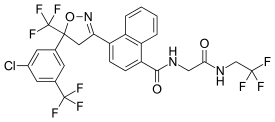
Afoxolaner
- Molecular FormulaC26H17ClF9N3O3
- Average mass625.870 Da
- A1443
- AH252723
1093861-60-9[RN]1-Naphthalenecarboxamide, 4-[5-[3-chloro-5-(trifluoromethyl)phenyl]-4,5-dihydro-5-(trifluoromethyl)-3-isoxazolyl]-N-[2-oxo-2-[(2,2,2-trifluoroethyl)amino]ethyl]-4-[5-[3-chloro-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-4H-1,2-oxazol-3-yl]-N-[2-oxo-2-(2,2,2-trifluoroethylamino)ethyl]naphthalene-1-carboxamide
Afoxolaner Merial
On 9 September 2021, the Committee for Medicinal Products for Veterinary Use (CVMP) adopted a positive opinion1, recommending the granting of a variation to the terms of the marketing authorisation for the veterinary medicinal product Frontpro. The marketing authorisation holder for this veterinary medicinal product is Boehringer Ingelheim Vetmedica GmbH. ,,,, https://www.ema.europa.eu/en/medicines/veterinary/summaries-opinion/frontpro-previously-known-afoxolaner-merial
Frontpro is currently authorised as chewable tablets for use in dogs. The variation concerns the change of legal status from prescription-only to non-prescription veterinary medicine. Additionally, the applicant is adding the list of local representatives to the package leaflet.
Detailed conditions for the use of this product are described in the summary of product characteristics (SPC), for which an updated version reflecting the changes will be published in the revised European public assessment report (EPAR) and will be available in all official European Union languages after the variation to the marketing authorisation has been granted by the European Commission.
| Name | Frontpro (previously known as Afoxolaner Merial) |
| Agency product number | EMEA/V/C/005126 |
| International non-proprietary name (INN) or common name | afoxolaner |
| Species | Dogs |
| Active substance | afoxolaner |
| Date opinion adopted | 09/09/2021 |
| Company name | Boehringer Ingelheim Vetmedica GmbH |
| Status | Positive |
| Application type | Post-authorisation |
| Medicine | Frontpro (previously known as Afoxolaner Merial) |
|---|---|
| Active Substance | afoxolaner |
| INN/Common name | afoxolaner |
| Pharmacotherapeutic Classes | Ectoparasiticides for systemic use |
| Status | This medicine is authorized for use in the European Union |
| Company | Boehringer Ingelheim Vetmedica GmbH |
| Market Date | 2019-05-20 |
European Medicines Agency (EMA)
| Medicine | Nexgard Spectra |
|---|---|
| Active Substance | afoxolaner, milbemycin oxime |
| INN/Common name | afoxolaner, milbemycin oxime |
| Pharmacotherapeutic Classes | Endectocides, Antiparasitic products, insecticides and repellents, milbemycin oxime, combinations |
| Status | This medicine is authorized for use in the European Union |
| Company | Boehringer Ingelheim Vetmedica GmbH |
| Market Date | 2015-01-15 |
| Medicine | NexGard |
|---|---|
| Active Substance | afoxolaner |
| INN/Common name | afoxolaner |
| Pharmacotherapeutic Classes | Isoxazolines, Ectoparasiticides for systemic use |
| Status | This medicine is authorized for use in the European Union |
| Company | Boehringer Ingelheim Vetmedica GmbH |
| Market Date | 2014-02-11 |
European Medicines Agency (EMA)
SYN WO2009126668,

SYN
IP .COM

PATENT
PATENT
https://patents.google.com/patent/WO2009126668A2/en
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017176948
A particularly active isoxazoline compound, 4-[5-[3-chloro-5-(trifluoromethyl)phenyl]-4,5-dihydro-5-(trifluoromethyl)-3-isoxazolyl]-N-[2-oxo-2-[(2,2,24rifluoroethyl)amino]ethyl]-l-naphthalenecarboxamide, is known by the nonproprietary name afoxolaner. Afoxolaner has the following chemical structure:
Afoxolaner
Other isoxazoline compounds that have been found to be highly active against parasitic insects and arachnids are known by the nonproprietary names fluralaner (see US 7,662,972, which is incorporated herein by reference), sarolaner (see US 8,466, 15, incorporated herein by reference) and lotilaner (see, for example US 8,383,659, incorporated herein by reference). The structures of these compounds are shown below:
In addition, published patent application nos. US 2010/0254960 Al, WO 2007/070606
A2, WO 2007/123855 A2, WO 2010/003923 Al, US7951828 & US7662972, US 2010/0137372 Al, US 2010/0179194 A2, US 2011/0086886 A2, US 2011/0059988 Al, US 2010/0179195 Al and WO 2007/075459 A2 and U.S. Patent No. 7,951,828 (all incorporated herein by reference) describe various other parasiticidal isoxazoline compounds.
It is known in the field that isoxazoline compounds having a chiral quaternary carbon atom such as the carbon atom adjacent to the oxygen on the isoxazoline ring of the compounds described above have at least two optical isomer (enantiomers) that are mirror images of each other. Furthermore, it is sometimes the case with biologically active compounds that one of the enantiomers is more active than the other enantiomer. In addition, it is sometimes the case that one enantiomer of a biologically active compound is less toxic than the other enantiomer.
