jain D. C, Bhakuni R. S, Saxena S, kumar, S and Vishwakarma, R. A.) the preparation of arteether from artemisinin comprises: Reduction of artemisinin into dihydroartemisinin. Isolation of dihydroartemisinin. Acylation of dihydroartemisinin by dissolving it in alcohol and adding trialkylorthoformate in the reaction mixture, which produce ethers in quantitative yield in 10 hours at 40 degree C.
The above mentioned methods carry some disadvantages being less cost effective and more time consuming as compared to the present invention. Moreover, benzene, a carcinogenic solvent, used in the previous methods is not acceptable according to the health standard. Further, all the above methods require at least two separate steps to convert artemisinin into ethers i.e. reduction of the artemisinin into dihydroartemisinin in the first pot followed by isolation of dihydroartemisinin and then comes the second step of conversion of dihydroartemisinin into different ethers in the second pot. However, the present invention provide an efficient method for conversion of artemisinin into artemether
EXAMPLE 1
Artemisinin (3 g.) was dissolved in dry methanol (40 ml) at room temperature. It was cooled to −5 degree C. Now sodium borohydride (700 mg) was added slowly for 30 minutes and the reaction mixture was stirred for about 1.5 hours. The reaction was monitored by TLC to check completion of the reduction step. Now cation exchange resin (8 g) was added slowly at cooling temperature and the reaction mixture was further stirred at room temperature for about 2 hours. Cooled water was added to the reaction mixture and the resin was filtered.
The filtrate was neutralized with 5% sodium bicarbonate solution followed by extracting with dichloromethane (3×50 ml). The dichloromethane extract was dried over anhydrous sodium sulphate and evaporation of the solvent yielded 3.21 g, of artemether along with some impurities. The impure artemether was purified over silica gel column (1:5 ratio) in hexane:ethyl acetate (96:4) furnished pure alpha and beta artemether 2.43 g (81% w/w). Small portion of artemether was separated by prep TLC into alpha and beta isomers and characterized by the analysis of their IR, Mass and 1H NMR data.
EXAMPLE 2
The experiment was carried out following example 1 except in place of solid acid catalyst in the second reaction. Liquid acid catalyst chlorotrimethylsilane was added at cooling temperature for methylation reaction. The overall yield of pure alpha, beta artemether after column chromatography was 2.46 gm (82% w/w).
EXAMPLE 3
Artemisinin (100 g.) was dissolved in dry methanol (3 ml). Added sodium borohydride (30 mg.) at −5° C. The reaction mixture was stirred for 2 hours. After completion of the reaction, trifluroacetic acid (0.5 ml) was added and the reaction mixture was stirred for 5 hours. The methylation was incompleted and after workup the artemether was purified by prep TLC to yield 46 mg (46%) pure alpha, beta artemether.
EXAMPLE 4
The experiment was carried following example 1 except before column chromatography, the beta isomer (40%) was recrystallized in hexane from impure artemether and remaining mother liquor was purified over silica gel column in 1:5 ratio to yield alpha and beta artemether in 80% w/w.
The earlier developed flow protocol for stoichiometric reduction of an important biologically derived pharmaceutical precursor, artemisinin, to dihydroartemisinin was extended to a sequential reaction to produce one of the final APIs, artemether. A highly active heterogeneous catalyst was found for the etherification reaction. The use of QuadraSil catalyst allows to eliminate one step of reaction workup. A comparative Life Cycle Assessment of both reactions has shown advantages of the flow process over the optimized literature batch protocols. Results of LCA highlight the significance of solvents in pharmaceuticals manufacture and the advantage of flow technology, enabling small solvent inventories to be used.
In a previous episode chemical company Sanofi was granted exclusive access to certain yeast cells that produce a precursor to anti-malarial drug artemisinin. One of the charities making this all possible is the Bill and Melinda Gates Foundation. Another charity that has apparently entered into the drug business is the Clinton Health Access Initiative. Bill together with Rodger Stringham and David Teager report on an improved process for the conversion of artemisinin to artemether in Organic Process Research & Development (DOI). Does the Clinton Health Access Initiative have a pilot-plant facility or even an organic lab? Unless it is all cramped in suite 400 on Dorchester Avenue in Boston, the article is not very explicit. The acknowledgements mention Mangalam Drugs and Organics. Case at hand: artemether has the carbonyl group replaced by a methoxy group in a two-step reduction – methylation. So far so good. The point is that principal supplier Novartis reports up to 68% overall yields but that many Indian and Chinese suppliers working with the procedure generously supplied by same Novartis, report considerably lower figures (58-62%). But Why? And how can the process be improved? Any organic chemist knows reported yields in the literature should be considered with caution. Chemists tend to be over-optimistic / self-delusionional but this scenario was not considered. No bottlenecks were encountered in step 1, the reduction with sodium borohydride. Only the beta form was isolated due to its poor solubility in the quench. Drying the product without heat prevented formation of one byproduct. Moving on to step two, the methylation with HCl in methanol was more troublesome. The byproducts lurking around the corner are the anomer and the elimination product. Co-solvent (co-reagent?) trimethyl orthoformate made all the difference. The critical element in the workup was first adding more methanol before adding the base quench otherwise you end up with a nasty gum. The new record yield for the improved synthesis is 72%. But what have all these suppliers been doing wrong with the existing Novartis procedure? The answer to that question, remains unclear. The Novartis yield for step two with co-solvent methylacetate (not the formate) was confirmed so no surprise there.
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Artemether is an antimalarial drug for uncomplicated malaria caused by P. falciparum (and chloroquine-resistant P. falciparum) or chloroquine-resistant P. vivax parasites.[8] Artemether can also be used to treat severe malaria.[2]
Artemether can also be used in treating and preventing trematode infections of schistosomiasis when used in combination with praziquantel.[11]
Artemether is rated category C by the FDA based on animal studies where artemisinin derivatives have shown an association with fetal loss and deformity. Some studies, however, do not show evidence of harm.[12][13]
Side effects
Possible side effects include cardiac effects such as bradycardia and QT interval prolongation.[14] Also, possible central nervous system toxicity has been shown in animal studies.[15][16]
Drug interactions
Plasma artemether level was found to be lower when the combination product was used with lopinavir/ritonavir.[16] There is also decreased drug exposure associated with concurrent use with efavirenz or nevirapine.[17][18]
Artemether/lumefantrine should not be used with drugs that inhibit CYP3A4.[19]
Hormonal contraceptives may not be as efficacious when used with artemether/lumefantrine.[19]
Artemether interact with ferriprotoporphyrin IX (heme) or ferrous ions in the acidic parasite food vacuole, and generates cytotoxic radical species
The accepted mode of action of the peroxide containing drug involve its interaction with heme (byproduct of hemoglobin degradation), derived from proteolysis of haemoglobin. This interaction results in the formation of toxic oxygen and carbon centered radicals.