Therefore, with optically active compounds it is desirable to utilize the enantiomer that is most active and less toxic (eutomer). However, isolating the most active enantiomer from a mixture can be costly and result in waste of up to half of the racemic mixture prepared.
Processes to prepare certain isoxazoline compounds enriched in an enantiomer using some cinchona alkaloid-derived phase transfer catalysts have been described. For example, US 2014/0206633 Al, US 2014/0350261 Al, WO 2013/116236 Al and WO 2014/081800 Al (incorporated herein by reference) describe the synthesis of certain isoxazoline active agents enriched in an enantiomer using cinchona alkaloid-based chiral phase transfer catalysts. Further, Matoba et al., Angew. Chem. 2010, 122, 5898-5902 describes the chiral synthesis of certain pesticidal isoxazoline active agents. However, these documents do not describe the processes and certain catalysts described herein.
Scheme 3
Example 7: Preparation of (S)-afoxolaner using chiral phase transfer catalyst (Ilia- 13-1):
(ΠΑ-1) (^-afoxolaner
1) Starting material (IIA-1) (200g, 1.Oeq, 94.0%) and DCM (6 L, 30 volumes) were placed into a 10 L reactor, the solid was dissolved completely.
2) The mixture was cooled to 0°C, and some starting material precipitated out.
3) The catalyst (Ilia- 13-1) (7.56g, 3% mol, 95.0%) was added to the mixture and the resulting mixture cooled further to -10° C.
4) Hydroxylamine (64.9 g, 3.0 eq, 50% solution in water) was added to a solution of NaOH (52.5g, 4. Oeq, in 5v water) in a separate reactor and stirred for 30 minutes.
5) The resulting hydroxylamine/NaOH solution was then added dropwise to the 10 L reactor containing (IIA-1) over about 4 hours.
6) The resulting mixture was stirred for 12 hours at -10°C and monitored for the extent of reaction until the amount of starting material was < 1.0% by HPLC.
7) The mixture was then warmed to 10°C, 1 liter of water was added and the mixture was stirred for 10 minutes.
8) The mixture was allowed to settle to separate the two phases, and the organic layer was collected.
9) The organic layer was then washed with 2 liters of water, the layers were allowed to separate again and the organic layer was collected.
10) The organic layer was washed with 1 liter of brine, the layers allowed to separate and the organic layer was collected and dried over Na2S04 (200 g).
11) The dried organic layer was concentrated under vacuum to about 2 volumes.
12) Toluene (2 L, 10 volumes) was charged to the concentrated mixture and concentration under vacuum was continued to about 5 volumes. Solvent exchange was repeated twice again.
13) The resulting solution was placed into a 2.0 L reactor and heated to 55-60°C.
14) Cyclohexane (300 ml, 1.5 volumes) was added at 55-60°C.
15) The mixture was then cooled to 40 °C over 1.5 hours and then stirred at 40°C for 3 hours.
16) The mixture was then cooled to 25 °C over 2 hours and stirred at 25°C for a further 3 hours.
17) The resulting mixture was cooled to 0-5 °C over 1 hour and stirred at 5 °C for 12 hours, at which time the mixture was filtered to isolate the product.
18) The filter cake was washed with cold toluene/ Cyclohexane (3 : 1, 1000 ml, 5 volumes).
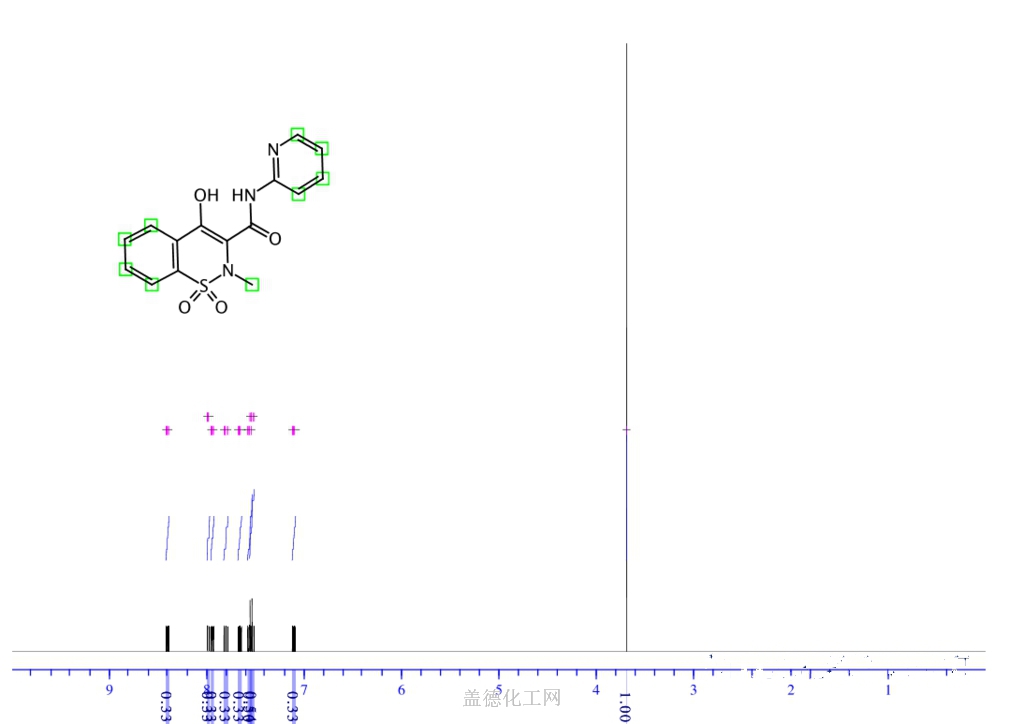
19) The product was obtained as a white solid. (171.5g, chiral purity > 99.0% by area using the chiral HPLC method described in Example 3, chemical purity > 99.0% by area (HPLC), yield: 83.6%, assay purity: 92%). The 1H NMR and LCMS spectra are consistent with the structure of (^-afoxolaner as the toluene solvate. Figure 3 shows the 1H NMR spectra of (S)-afoxolaner in DMSO-d6 and Figure 4 shows the 1H NMR spectra of afoxolaner (racemic) for comparison. The chiral purity of the product was determined using the chiral HPLC method described in Example 3. Figure 5 shows the chiral HPLC chromatogram of afoxolaner (racemic) and Figure 6 shows the chiral HPLC chromatogram of the product (^-afoxolaner showing one enantiomer.
Example 8: Alternate Process to prepare (^-afoxolaner
An alternate process for the preparation of (S)-afoxolaner was conducted. Some of the key variations in the alternate process are noted below.
1. 1 kilogram of compound (IIA-1) (1 eq.) and 9 liters of DCM are charged to a reactor and stirred to dissolve the compound.
2. The mixture is cooled to about 0° C and 50 grams (5 mole %) of the chiral phase transfer catalyst (Ilia- 13-1) and 1 liter of DCM are charged and the resulting mixture is cooled to about -13° C.
3. A solution of 19% (w/w) hydroxylamine sulfate (294 g, 1.1 eq.) (made with 294 grams of ( H2OH)H2S04 and 141 grams of NaCl in 1112 mL of water) and 4.4 equivalents of NaOH as a 17.6% (w/w) solution (286 grams NaOH and 158 grams of NaCl in 1180 mL water) are charged to the reaction mixture simultaneously.
4. The resulting reaction mixture was aged about 20 hours at about -13° C and then checked for reaction conversion by HPLC (target < 0.5% by area);
5. After completion of the reaction, water (3 vol.) was added at about 0° C. Then, a solution of 709 g of KH2P04 in 4.2 liters of water are added to the mixture to adjust the pH (target 7-8) and the resulting mixture is stirred at about 20° C for 30 minutes.
6. The layers are allowed to settle, the aqueous layer is removed and the organic layer is washed with 3 liters of water twice.
Crystallization of Toluene Solvate
1. After the extraction/washing step, the dichloromethane is removed by distillation under vacuum to about 1-2 volumes and toluene (about 5-10 volumes) is added.
2. The volume is adjusted by further distillation under vacuum and/or addition of more toluene to about 5-6 volumes. The mixture is distilled further while maintaining the volume to completely remove the dichloromethane reaction solvent.
3. The mixture is then cooled to about 10° C and seeded with afoxolaner (racemic compound) and stirred at the same temperature for at least 2 hours;
4. The mixture is heated to about 55-65° C, aged for at least 17 hours and then the solid is filtered off. The filtered solid is washed with toluene;
5. The combined filtrate and wash is adjusted to a volume of about 5-6 volumes by
distillation under vacuum and/or toluene addition;
6. The resulting mixture is cooled to about 10° C and aged for at least 5 hours then filtered.
The cake is washed with toluene.
7. The cake is dried at 50° C under vacuum to obtain a toluene solvate of (S)-afoxolaner containing between about 6% and 8% toluene.
Re-crystallization from cyclohexane/ethanol
The toluene solvate of (S)-afoxolaner was subsequently re-crystallized from a mixture of cyclohexane and ethanol to remove the associated toluene and to further purify the product.
1. 591 grams of the (S)-afoxolaner toluene solvate were charged to a vessel along with 709 mL of ethanol (1.2 vol.) and 1773 mL of cyclohexane (3 vol.) and the mixture heated to about 60° C.
2. To the resulting mixture was added an additional 6383 mL of cyclohexane with stirring.
3. The resulting mixture was cooled to about 30° C and then heated again to 60° C. This process was repeated once.
4. The mixture was slowly cooled to 10° C and stirred for at least 5 hours.
5. The resulting slurry was filtered and the cake washed with cyclohexane.
6. The cake was dried at 50° C under vacuum to provide 453.7 grams of (S)-afoxolaner
Example 9: Comparative selectivity of benzyloxy-substituted chiral phase transfer catalyst (Illa-13) with other cinchona alkaloid-based chiral phase transfer catalysts.
The selectivity of the formation of (S)-afoxolaner from compound IIA-1 as shown above was studied with sixteen chiral phase transfer catalysts (PTC) of different structures. The reaction was conducted using conditions similar to those of example 7. The ratio of (^-afoxolaner and (R)-afoxolaner in the reaction mixture was determined by chiral HPLC using the method described in Example 3. The results of the study are provided in Table 2 below.
Table 2
No. Chiral PTC Ratio of (S)- to (R)-afoxolaner
16 50% : 50%
As shown in the table, the catalyst in which the group R in the structure of formula (Ilia) is 3,4,5-tribenzyloxy phenyl results in a surprising improved selectivity for the (S)-enantiomer compared with other quinine-based phase transfer catalysts in which the group corresponding to R in formula (Ilia) is another group.