One of the proposed mechanisms is that through inhibiting anti-oxidant and metabolic enzymes, artemisinin derivatives inflict oxidative and metabolic stress on the cell. Some pathways affected may concern glutathione and glucose metabolism. As a consequence, lesions and reduced growth of the parasite may result.[20]
Another possible mechanism of action suggests that arteristinin drugs exert their cidal action through inhibiting PfATP6. Since PfATP6 is an enzyme regulating cellular calcium concentration, its malfunctioning will lead to intracellular calcium accumulation, which in turns causes cell death.[21]
Pharmacokinetics
Absorption of artemether is improved 2- to 3-fold with food. It is highly bound to protein (95.4%). Peak concentrations of artemether are seen 2 hours after administration.[4]
Artemether is metabolized in the human body to the active metabolite, dihydroartemisinin, primarily by hepatic enzymes CYP3A4/5.[4] Both the parent drug and active metabolite are eliminated with a half-life of about 2 hours.[4]
Chemistry
Artemether is a methylether derivative of artemisinin, which is a peroxide-containing lactone isolated from the antimalarial plant Artemisia annua. It is also known as dihydroartemisinin methyl ether, but its correct chemical nomenclature is (+)-(3-alpha,5a-beta,6-beta,8a-beta, 9-alpha,12-beta,12aR)-decahydro-10-methoxy-3,6,9-trimethyl-3,12-epoxy-12H-pyrano(4,3-j)-1,2-benzodioxepin. It is a relatively lipophilic and unstable drug,[22] which acts by creating reactive free radicals in addition to affecting the membrane transport system of the plasmodium organism.[14]
^“Artemether and Lumefantrine”. The American Society of Health-System Pharmacists. Archived from the original on 20 December 2016. Retrieved 28 November 2016.
^World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. 2019. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^ Piola P, Nabasumba C, Turyakira E, et al. (2010). “Efficacy and safety of artemether—lumefantrine compared with quinine in pregnant women with uncomplicated Plasmodium falciparum malaria: an open-label, randomised, non-inferiority trial”. Lancet Infect Dis. 10 (11): 762–769. doi:10.1016/S1473-3099(10)70202-4. hdl:10144/116337. PMID20932805.
^ Kiang, Tony K. L.; Wilby, Kyle J.; Ensom, Mary H. H. (2013-10-26). “Clinical Pharmacokinetic Drug Interactions Associated with Artemisinin Derivatives and HIV-Antivirals”. Clinical Pharmacokinetics. 53 (2): 141–153. doi:10.1007/s40262-013-0110-5. ISSN0312-5963. PMID24158666. S2CID1281113.
^ Saeed, ME; Krishna, S; Greten, HJ; Kremsner, PG; Efferth, T (August 2016). “Antischistosomal activity of artemisinin derivatives in vivo and in patients”. Pharmacological Research. 110: 216–26. doi:10.1016/j.phrs.2016.02.017. PMID26902577.
The most common side effects include respiratory tract infection, viral infection, diarrhea, dyspepsia (indigestion), cough, arthralgia (joint stiffness), arthritis, and swelling in the lower legs and hands.[2]
Sutimlimab prevents complement-enhanced activation of autoimmune human B cells in vitro.[4]
This drug is being developed by Bioverativ, a Sanofi company.[5] Sutimlimab was approved for medical use in the United States in February 2022.[2][6]
Sutimlimab-jome, a classical complement inhibitor, is a humanized monoclonal antibody expressed by recombinant in Chinese hamster ovary (CHO) cells and produced in vitro using standard mammalian cell culture methods. Sutimlimab-jome is composed of two heterodimers. Each heterodimer is composed of a heavy and a light polypeptide chain. Each heavy chain (H-chain) is composed of 445 amino acids and each light chain (L-chain) contains 216 amino acids. Sutimlimab-jome has a molecular weight of approximately 147 kDa.
ENJAYMO (sutimlimab-jome) injection is a sterile, clear to slightly opalescent, colorless to slightly yellow, preservative-free solution for intravenous use. Each single-dose vial contains 1,100 mg sutimlimab-jome at a concentration of 50 mg/mL with a pH of 6.1. Each mL contains 50 mg of sutimlimab-jome and also contains polysorbate 80 (0.2 mg), sodium chloride (8.18 mg), sodium phosphate dibasic heptahydrate (0.48 mg), sodium phosphate monobasic monohydrate (1.13 mg), and Water for Injection, USP. https://www.rxlist.com/enjaymo-drug.htm#clinpharm
Medical uses
Sutimlimab is indicated to decrease the need for red blood cell transfusion due to hemolysis (red blood cell destruction) in adults with cold agglutinin disease (CAD).[1][2]
History
The effectiveness of sutimlimab was assessed in a study of 24 adults with cold agglutinin disease who had a blood transfusion within the past six months.[2] All participants received sutimlimab for up to six months and could choose to continue therapy in a second part of the trial.[2] Based on body weight, participants received either a 6.5g or 7.5g infusion of sutimlimab into their vein on day 0, day 7, and every 14 days through week 25.[2]
In total, 54% of participants responded to sutimlimab.[2] The response was defined in the study as an increase in hemoglobin (an indirect measurement of the amount of red blood cells that are not destroyed) of 2 g/dL or greater (or to 12 g/dL or greater), and no red blood cell transfusions after the first five weeks of treatment; and no other therapies for cold agglutinin disease as defined in the study.[2]
FDA approves Enjaymo (sutimlimab-jome), first treatment for use in patients with cold agglutinin disease
Enjaymo is the only approved treatment to decrease the need for red blood cell transfusion due to hemolysis, the destruction of red blood cells, in adults with cold agglutinin disease (CAD)
Enjaymo addresses a serious and chronic unmet medical need for adults living with CAD, a rare blood disorder
Paris, February 4, 2022. The U.S. Food and Drug Administration (FDA) has approved Enjaymo (sutimlimab-jome) to decrease the need for red blood cell transfusion due to hemolysis in adults with cold agglutinin disease (CAD). Enjaymo is the first and only approved treatment for people with CAD and works by inhibiting the destruction of red blood cells (hemolysis).
Bill Sibold Executive Vice President, Head of Specialty Care “Until now, people living with cold agglutinin disease haven’t had an approved treatment option to manage the constant destruction of red blood cells. Without healthy, viable red blood cells, a chain reaction of debilitating signs and symptoms can be triggered, starting with severe anemia. Enjaymo is the only approved treatment to inhibit red blood cell destruction in CAD and help stop the chain reaction from the start.”
CAD, a rare autoimmune hemolytic anemia, is caused by antibodies called cold agglutinins binding to the surface of red blood cells, which starts a process that causes the body’s immune system to mistakenly attack healthy red blood cells and cause their rupture (hemolysis). As red blood cells have the vital job of carrying oxygen throughout the body, patients with CAD may experience severe anemia, which can result in fatigue, weakness, shortness of breath, light-headedness, chest pain, irregular heartbeat, and other potential complications. CAD is a chronic and rare blood disorder that impacts the lives of an estimated 5,000 people in the U.S.
Enjaymo, targeting C1s in the classical complement pathway
Enjaymo is a humanized monoclonal antibody that is designed to selectively target and inhibit C1s in the classical complement pathway, which is part of the innate immune system. By blocking C1s, Enjaymo inhibits the activation of the complement cascade in the immune system and inhibits C1-activated hemolysis in CAD to prevent the abnormal destruction of healthy red blood cells. Enjaymo does not inhibit the lectin and alternative pathways.
Enjaymo Phase 3 pivotal CARDINAL study results supporting approval
The approval of Enjaymo in the U.S. is based on positive results from the 26-week open label, single arm pivotal Phase 3 study in patients with CAD (n=24) who have a recent history of blood transfusion, also known as the CARDINAL study.
Catherine Broome, MD Associate professor of medicine at Georgetown University Lombardi Comprehensive Cancer Center, and a principal investigator in the CARDINAL study “For people living with cold agglutinin disease, it is as if their body’s immune system is waging a war on itself. The relentless destruction of healthy red blood cells is a daily, silent reality for people with CAD.For the first time, we have a treatment that targets complement-mediated hemolysis, which is the underlying cause of the red blood cell destruction in many CAD patients. In thepivotal study, patients treated with sutimlimab had an improvement in anemia as measured by hemoglobin and bilirubin levels during the 26-week study.”