Example 10: Improvement of Chiral Purity of (<S)-afoxolaner by Crystallization from Toluene
A sample of reaction mixture containing a ratio (HPLC area) of 92.1 :7.9, (^-afoxolaner to (R)-afoxolaner, was concentrated to dryness and the residue was crystallized from toluene and from ethanol/cyclohexane using a process similar to that described in Example 8. The isolated crystalline solid was analyzed by chiral HPLC to determine the relative amounts of (S)-afoxolaner and (R)-afoxolaner (HPLC method: column – Chiralpak AD-3 150 mm x 4.6 mm x 3.0 μηι, injection volume – 10 μΐ., temperature – 35° C, flow – 0.8 mL/minute, mobile phase -89% hexane/10% isopropanol/1% methanol, detection – 312 nm). The ratio of (^-afoxolaner to (R)-afoxolaner in the solid isolated from the toluene crystallization was found to be 99.0 : 1.0 while the ratio of (S)-afoxolaner to (R)-afoxolaner in the solid crystallized from ethanol/cyclohexane was found to be 95.0 : 5.0.
The example shows that the crystallization (^-afoxolaner from an aromatic solvent such as toluene results in a significant improvement of chiral purity of the product. This is very unexpected and surprising.
Example 1 1 : Comparative selectivity of benzyl oxy vs. alkoxy-substituted chiral phase transfer catalyst of Formula (Ilia- 13)
Three chiral phase transfer catalysts of Formula (IIIa-13), wherein the phenyl ring is substituted with three alkoxy groups and three benzyloxy groups (R = methyl, ethyl and benzyl); R’=OMe, W=vinyl and X=chloro were evaluated in the process to prepare of (,S)-IA from compound IIA-1
as shown below.
The amount of solvents and reagents and the reaction and isolation conditions were as described in Example 7 above. The same procedure was used for each catalyst tested. It was found that the selectivity of the tri-benzyloxy catalyst was surprisingly significantly better than the two alkoxy-substituted catalysts, as shown by the chiral purity of the product. Furthermore, it was found that using the tri-benzyloxy substituted phase transfer catalyst the resulting chemical purity was also much better. The superior selectivity of the benzyloxy-substituted catalyst is significant and surprising and cannot be predicted. Chiral phase transfer catalysts containing a phenyl substituted with benzyloxy and alkoxy groups were found to be superior to catalysts substituted with other groups such as electron-withdrawing groups and alkyl groups. The chiral purity and chemical purity of the product produced from the respective phase-transfer catalysts is shown in the Table 3 below:
Table 3
PATENT
WO 2009002809
WO 2009025983
WO 2009126668
WO 2017176948
WO 2018117034
CN 109879826
JP 2020023442
WO 2020158889
WO 2020171129
WO 2021013825
CN 112457267
CN 112679338
PAPER
IP.com Journal (2009), 9(9B), 35.
Afoxolaner (INN)[2] is an insecticide and acaricide that belongs to the isoxazoline chemical compound group.
It acts as an antagonist at ligand-gated chloride channels, in particular those gated by the neurotransmitter gamma-aminobutyric acid (GABA-receptors). Isoxazolines, among the chloride channel modulators, bind to a distinct and unique target site within the insect GABA-gated chloride channels, thereby blocking pre-and post-synaptic transfer of chloride ions across cell membranes. Prolonged afoxolaner-induced hyperexcitation results in uncontrolled activity of the central nervous system and death of insects and acarines.[3]
Marketing
Afoxolaner is the active principle of the veterinary medicinal products NexGard (alone) and Nexgard Spectra (in combination with milbemycin oxime).[4][5][6] They are indicated for the treatment and prevention of flea infestations, and the treatment and control of tick infestations in dogs and puppies (8 weeks of age and older, weighing 4 pounds (~1.8 kilograms) of body weight or greater) for one month.[7] These products are administered orally and poisons fleas once they start feeding.
The marketing authorization was granted by the European Medicines Agency in February 2014, for NexGard and January 2015, for Nexgard Spectra, after only 14[8] and 12[9] months of quality, safety and efficacy assessment performed by the Committee for Medicinal Products for Veterinary Use (CVMP).[10] Therefore, long-term effects are not known.
List of excipients
In NexGard[11] and NexGard Spectra:[3]
- Maize starch
- Soy protein fines
- Beef braised flavouring
- Povidone (E1201)
- Macrogol 400 (reputed laxatives)
- Macrogol 4000 (reputed laxatives)
- Macrogol 15 hydroxystearate (reputed laxatives)
- Glycerol (E422)
- Triglycerides, medium-chain
Additionally in NexGard Spectra:
Safety
Dosage
Afoxolaner is recommended to be administered at a dose of 2.7–7 mg/kg dog’s body weight.[11]
Toxicity for mammals
According to clinical studies performed prior marketing:
- The oral toxicity profile of afoxolaner consists of a diuretic effect (rats only), effects secondary to a reduction in food consumption (rats and rabbits only) and occasional vomiting and/or diarrhoea (dogs, 120 and 200 mg/kg bodyweight (bw)) following high oral doses. No treatment-related effects on vomiting or diarrhoea were noted following oral doses of up to 31.5 mg/kg bw in the pivotal target animal safety study, nor in the EU field trial.[9]
- mild gastrointestinal effects (vomiting, diarrhoea), pruritus, lethargy, anorexia, and neurological signs (convulsions, ataxia and muscle tremors) have been reported in less than 0.1% of 10,000 animals treated, including isolated reports, most reported adverse reactions being self-limiting and of short duration,[11]
- (in combination with milbemycin oxime): vomiting, diarrhoea, lethargy, anorexia, and pruritus were observed in 0.2 to 1% of 10,000 animals treated and were generally self-limiting and of short duration,[3]
- In vitro studies reported that afoxolaner can bind to dopamine and norepinephrine cellular transport receptor systems and the CB1 receptor; inhibition of these catecholaminergic systems and certain types of competitive binding at CB1 receptors may mediate pharmacodynamic effects of diuresis, decreased food consumption, and decreased body weight in animals.[9]
According to post-marketing safety experience:
- (in combination with milbemycin oxime): erythema and neurological signs (convulsions, ataxia and muscle tremors) have been reported in less than 0.1% of 10,000 animals treated, including isolated reports,[3]
- The US FDA reports[12] that some drugs in this class (isoxazolines), including afoxalaner, can have adverse neurologic effects on some dogs, such as muscle tremors, ataxia, and seizures.
- Extralabel use of afoxolaner in a pet pig has been described without any adverse effects.[13] Experimental use in commercial pigs also did not result in any adverse effects.[14]
Selectivity in insects over mammalians
In vivo studies (repeat-dose toxicology in laboratory animals, target animal safety, field studies) provided by MERIAL, the company that produces afoxolaner-derivative medicines, did not show evidence of neurological or behavioural effects suggestive of GABA-mediated perturbations in mammals. The Committee for Medicinal Products for Veterinary Use (CVMP) therefore concluded that binding to dog, rat or human GABA receptors is expected to be low for afoxolaner.[9]
Selectivity for insect over mammalian GABA-receptors has been demonstrated for other isoxazolines.[15] The selectivity might be explained by the number of pharmacological differences that exist between GABA-gated chloride channels of insects and vertebrates.[16]
GEN REF
- Shoop WL, Hartline EJ, Gould BR, Waddell ME, McDowell RG, Kinney JB, Lahm GP, Long JK, Xu M, Wagerle T, Jones GS, Dietrich RF, Cordova D, Schroeder ME, Rhoades DF, Benner EA, Confalone PN: Discovery and mode of action of afoxolaner, a new isoxazoline parasiticide for dogs. Vet Parasitol. 2014 Apr 2;201(3-4):179-89. doi: 10.1016/j.vetpar.2014.02.020. Epub 2014 Mar 14. [Article]
References
- ^ Jump up to:a b c “Frontline NexGard (afoxolaner) for the Treatment and Prophylaxis of Ectoparasitic Diseases in Dogs. Full Prescribing Information” (PDF) (in Russian). Sanofi Russia. Retrieved 14 November 2016.
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 70” (PDF). World Health Organization. pp. 276–7. Retrieved 14 November 2016.
- ^ Jump up to:a b c d “NexGard Spectra product information – Annex I “Summary of product characteristics”” (PDF). European Medicines Agency. Retrieved 13 November 2019.
- ^ Shoop WL, Hartline EJ, Gould BR, Waddell ME, McDowell RG, Kinney JB, et al. (April 2014). “Discovery and mode of action of afoxolaner, a new isoxazoline parasiticide for dogs”. Veterinary Parasitology. 201 (3–4): 179–89. doi:10.1016/j.vetpar.2014.02.020. PMID 24631502.
- ^ Beugnet F, deVos C, Liebenberg J, Halos L, Fourie J (25 August 2014). “Afoxolaner against fleas: immediate efficacy and resultant mortality after short exposure on dogs”. Parasite. 21: 42. doi:10.1051/parasite/2014045. PMC 4141545. PMID 25148564.
- ^ Beugnet F, Crafford D, de Vos C, Kok D, Larsen D, Fourie J (August 2016). “Evaluation of the efficacy of monthly oral administration of afoxolaner plus milbemycin oxime (NexGard Spectra, Merial) in the prevention of adult Spirocerca lupi establishment in experimentally infected dogs”. Veterinary Parasitology. 226: 150–61. doi:10.1016/j.vetpar.2016.07.002. PMID 27514901.
- ^ “Boehringer-Ingelheim companion-animals-product NexGard (afoxolaner)”. Boehringer Ingelheim International GmbH. Retrieved 13 November 2019.
- ^ “CVMP Assessment Report for NEXGARD SPECTRA(EMEA/V/C/003842/0000)” (PDF). European Medicines Agency. Retrieved 14 November 2019.
- ^ Jump up to:a b c d “CVMP assessment report for NexGard (EMEA/V/C/002729/0000)” (PDF). European Medicines Agency. Retrieved 14 November 2019.
- ^ “Committee for Medicinal Products for Veterinary Use (CVMP) – Section “Role of the CVMP””. European Medicines Agency. Retrieved 14 November 2019.
- ^ Jump up to:a b c “NexGard product information – Annex I “Summary of product characteristics”” (PDF). European Medicines Angency. Retrieved 14 November 2019.
- ^ Medicine, Center for Veterinary. “CVM Updates – Animal Drug Safety Communication: FDA Alerts Pet Owners and Veterinarians About Potential for Neurologic Adverse Events Associated with Certain Flea and Tick Products”. http://www.fda.gov. Retrieved 2018-09-22.
- ^ Smith, Joe S.; Berger, Darren J.; Hoff, Sarah E.; Jesudoss Chelladurai, Jeba R. J.; Martin, Katy A.; Brewer, Matthew T. (2020). “Afoxolaner as a Treatment for a Novel Sarcoptes scabiei Infestation in a Juvenile Potbelly Pig”. Frontiers in Veterinary Science. 7: 473. doi:10.3389/fvets.2020.00473. PMC 7505946. PMID 33102538.