In the study, Enjaymo met its primary efficacy endpoint, which was a composite endpoint defined as the proportion of patients who achieved normalization of hemoglobin (Hgb) level ≥12 g/dL or demonstrated an increase from baseline in Hgb level ≥2 g/dL at the treatment assessment time point (mean value from weeks 23, 25, and 26) and no blood transfusion from weeks 5 through 26 or medications prohibited per the protocol from weeks 5 through 26. Secondary endpoints were also met, including improvements in hemoglobin and normalization of bilirubin.
The majority of patients (54%; n=13) met the composite primary endpoint criteria with 63% (n=15) of patients achieving a hemoglobin ≥ 12 g/dL or an increase of at least 2 g/dL; 71% (n=17) of patients remaining transfusion-free after week five; and 92% (n=22) of patients did not use other CAD-related treatments.
For the secondary measures on disease process, patients enrolled experienced a mean increase in hemoglobin level of 2.29 g/dL (SE: 0.308) at week 3 and 3.18 g/dL (SE: 0.476) at the 26-week treatment assessment timepoint from the mean baseline level of 8.6 g/dL. The mean reduction in bilirubin levels (n=14) was by -2.23 mg/dL (95% CI: -2.49 to -1.98) from a mean baseline level of 3.23 mg/dL (2.7-fold ULN).
In the CARDINAL study, the most common adverse reactions occurring in 10 percent or more of patients were respiratory tract infection, viral infection, diarrhea, dyspepsia, cough, arthralgia, arthritis, and peripheral edema. Serious adverse reactions were reported in 13 percent (3/24) of patients who received Enjaymo. These serious adverse reactions were streptococcal sepsis and staphylococcal wound infection (n=1), arthralgia (n=1), and respiratory tract infection (n=1). None of the adverse reactions led to discontinuation of Enjaymo in the study. Dosage interruptions due to an adverse reaction occurred in 17 percent (4/24) of patients who received Enjaymo.
Following the completion of the 26-week treatment period of CARDINAL (Part A), eligible patients continued to receive Enjaymo in an extension study.
The recommended dose of Enjaymo is based on body weight (6,500 mg for people 39-75 kg and 7,500 mg for people >75 kg). Enjaymo is administered intravenously weekly for the first two weeks with administration every two weeks thereafter.
Enjaymo is expected to be available in the U.S. in the coming weeks. The U.S. list price, or wholesale acquisition cost, of Enjaymo is $1,800 per vial. Actual costs to patients are generally anticipated to be lower as the list price does not reflect insurance coverage, co-pay support, or financial assistance from patient support programs. As part of our commitment to ensure treatment access and affordability for innovative therapies, Enjaymo Patient Solutions provides disease education, financial and co-pay assistance programs and other support services to eligible patients. For more information, please call 1-833-223-2428.
Enjaymo received FDA Breakthrough Therapy and Orphan Drug designation, and priority review, which is reserved for medicines that, if approved, would represent significant improvements in safety or efficacy in treating serious conditions. Outside of the U.S., sutimlimab has been submitted to regulatory authorities in Europe and Japan and reviews are ongoing.
About Sanofi We are an innovative global healthcare company, driven by one purpose: we chase the miracles of science to improve people’s lives. Our team, across some 100 countries, is dedicated to transforming the practice of medicine by working to turn the impossible into the possible. We provide potentially life-changing treatment options and life-saving vaccine protection to millions of people globally, while putting sustainability and social responsibility at the center of our ambitions. Sanofi is listed on EURONEXT: SAN and NASDAQ: SNY
^ Tvedt TH, Steien E, Øvrebø B, Haaverstad R, Hobbs W, Wardęcki M, et al. (February 2022). “Sutimlimab, an investigational C1s inhibitor, effectively prevents exacerbation of hemolytic anemia in a patient with cold agglutinin disease undergoing major surgery”. American Journal of Hematology. 97 (2): E51–E54. doi:10.1002/ajh.26409. PMID34778998. S2CID244116614.
^World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 80”. WHO Drug Information. 32 (3). hdl:10665/330907.
External links
“Sutimlimab”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03347396 for “A Study to Assess the Efficacy and Safety of BIVV009 (Sutimlimab) in Participants With Primary Cold Agglutinin Disease Who Have a Recent History of Blood Transfusion (Cardinal Study)” at ClinicalTrials.gov
//////////////Sutimlimab-jome, Enjaymo, FDA 2022, APPROVALS 2022, agglutinin disease, BIVV009, TNT009, UNII-GNWE7KJ995, WHO 10757, PEPTIDE, MONOCLONAL ANTIBODY, スチムリマブ (遺伝子組換え),
Fabomotizole dihydrochloride CAS#: 189638-30-0 (2HCl) Chemical Formula: C15H23Cl2N3O2S
Molecular Weight: 380.33
Fabomotizole (also known as Afobazole) is a selective non-benzodiazepine anxiolytic which was developed in Russia and launched in 2006. The drug is used for the treatment of wide range of diseases: generalized anxious disorders, neurasthenia, adaptation disorders, sleep disorders, for alleviation of withdrawal syndrome. According to the drug label (in Russian), its action is related to the interaction with sigma-1 receptors.
Fabomotizole (INN;[1] brand name Afobazole) is an anxiolyticdrug launched in Russia in the early 2000s. It produces anxiolytic and neuroprotective effects without any sedative or muscle relaxant actions.[citation needed] Its mechanism of action remains poorly defined however, with GABAergic, NGF– and BDNF-release-promoting, MT1 receptor agonism, MT3 receptor antagonism, and sigma agonism suggested as potential mechanisms. Fabomotizole was shown to inhibit MAO-A reversibly and there might be also some involvement with serotonin receptors.[2][3][4][5][6] Clinical trials have shown fabomotizole to be well tolerated and reasonably effective for the treatment of anxiety.[7]
Experiments of mice have shown antimutagenic and antiteratogenic properties.[8]
Fabomotizole has found little clinical use outside Russia and has not been evaluated by the FDA.
A ligand-based virtual screening study to search for giardicidal compounds on a 6551 ChEMBL drugs database was carried out using molecular similarity. Three fingerprints implemented in MayaChemTools with different design and validated by ROC curves, were used. Twelve compounds were retrieved from this screening, from which, four representative compounds were selected to carry out biological assays. Whereas two compounds were commercially available, the additional two compounds were synthesized during the development of this work. The biological assays revealed that the compounds possess in vitro activity against five strains of Giardia intestinalis, each with different susceptibility/resistance rates to metronidazole, albendazole and nitazoxanide. Particularly, tenatoprazole showed the best effect against the WB and IMSS strains. Furthermore, fabomotizole, tenatoprazole and ipriflavone showed a higher activity against resistant strains than the reference drugs: metronidazole, albendazole and nitazoxanide.
Graphical abstract
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
^ Neznamov, GG; Siuniakov, SA; Chumakov, DV; Bochkarev, VK; Seredenin, SB (2001). “Clinical study of the selective anxiolytic agent afobazol”. Eksperimental’naia i Klinicheskaia Farmakologiia. 64 (2): 15–9. PMID11548440.
^ Silkina, IV; Gan’shina, TC; Seredin, SB; Mirzoian, RS (2005). “Gabaergic mechanism of cerebrovascular and neuroprotective effects of afobazole and picamilon”. Eksperimental’naia i Klinicheskaia Farmakologiia. 68 (1): 20–4. PMID15786959.
^ Seredin, SB; Melkumian, DS; Val’dman, EA; Iarkova, MA; Seredina, TC; Voronin, MV; Lapitskaia, AS (2006). “Effects of afobazole on the BDNF content in brain structures of inbred mice with different phenotypes of emotional stress reaction”. Eksperimental’naia i Klinicheskaia Farmakologiia. 69 (3): 3–6. PMID16878488.
^ Antipova, TA; Sapozhnikova, DS; Bakhtina, LIu; Seredenin, SB (2009). “Selective anxiolytic afobazole increases the content of BDNF and NGF in cultured hippocampal HT-22 line neurons”. Eksperimental’naia i Klinicheskaia Farmakologiia. 72 (1): 12–4. PMID19334503.
^ Seredenin, SB; Antipova, TA; Voronin, MV; Kurchashova, SY; Kuimov, AN (2009). “Interaction of afobazole with sigma1-receptors”. Bulletin of Experimental Biology and Medicine. 148 (1): 42–4. doi:10.1007/s10517-009-0624-x. PMID19902093. S2CID37411324.
^ Medvedev, VE; Trosnova, AP; Dobrovol’skiĭ, AV (2007). “Psychopharmacotherapy of anxiety disorders in patients with cardio-vascular diseases: the use of aphobazole”. Zh Nevrol Psikhiatr Im S S Korsakova. 107 (7): 25–9. PMID18379478.
Conventionally, 0-aryl N- (6-alkoxy-2-pyridyl) -N-alkylthio-force rubamate has generally been produced by a method using thiophosgen. For example, in Patent Document 1, 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N- represented by the following reaction formula 0 or ii) A method for producing methylthiolbamate (4) is disclosed.
To methanol (10 ml), 0.54 g (10.0 mmol) of sodium methoxide was added, and the mixture was stirred at room temperature. There, 1.50 g (10.0 mmol) of 5,6,7,8-tetrahydro-2-naphthol was added and he stirred for 1 hour at room temperature. The solvent was distilled off under reduced pressure to obtain 3.75 g ( q uant.) Of white powder. I left it overnight in a desiccator.
To ethyl acetate (30 ml), 2.07 g (15.0 mmol) of 6-methoxy-2-methylaminoviridin and 2.67 g (15.0 mmol) of 1,1, -thiocarboldiimidazole were added, and the mixture was heated under reflux for 2 hours. After allowing to cool, the solvent was distilled off under reduced pressure to obtain 3.70 g of brown oil. (Yield 99.3%). If necessary, further purification was performed by silica gel column chromatography (hexane: ethyl acetate = 10: 1) to obtain pale yellow crystals.
Dissolve 2- [N- (1-imidazolithiocarbol) -N-methyl] amino-6-methoxypyridin 250 mg (1.0 mmol) in N, N-dimethylformamide (4 ml), and then dissolve. At room temperature, Natrium 5, 6, 7, 8-tetrahydro-2-naphthoside 360 mg (2.0 mmol) was added. -After stirring at room temperature, the reaction solution was extracted with ethyl acetate (10 mlx2), and the insoluble material was filtered off on the way. The organic layer was washed with saturated brine, dried over magnesium sulfate, filtered off magnesium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (eco-gel C-200, hexane: ethyl acetate = 10: 1) gave the title compound 266.6 mg (yield 81.3%).
1.34 g (33.6 mmol) of 60% sodium hydride was added to N, N-dimethylformamide (20 ml), followed by the addition of 5, 6, 7, 8-tetrahydro-2-naphthol 4.65 g (30.5 mmol). After gas generation is complete, add 2- [N- (1-imidazolthiocarbonyl) -N-methyl] amino-6-methoxypyridin 7.45 g (30.0 mmol) and zinc chloride 2.05 g (15.0 mmol). rice field. After heating and stirring at 60 ° C for 3 hours and allowing to cool, the reaction solution was extracted with ethyl acetate (150 mlx2), and the insoluble material was filtered off on the way. The organic layer is washed with saturated brine, dried over magnesium sulfate, and filtered through magnesium sulfate.
Separately, the solvent was distilled off under reduced pressure. The obtained crystals were purified by one of the following methods.
[0028] A) Purification was performed by silica gel column chromatography (eco-gel C 200, hexane: ethyl silicate = 10: 1) to obtain 9.80 g of the indicated compound (yield 99.5%).
B) Suspended in hexane (10 ml), stirred for 30 minutes, and then the crystals were collected by filtration to obtain 9.65 g of crystals. Further, the mixture was suspended in methanol (10 ml), stirred for 30 minutes, and then the crystals were collected by filtration to obtain 8.62 g (yield 87.5%) of the indicated compound.
The physics and physics data of the obtained compound were consistent with the compounds obtained in the examples.
(Example 3)
1) Synthesis of 2- [N- [1-2 (1H) -pyridonylthiocarbol] -N-methyl] amino-6-methoxypyridine
[Chemical 10]
OMe
Add 6-methoxy-2-methylaminoviridin 690 mg (5.0 mmol) and 1, 1, -thiocarbol-di-2 (1H) -pyridone 1.16 g (5.0 mmol) to ethyl acetate (15 ml). Heated and refluxed for 1 hour. After allowing to cool, the solvent was distilled off under reduced pressure, and purification was performed by silica gel column chromatography (hexane: ethyl acetate = 10: 1)! ヽ, 297.4 mg of brown oil was obtained. (Yield 21.6%).
N, N-dimethylformamide (2 ml), 2- [N- [1-2 (1H) -pyridonylthiocarbol] –N-methyl] amino-6-methoxypyridin 297 mg (1.08 mmol) and sodium 5 , 6, 7, 8-Tetrahydro-2-naphthoside 390 mg (2.16 mmol) was added and stirred overnight at room temperature. The reaction mixture was extracted with ethyl acetate (50 mlx2), the organic layer was washed with saturated brine, dried over magnesium sulfate, magnesium sulfate was filtered off, and the solvent was distilled off under reduced pressure. The obtained crystals were purified by silica gel column chromatography (eco-gel C-200, hexane: ethyl acetate = 10: 1) to obtain the title compound 288.2 mg (81.4%).
Liranafate is a new-generation antifungal drug, a squalene cyclooxygenase inhibitor and a cell wall synthesis inhibitor, with the chemical name of 6-methoxy-2-N-methyl-pyridylamino-thio Formic acid-(5,6,7,8-tetrahydro)-β-naphthyl ester. A new type of antifungal drug jointly developed by Tosoh Corporation of Japan and Zenyaku Kogyo Corporation was first listed in Japan by Torii Corporation in August 2000. The antifungal drug exerts antifungal activity by inhibiting the squalene epoxidation reaction of fungal cells and inhibiting the synthesis of ergosterol, a component of cell membranes. effect is particularly evident. Today, with the increasing concern of the world about environmental pollution, the development of new green and effective drug synthesis methods is an important task faced by the research of drug synthesis. In recent years, room temperature ionic liquids have been widely used in various organic synthesis reactions as a new type of environmentally friendly reaction media. Compared with traditional organic solvents, ionic liquids have many advantages, such as extremely low vapor pressure, non-flammability, good thermal stability and recyclability.
At present, the main synthetic route of liranaftate is as follows:
Among the four synthetic routes, the pyridine derivative intermediates of routes C and D need to be prepared through multi-step reactions, the routes are long, the steps are cumbersome, the actual operation is cumbersome, the cost is high, and they are not suitable for industrialized large-scale production. Although route A has simple steps, the yield of pyridine derivatives is low. Each intermediate structure in route B is relatively simple and easy to prepare, but this route uses 6-methoxy-2-methylaminopyridine and 5,6,7,8-tetrahydro-2-naphthoxysulfuryl chloride as raw materials to synthesize the In the process of lanaphthalate, isopropanol-water is used as the reaction medium, and the experiment shows that with the progress of the reaction, the reaction solution becomes viscous, and the reaction is difficult to complete.