- ^ Bernigaud, C.; Fang, F.; Fischer, K.; Lespine, A.; Aho, L. S.; Mullins, A. J.; Tecle, B.; Kelly, A.; Sutra, J. F.; Moreau, F.; Lilin, T.; Beugnet, F.; Botterel, F.; Chosidow, O.; Guillot, J. (2018). “Efficacy and Pharmacokinetics Evaluation of a Single Oral Dose of Afoxolaner against Sarcoptes scabiei in the Porcine Scabies Model for Human Infestation”. Antimicrobial Agents and Chemotherapy. 62 (9). doi:10.1128/AAC.02334-17. PMC 6125498. PMID 29914951.
- ^ Casida JE (April 2015). “Golden age of RyR and GABA-R diamide and isoxazoline insecticides: common genesis, serendipity, surprises, selectivity, and safety”. Chemical Research in Toxicology. 28 (4): 560–6. doi:10.1021/tx500520w. PMID 25688713.
- ^ Hosie AM, Aronstein K, Sattelle DB, ffrench-Constant RH (December 1997). “Molecular biology of insect neuronal GABA receptors”. Trends in Neurosciences. 20 (12): 578–83. doi:10.1016/S0166-2236(97)01127-2. PMID 9416671. S2CID 5028039.
| Clinical data | |
|---|---|
| Pronunciation | /eɪˌfɒksoʊˈlænər/ ay-FOK-soh-LAN-ər |
| Trade names | NexGard, Frontpro |
| Other names | 4-[(5RS)-5-(5-Chloro-α,α,α-trifluoro-m-tolyl)-4,5-dihydro-5-(trifluoromethyl)-1,2-oxazol-3-yl]-N-[2-oxo-2-(2,2,2-trifluoroethylamino)ethyl]naphthalene-1-carboxamide |
| License data | US DailyMed: Afoxolaner |
| Routes of administration | By mouth (chewables) |