Example 1
(1) Ionic liquid [bmim]BF 4 Synthesis
Add N-methylimidazole (14.8g, 0.18mol) and trichloroethane (80mL) to a dry 250mL three-neck flask, stir to make the mixture uniform, add 20.4mL of freshly distilled n-bromine to the dropping funnel Butane (26.03g, 0.19mol) was added dropwise for about 30min, and the reaction was refluxed for 4-5h (the reflux temperature was about 78±1℃). With the progress of the reaction, the reaction solution changed from colorless and transparent to white turbidity, light yellow turbidity, and the color gradually became darker until brownish red. After the reaction is completed, the liquids are separated into layers, the upper layer is lighter in color, which is the trichloroethane layer, and the lower layer is darker in color (brown red), which is the ionic liquid [bmim]Br layer. The prepared ionic liquid [bmim]Br and trichloroethane were separated, and the ionic liquid [bmim]Br was washed twice with trichloroethane, and then the trichloroethane in the ionic liquid [bmim]Br was washed with a water pump. The alkane was pumped away until the ionic liquid [bmim]Br liquid was no longer turbid, and then dried in a vacuum drying oven at 90 °C for 10-12 h to obtain relatively pure ionic liquid [bmim]Br.
Then prepare 0.03mol NaBF 4 of aqueous solution. Add 6.58g (about 0.03mol) ionic liquid [bmim]Br and 5-10mL water to a 100mL round-bottomed single diameter flask, stir, ice-water bath, and dropwise add NaBF 4 The solution (completed dropwise addition in about 5min), continue to stir for 10-20min, the solution is yellow and transparent, pour it into a separatory funnel, extract twice with dichloromethane, combine the dichloromethane layers, and wash the dichloromethane layer 2 with 50 mL of water times, and then the dichloromethane layer was washed with anhydrous MgSO 4 Dry, filter, evaporate the dichloromethane under normal pressure in a water bath (50-52°C), and dry the remaining dark yellow viscous liquid in a vacuum drying oven at 90°C for 10-12h to obtain the ionic liquid [bmim]BF 4 。
(2) Synthesis of 6-methoxy-2-chloropyridine 2
2,6-dichloropyridine (10g, 0.068mol) and sodium methoxide (24.5g, 0.136mol) were put into the reaction flask, heated under reflux for 4-5h, and the reaction was completed by TLC (ethyl acetate: petroleum ether=1 : 15), concentrated to remove methanol, added 100 mL of water, extracted with ethyl acetate, combined the organic phases, washed with saturated brine, dried, filtered, and the filtrate was concentrated to obtain 9 g of a crude colorless oily product with a yield of 92.5%. used for the next reaction.
(3) Synthesis of 6-methoxy-2-methylaminopyridine 3
Take 6-methoxy-2-chloropyridine 2 (9g, 0.127mol), cuprous chloride (1.72g, 0.0017mol) and methylamine aqueous solution (29mL, mass concentration is 25%-30%) and add it to the autoclave , sealed and heated to 120 °C for 7 h, the reaction was stopped, ethyl acetate was added for extraction, the organic phases were combined, washed with saturated brine, dried, and the filtrate was concentrated to obtain 6.18 g of brown oil, the yield was 71.2%, and the HPLC purity was 98% .
(4) Synthesis of 5,6,7,8-tetrahydro-2-naphthyloxysulfuryl chloride 4
Mix 50 mL of ethyl acetate, thiophosgene (4.25 mL, 0.056 mol) and 5,6,7,8-tetrahydro-2-naphthol (6.3 g, 0.0425 mol), and cool it in an ice-salt bath to below 0 °C. Add 10 mL of potassium carbonate (3 g, 0.022 mol) solution, continue to stir the reaction after the dropwise addition, and check by TLC (developing solvent: petroleum ether) that the reaction is complete, add 100 mL of water, extract with ethyl acetate, wash the organic phase with saturated brine, Dry, filter, and concentrate the filtrate to obtain 8.7 g of yellow oil with a yield of 90.4%, which can be directly used in the next reaction without purification.
(5) Synthesis of Liranaftate 1
The prepared ionic liquid [bmim]BF 4 (100mL), 6-methoxy-2-methylaminopyridine 3 (5.7g, 0.0413mol) and potassium carbonate (5.7g, 0.0413mol) were mixed, cooled with ice water, and slowly added dropwise 5,6,7,8 -Tetrahydro-2-naphthyloxysulfuryl chloride 4 (8.7g, 0.0385mol) was added dropwise for 4h, slowly added 150mL of water under full stirring, continued to stir for 20min, filtered, washed with deionized water to obtain 12.2g of crude product, collected The yield was 96.81%, and acetone was recrystallized to obtain 11 g of white crystalline powder, the yield was 90%, and the HPLC purity was 99.7%. mp: 98.8-99.5°C, IR (2973cm -1 , 2930cm -1 , 2852cm -1 , 1416cm -1 , 1264cm -1 , 1037cm -1 ), 1 HNMR: 1.8 (m, 4H); 6.68(d, 1H) ;6.86(dd,1H);3.78(s,3H);3.98(s,3H);6.68(d,1H);6.86(dd,1H);7.05(d,1H);7.10(d.1H); 7.65 (dd, 1H), MS (m/z: 328, 181, 165, 108).
Example 2
Under the same conditions, the ionic liquid 1-n-butyl-3-methylimidazolium tetrafluoroborate ([bmim]BF 4 ), N-ethylpyridine tetrafluoroborate ([EPy]BF 4 ), 1-n-butyl-3-methylimidazolium hexafluorophosphate ([bmim]PF 6 ), 1-hydroxyethyl-2,3-dimethylimidazolium chloride (LOH), 1-cyanopropyl-3-methylimidazolium chloride (LCN), 1-carboxyethyl-3-methylimidazole Chloride salt (LOOH), [Hnmp]HSO 4 The effects of and [bmim]OH on the synthesis of liranaftate are shown in Table 1. The results show that different ionic liquids have little effect on the yield of the synthesis and the yields are relatively high.
Table 1 Effects of different ionic liquids on the reaction yield
Whether the reaction medium used can be recovered and reused is an important content of “green chemistry”. This example specifically examines the reuse of ionic liquid for synthesizing liranaftate. After 5 times of use of ionic liquid, the product yield It just started to decrease, which shows that the ionic liquid can be recovered and reused effectively, and the reuse performance is good. It is a recyclable green solvent.
Put 10 g of 2,6-dichloropyridine, 100 ml of methanol, and 15 g of sodium methoxide into a reaction flask, heat under reflux for about 4 to 5 hours, concentrate to remove methanol, add 150 ml of water, extract with ethyl acetate, and concentrate under reduced pressure to remove ethyl acetate. 6-Methoxy2-chloropyridine was obtained as a colorless oil.
9 g of 6-methoxy 2-chloropyridine, 1.72 g of cuprous chloride, and 29 ml of 30% methylamine aqueous solution were put into the reaction flask, heated and added with a mass fraction of 11.6 g of cuprous chloride, and the temperature was kept at 120 ° C for the reaction 8h, extracted three times with 150 ml of ethyl acetate, washed with saturated brine, concentrated under reduced pressure to remove the ethyl acetate to obtain 6.18 g of 6-methoxy-2-methylaminopyridine as a brown oily product. The two-step yield was 71.2%.
50ml of carbon tetrachloride, 4.25g of thiophosgene, 6.3g of 5,6,7,8-tetrahydro-2-naphthol were added to the reaction flask, the ice-salt bath was lowered to below 0°C, and 10ml of 3g potassium carbonate aqueous solution was added dropwise. , Continue the reaction at 0°C after the dropwise addition, and detect by TLC (developing solvent: petroleum ether) after the reaction is completed, separate the organic phase, wash three times with saturated brine, and concentrate under reduced pressure to obtain red oily products 5, 6, 7 , 8.7g of 8-tetrahydro-2-naphthyloxysulfuryl chloride was directly used in the next reaction.
100ml of acetone, 5.7g of 6-methoxy-2-methylaminopyridine and 5.7g of potassium carbonate were added to the reaction flask, cooled with ice water, and 5,6,7,8-tetrahydro-2-naphthyloxysulfuryl chloride was added dropwise 8.7g, continue to stir and react for 4h after dropping, add 150ml of water, continue to stir for 30min, and filter to obtain the crude product. The crude product was recrystallized with acetone to obtain 11 g of off-white crystalline powder. The weight yield was 174.6% based on 5,6,7,8-tetrahydro-2-naphthol. The maximum single impurity content determined by HPLC was 1.5%, which did not meet the requirements of the Pharmacopoeia.
A preparation method of liranaftate of the present invention comprises the following steps:
(1) preparation of Liranaftate crude product:
Feeding: 250g of absolute ethanol was added to the reaction flask, 12.5g of 2-methoxy-6-methylaminopyridine, 8.8g of anhydrous sodium carbonate and 31.3g of purified water were added to the reaction flask in turn, stirred for 30 minutes, slowly 18.8 g of 2-(5,6,7,8-tetrahydronaphthyloxy) thioformate chloride was added, and the addition was completed in 2 hours;
Reaction: control the temperature at 20°C for 2 hours, add 125.0g of purified water, and stir for 30 minutes;
Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, and the consumption of purified water was 25.0 g each time;
Drying: put the wet product into a drying box, control the temperature to 45 ℃ and dry for 4 hours, to obtain 24 g of the crude product of lira naphthate;
The synthesis yield is 81%;
(2) preparation of Liranaftate fine product:
Impurity removal: put 23g of Liranaftate crude product and 115g of absolute ethanol into the reaction flask, add 1.38g of medicinal charcoal, decolorize at 55°C under temperature control, remove impurities for 30 minutes, filter, transfer the filtrate to the reaction flask, control the temperature Crystallize at 55°C, centrifuge, dry, pulverize, and pack to obtain 22g of Lira naphthate fine product.
The purification yield was 92%.
Example 2
A preparation method of liranaftate of the present invention comprises the following steps:
(1) preparation of Liranaftate crude product:
Feeding: 500g of absolute ethanol was added to the reaction flask, 25g of 2-methoxy-6-methylaminopyridine, 17.6g of anhydrous sodium carbonate and 62.6g of purified water were added to the reaction flask in turn, stirred for 30 minutes, and slowly added 2-(5,6,7,8-tetrahydronaphthyloxy) chlorothioformate 37.6g, added in 2.5 hours;
Reaction: control the temperature at 25°C for 2.5 hours, add 250 g of purified water, and stir for 30 minutes;
Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, 50 g each time;
Drying: put the wet product into a drying box, control the temperature to 55 ℃ and dry for 4 hours to obtain 49 g of the crude product of lira naphthate;
The synthesis yield is 82%;
(2) preparation of Liranaftate fine product:
Impurity removal: put 49g of Liranaftate crude product and 245g of absolute ethanol into the reaction flask, add 2.9g of medicinal charcoal, decolorize at 55~65 ℃ of temperature, remove impurities for 30 minutes, filter, and transfer the filtrate to the reaction flask, The temperature was controlled at 65°C for crystallization, centrifugation, drying, pulverization, and packaging to obtain 45g of fine lanaftate.
The purification yield was 92%.
Example 3
A preparation method of liranaftate of the present invention comprises the following steps:
(1) preparation of Liranaftate crude product:
Feeding: 250g of absolute ethanol was added to the reaction flask, 12.5g of 2-methoxy-6-methylaminopyridine, 8.8g of anhydrous sodium carbonate and 31.3g of purified water were added to the reaction flask in turn, stirred for 30 minutes, slowly 18.8 g of 2-(5,6,7,8-tetrahydronaphthyloxy) thioformate chloride was added, and the addition was completed in 2 hours;
Reaction: control the temperature at 20°C for 2 hours, add 125.0g of purified water, and stir for 30 minutes;
Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, 25.0 g each time;
Drying: put the wet product into a drying oven, control the temperature to 45~55 ℃ and dry for 4 hours, to obtain the crude product, 23.3 g of the crude liranaftate;
The synthesis yield is 82%;
(2) preparation of Liranaftate fine product:
Removal of impurities: 140g of absolute ethanol was added to the reaction flask, 23.3g of crude liranaftate was added, the temperature was controlled at 50°C and stirred for 30 minutes, 1.5g of medicinal charcoal was added, the temperature was controlled at 60°C for decolorization for 30 minutes, filtered, and the temperature was controlled Crystallize at 60°C, centrifuge, dry, pulverize, and package to obtain 23g of Lira naphthate fines.
The purification yield was 92%.
SYN
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
^ Koga H, Nanjoh Y, Makimura K, Tsuboi R (2009). “In vitro antifungal activities of luliconazole, a new topical imidazole”. Medical Mycology. 47 (6): 640–7. doi:10.1080/13693780802541518. PMID19115136.
Percent Composition: C 52.15%, H 5.47%, N 7.60%, O 17.37%, S 17.40%
Literature References: Prepn: Zima, Schorre, US3010966 (1961 to E. Merck); Iwanami et al.,Bitamin36, 122 (1967); J. Vitaminol.14, 321, 326 (1968). HPLC determn in urine: K. Kitao et al.,Chem. Pharm. Bull.25, 1335 (1977). Pharmacokinetics and metabolism: Darge et al.,Arzneim.-Forsch.19, 5, 9, (1969); Nowak, Schorre, ibid. 11. Clinical trial in dementia: S. Hoyer et al.,ibid.27, 671 (1977); A. J. Cooper, R. V. Magnus, Pharmacotherapeutica2, 317 (1980); in cerebrovascular disorders: Y. Tazaki et al.,J. Int. Med. Res.8, 118 (1980).
Pyritinol has been used in trials studying the treatment of Dementia, Depression, Schizophrenia, Anxiety Disorders, and Psychosomatic Disorders.
Pyritinol also called pyridoxine disulfide or pyrithioxine (European drug names Encephabol, Encefabol, Cerbon 6) is a semi-synthetic water-soluble analog of vitamin B6 (Pyridoxine HCl). It was produced in 1961 by Merck Laboratories by bonding 2 vitamin B6 compounds (pyridoxine) together with a disulfide bridge. Since the 1970s, it has been a prescription and OTC drug in several countries for cognitive disorders, rheumatoid arthritis,[1] and learning disorders in children. Since the early 1990s it has been sold as a nootropicdietary supplement in the United States.
Pyritinol, it is the derivative of vitamin B6, for nootropic agents, can promote glucose and amino acid metabolism in brain, improve whole body assimilation, increase Flow of carotid artery, improve cerebral blood flow (CBF), be applicable to the dizzy distending pain, insomnia, hypomnesis of cerebral trauma sequela, encephalitis and meningitis sequela etc., the improvement of absent minded, emotional change; Also for cerebral arteriosclerosis, senile dementia mental symptom etc.