| ATCvet code | QP53BE01 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyEU: Rx-onlyOTC (RU)[1] |
| Pharmacokinetic data | |
| Bioavailability | 74% (Tmax = 2–4 hours)[1] |
| Elimination half-life | 14 hours[1] |
| Excretion | Bile duct (major route) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1093861-60-9 |
| PubChem CID | 25154249 |
| DrugBank | DB11369 |
| ChemSpider | 28651525 |
| UNII | 02L07H6D0U |
| KEGG | D10361 |
| ChEMBL | ChEMBL2219412 |
| CompTox Dashboard (EPA) | DTXSID50148921 |
| Chemical and physical data | |
| Formula | C26H17ClF9N3O3 |
| Molar mass | 625.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| showSMILES | |
| showInChI |
///////////// afoxolaner, A1443, AH252723
FC(F)(F)CNC(=O)CNC(=O)C1=C2C=CC=CC2=C(C=C1)C1=NOC(C1)(C1=CC(=CC(Cl)=C1)C(F)(F)F)C(F)(F)F

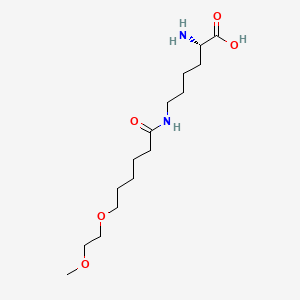







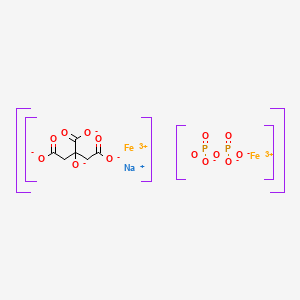


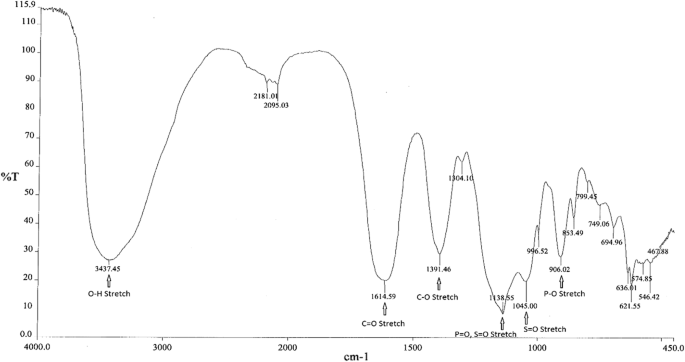
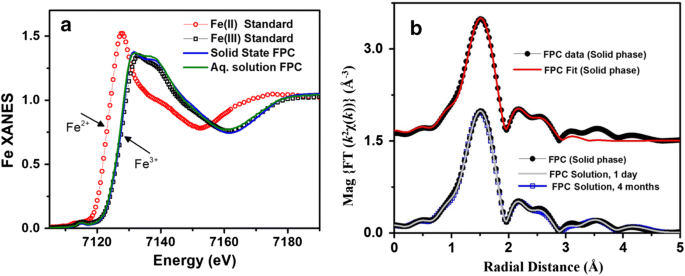

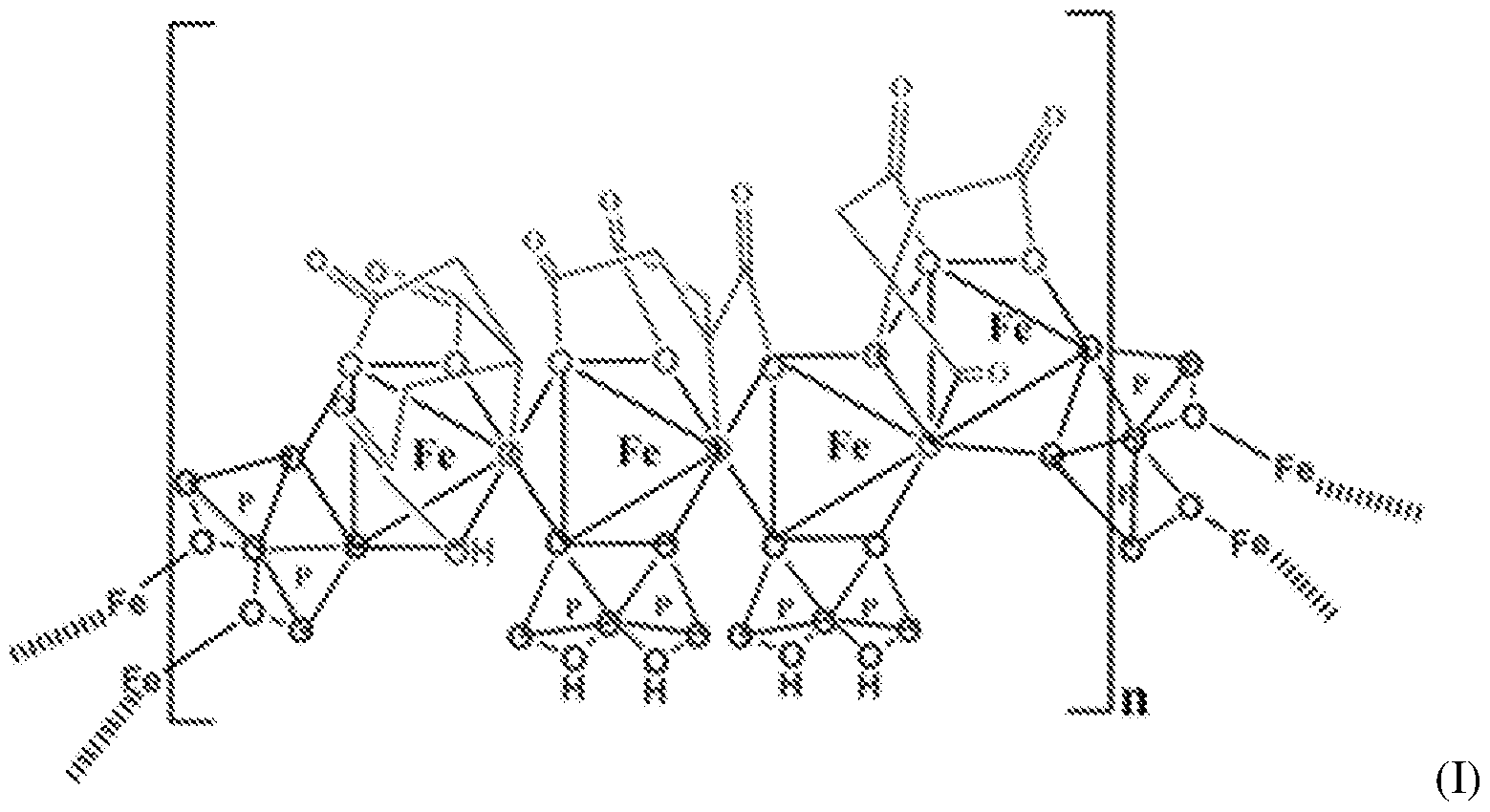



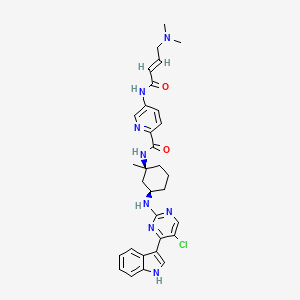
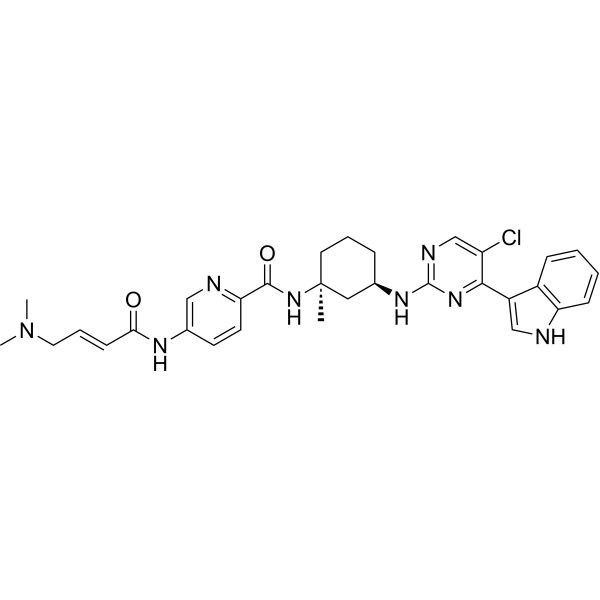











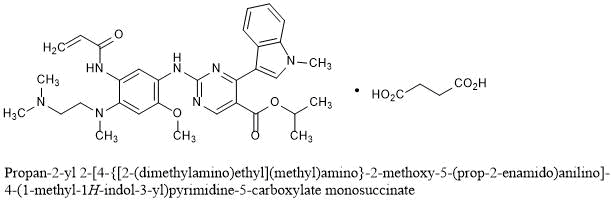
 C4H6O4 : 703.78
C4H6O4 : 703.78 (mobocertinib) Approved by U.S. FDA as the First Oral Therapy Specifically Designed for Patients with EGFR Exon20 Insertion+ NSCLC……..