The pyritinol of applying clinically at present, it is pyritinol hydrochloride, be specially the monohydrate of hydrochloride, its chemical name is 3,3-(dithio methylene radical) two (5-hydroxyl-6-methyl-pyridine methane) dihydrochloride monohydrate, has recorded in < < Chinese Pharmacopoeia version > > in 2010.The preparation of this product listing has sheet, capsule and sterile powder injection, and its injection easily causes venous stimulation when clinical application, has greatly limited clinical application.The powder injection of pyritinol hydrochloride easy caking after standing storage, not soluble or dissolve and thoroughly cause liquid unclarity, particulate matter to exceed standard and easily cause the untoward reactions such as Microembolization during use.
CN101003509A discloses hydrobromate and the mesylate of pyritinol, record its stability having had, solvability and bland advantage, but in fact, Hydrogen bromide pyritinol, methylsulfonic acid pyritinol store easy moisture absorption under normal condition, in purification refine, be difficult to separate out with conventional crystallization method, need loaded down with trivial details aftertreatment technology, Hydrogen bromide and methylsulfonic acid have strong corrodibility in addition, comparatively difficult to its suitability for industrialized production.
CN101066266A discloses organic acid salt of pyritinol and preparation method thereof, wherein preferred pyritinol nicotinate.Yet, in nicotinic acid pyritinol water solvability a little less than, and nicotinic acid pyritinol preparation technology used dry-out benzene, toxicity is larger, and aftertreatment technology is complicated, is not suitable for suitability for industrialized production.
Yet, existing pyritinol or its salt, or pyritinol salt exists defect in the use, or the production technique that obtains this pyritinol salt is unsuitable for suitability for industrialized production.For this reason, need to provide a kind of safe, pyritinol salt and production method thereof of stablizing, meeting industrialization production requirements.
Embodiment 1: pyritinol maleate synthetic
Get 5.0g pyritinol powder, drop in reaction flask, add 100ml purified water, then under agitation add toxilic acid 3.8g, finish, be heated to 60-65 ℃ and stir 30min and all dissolve to solid, remove heating fluid, stirred crystallization under room temperature, separate out a large amount of white solids, use a small amount of cold water washing, 45 ℃ of vacuum-dryings, obtain white powder 5.97g, yield 72.9%.Purity: 99.5%; M.p.:134~137 ℃; Ultimate analysis (C16H20N2O4S22C4H4O4): C:47.9%, H:4.8%, N:4.6%, S:10.6%, O:32.1% (theory: C:48.0%, H:4.7%, N:4.7%, S:10.7%, O:32.0%); 1H-NMR (600MHz, DMSO) δ: 2.39 (6H, s), 3.93 (4H, s), 4.76 (4H, s), 6.18 (4H, s), 7.87 (2H, s).By the 1H-NMR (Fig. 2) of toxilic acid pyritinol and the 1H-NMR (Fig. 1) of pyritinol contrast, in a part toxilic acid pyritinol, contain 2 molecule toxilic acids.
Embodiment 2: pyritinol maleate synthetic
Get 5.0g pyritinol powder, drop in reaction flask, add 100ml ethanol, then under agitation add toxilic acid 3.0g, finish, be heated to return stirring 30min and all dissolve to solid, remove heating fluid, stirred crystallization under room temperature, separate out a large amount of white solids, use a small amount of cold water washing, 45 ℃ of vacuum-dryings, obtain white powder 5.50g, yield 67.5%.After measured, the toxilic acid pyritinol that structure makes with embodiment 1.
Toxilic acid 3.8g is dissolved in 100ml ethanol, be warming up to 60 DEG C clearly molten, add pyritinol 5.0g, stir clearly molten, react 1 hour, cooling crystallization, filter, solid is drying under reduced pressure at 50 DEG C, obtains white crystalline solid toxilic acid pyritinol crystal form A 4.9g.X-ray powder diffraction analysis, as Fig. 1, its 2 θ value is as following table.
Embodiment 2
Toxilic acid 3.8g is dissolved in 100ml acetone, be warming up to 45 DEG C clearly molten, add pyritinol 5.0g, stir clearly molten, react 1.5 hours, cooling crystallization, filter, solid is drying under reduced pressure at 50 DEG C, obtains white crystalline solid 5.2g.It is toxilic acid pyritinol crystal form A that dry product does X-ray powder diffraction.
Embodiment 3
Toxilic acid 3.8g is dissolved in and adds 100ml Virahol, be warming up to 60 DEG C clearly molten, add pyritinol 5.0g, stir clearly molten, react 2 hours, cooling crystallization, filter, solid is drying under reduced pressure at 50 DEG C, obtains white crystalline solid 5.1g.It is toxilic acid pyritinol crystal form A that dry product does X-ray powder diffraction.
Embodiment 1: nicotinic acid pyritinol salt synthetic
Get nicotinic acid 24.6g, fully be dissolved in the 300ml anhydrous benzene, heated and stirred is to molten entirely, under complete molten state, add pyritinol 40.5g, reflux mixture 3 hours, TLC thin layer identification (developing solvent: ethyl acetate: ethanol: glacial acetic acid=5: 6: 0.6) fully, the cooling back adds the 200ml dehydrated alcohol slightly, mixture is put into refrigerator fully cool off, sucking filtration is separated out white crystals, with a small amount of cold absolute ether washing solid.65 ℃ of vacuum dryings get 62.1g nicotinic acid pyritinol salt, yield 89.7%.Determination of acid-basetitration nicotinic acid and pyritinol content are measured moisture with the karl Fischer method.The result is: nicotinic acid 37.2%, and pyritinol 62.0%, water 5.8%, approaching with theoretical value, contain 2 water of crystallization.Elementary analysis: theoretical value C52.8% H5.3% O25.2%N6.6% S10.1%; Measured value C52.4% H5.2% O25.1%N6.5% S10.0%.
Embodiment 2: fumaric acid pyritinol salt synthetic
Get fumaric acid 11.6g, fully be dissolved in the 300ml anhydrous benzene, heated and stirred is to molten entirely, under complete molten state, add pyritinol 40.5g, reflux mixture 3 hours, TLC thin layer identification (developing solvent: ethyl acetate: ethanol: glacial acetic acid=5: 4: 0.8) fully, the cooling back adds the 200ml dehydrated alcohol slightly, mixture is put into refrigerator fully cool off, sucking filtration is separated out white crystals, with a small amount of cold absolute ether washing solid.65 ℃ of vacuum dryings get 49.9g fumaric acid pyritinol salt, yield 88.9%.Determination of acid-basetitration fumaric acid and pyritinol content are measured moisture with the karl Fischer method.The result is: fumaric acid 20.8%, and pyritinol 72.7%, water 6.5%, approaching with theoretical value, contain 2 water of crystallization.Elementary analysis: theoretical value C49.6% H5.0%O26.4% N5.8% S13.2%; Measured value C49.4% H5.2% O26.5% N5.9%S13.1%.
With Pyrithioxine hydrochloride 10g, be dissolved in the 20ml pyridine, slowly drip POCl3 solution 10ml under the room temperature; Drip and finish, stirring at room reaction 12 hours slowly adds the 100g frozen water and stirred hydrolysis reaction 2 hours; Toluene gradation extraction 30ml * 3, water layer evaporated under reduced pressure, Virahol dissolution residual substance; Filter, evaporate to dryness gets compd A 4.2g.
Embodiment 2: the preparation of compd B
With Pyrithioxine hydrochloride 10g, be dissolved in the 40ml THF, add 4gNaH, 30 ℃ were stirred 2 hours; Add the 20ml POCl3, stirring reaction 16 hours slowly adds the 100g frozen water and stirred hydrolysis reaction 2 hours; ETHYLE ACETATE gradation extraction 30ml * 3, the water layer evaporated under reduced pressure adds 80ml Virahol dissolution residual substance; Add 40ml water, freezing crystallization gets compd B 5.6g.
Embodiment 3: the preparation of Compound C
With Pyrithioxine hydrochloride 10g, be dissolved in the 40ml THF, add 4gNaH, 30 ℃ were stirred 2 hours; Add the 20ml chloroiodomethane, stirring reaction 16 hours, 60 ℃ of evaporated under reduced pressure add 20ml acetonitrile dissolution residual substance; As midbody, other gets triethylamine 9ml and is dissolved in the 10ml acetonitrile, drips 3.6ml phosphoric acid, after dropping finishes; Stir down and slowly splash into midbody, continued 60 ℃ of stirring reactions 12 hours, steaming desolventizes; Residue adds water 20ml dissolving, and water layer filters clarification, and freeze-drying promptly gets compd B 6.7g.
Embodiment 4: the preparation of Compound D
Serine 3 grams, ethylene bromohyrin 2.5g, N with the BOC protection; N-Dimethylamino pyridine 3g and NSC 57182 3g are dissolved in the THF; Stirring at room 10 hours, vacuum concentration is with the thick product of chromatography purification (with the ETHYLE ACETATE/normal hexane wash-out of normal hexane to 30%); Merging filtrate, evaporate to dryness gets intermediate A; Pyrithioxine hydrochloride 2g and intermediate A 2.5g are dissolved with THF 30ml, add triphenyl phosphorus 2g, slowly drip diethyl azodiformate solution 2ml, room temperature reaction 5 hours; Reaction is finished, and evaporated under reduced pressure adds ETHYLE ACETATE 50ml dissolving, filters insolubles; With the thick product of chromatography purification (with the ETHYLE ACETATE/normal hexane wash-out of normal hexane to 10%), merging filtrate, evaporate to dryness dissolves with methylene dichloride 20ml then; Feed hydrogen chloride gas to saturated, stirring reaction 5 hours filters; Get the hydrochloride of Compound D, transferring pH behind the use dissolved in distilled water is about 8, and the water layer lyophilize gets Compound C 0.27g.
Embodiment 5: the preparation of compd E
Get compd A 10g, be dissolved in the 30ml Virahol, add 25gBoc-Ser-OBZL in batches, 50 ℃ of stirring reactions; HPLC monitoring react to compd B less than 5%, add 0.1M hydrochloric acid soln 20ml, 60 ℃ of heating hydrolysis 5 hours are regulated pH to 7; Evaporated under reduced pressure adds anhydrous alcohol solution, removes by filter insolubles, evaporated under reduced pressure; Add the 5ml water dissolution, filtering, lyophilize get compd E 6.9g
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
It is approved for “symptomatic treatment of chronically impaired brain function in dementia syndromes” and for “supportive treatment of sequelae of craniocerebral trauma” in various European countries, including Austria, Germany, France, Italy, Portugal, and Greece. In France it is also approved for rheumatoid arthritis as a disease modifying drug, on the basis of the results of clinical trials. In many countries it is available over the counter and is widely advertised on the internet as being for “memory disturbances.”
Effects
review refs needed
Adverse effects
Adverse effects include nausea, headache,[2] and rarely allergic reaction (mild skin reactions).[3] A 2004 survey of six case reports suggested a link between pyritinol and severe cholestatichepatitis when on several drugs for certain diseases.[4]
Other rare side effects: acute pancreatitis[5] and photoallergic eruption.[6]
References
^ Lemmel EM (May 1993). “Comparison of pyritinol and auranofin in the treatment of rheumatoid arthritis. The European Multicentre Study Group”. British Journal of Rheumatology. 32 (5): 375–82. doi:10.1093/rheumatology/32.5.375. PMID8495257.
^ Nachbar F, Korting HC, Vogl T (1993). “Erythema multiforme-like eruption in association with severe headache following pyritinol”. Dermatology. 187 (1): 42–6. doi:10.1159/000247196. PMID8324277.
^ de Groot, Anton C.; Nater, Johan Pieter; Weyland, J. Willem. Unwanted Effects of Cosmetics and Drugs Used in Dermatology.[full citation needed][page needed]
^ Straumann A, Bauer M, Pichler WJ, Pirovino M (August 1998). “Acute pancreatitis due to pyritinol: an immune-mediated phenomenon”. Gastroenterology. 115 (2): 452–4. doi:10.1016/S0016-5085(98)70212-4. PMID9679051.































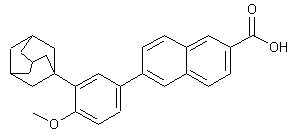

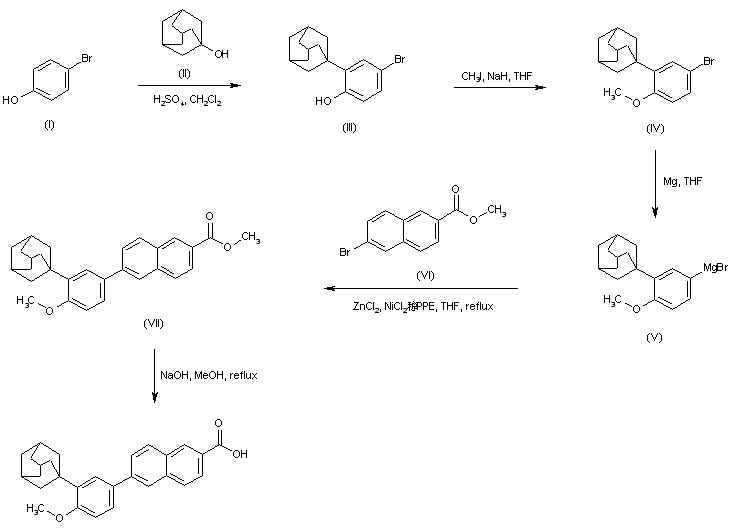
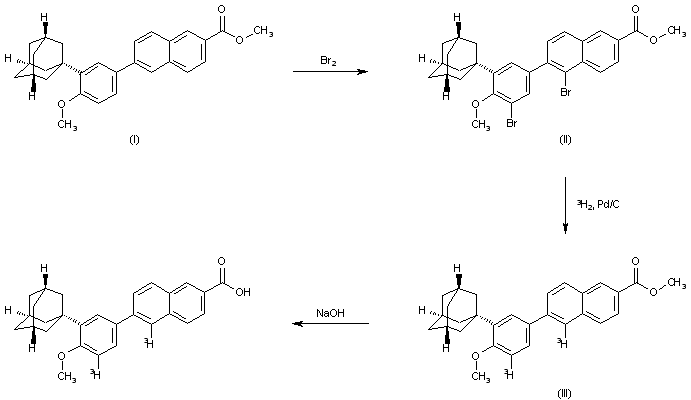







































































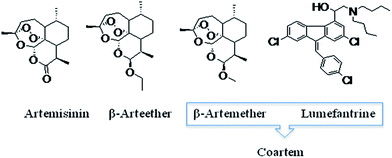








 (sutimlimab-jome), first treatment for use in patients with cold agglutinin disease
(sutimlimab-jome), first treatment for use in patients with cold agglutinin disease