(mobocertinib) Approved by U.S. FDA as the First Oral Therapy Specifically Designed for Patients with EGFR Exon20 Insertion+ NSCLC…….. 



































 Dewang")














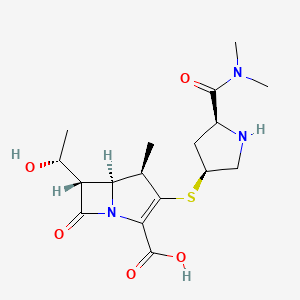
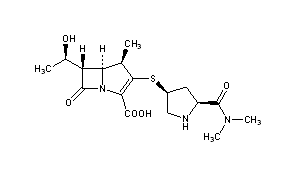
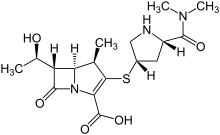





 In a 500 ml reaction flask, 40; 7 g (0.0622 mol) of (3R, 4S)-3-[(R)-l-(tert-butyldimethylsilyloxy) was added. Ethyl]-4-[(2,S,4,R)-1-(indolyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinylsulfate]small (sweetoxypropanoyl)-2-nitrogen Heterocyclic butanone (III) and 150 ml of toluene, 22 ml of trimethyl phosphite (furrowing lg of hydroquinone) were added under nitrogen. After reacting at 60 ° C for 16 hours, the solvent was evaporated under reduced pressure. It was recrystallized by adding 300 ml of ethyl acetate, and the solid was collected, and vacuum-dried at 40 ° C to obtain 32.8 g (0.0528 mol, yield: 85.0%) (5R, 6S, 8R, 2’S, 4,S)-[(R)- 1-(tert-Butyldimethylsilyloxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-ene Propoxycarbonyl ethoxy) small azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (oxime).1H-NMR (300 MHz, CDC 13):0.82(9H, s), 1.24(6H, d), 1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.69(1 H, m), 2.97-3.11(6H, m ), 3.15-3.74(4H, m), 4.35(2H,m), 4.37-4.67(5H, m), 5.24-5.28(4H, m), 5.84(1H, m). Example 44) (5R, 6S, 8R, 2, S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyrrolidinyl Synthesis of thio]-6-(1-allyloxycarbonylethoxy)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (Vffl) at room temperature , in a 2000ml reaction flask, add 32.8g (0.0528mol) (5R,6S,8R,2’S,4,S)-[(R)-1-(tert-butyldimethylsilyloxy)ethyl] 3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-indolyloxycarbonylethoxy)-1-azabicyclo[3.2.0 -Hept-2-ene-7-one-2-carboxylate (W), 27.4 ml of acetic acid, 41.3 g of fluorohydrogenamine and 1000 ml of dichloromethane, stirred at room temperature for 48 h. After completion of the reaction, 500 ml of a saturated aqueous solution of sodium hydrogencarbonate was added to the reaction mixture, and the mixture was stirred for 10 minutes, and the methylene chloride layer was separated and dried over anhydrous magnesium sulfate to give a white solid (26.2 g (0.0517 mol, yield 98.0). %) (5R, 6S, 8R, 2’S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyr Rhodium thio] -6-(l-allyloxycarbonylethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (ring The product was directly charged to the next step without further purification.1H-NMR (300 MHz, CDC 13):1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.67(1H, m), 2.97-3.11(6H, m), 3.2-3.7(4H, m) ; 4.25(2H, m), 4.47-4.87 (5H, m), 5.15-5.50 (4H, m), 5.94 (2H, m). Example 55) (5R,6S,8R,2,S,4,S)-3-[4-dimethylaminocarbonyl)pyrrolidinyl]-6-(l-hydroxyethyl)-1-aza Synthesis of bicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) (5R, 6S, 8R, 2’S, 4’S) was added. – [(R)-l-(hydroxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-allyloxy Carbonyl ethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (VDI), 21.3 g (0.152 mol) dimethylcyclohexane The ketone and 550 ml of ethyl acetate were heated to 30 ° C, and a solution of 1.0 g (0.865 mmol) of tetratriphenylphosphine palladium in 150 ml of dichloromethane was added dropwise thereto, and the mixture was reacted at room temperature for 3 h under nitrogen atmosphere. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, the aqueous layer was washed with ethyl acetate, and then, 500 ml of tetrahydrofuran was added dropwise with stirring in an ice bath, and the crystals were stirred, and the crystals were collected and dried in vacuo to give pale yellow crystals of 13.4 g (0.0352 md, Yield 68.1%) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl) 1-Azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (I)-Meropectin.IR max KBr cm- 1 : 1755, 1627, 1393, 1252, 1130NMR (D20, 300Hz): 1.25 (3H, d), 1.81-1.96 (1H, m), 2.96 (3H, s), 3.03 (3H, s), 3.14-3.20 (3H, m), 3.31-3.41 (2H, m), 3.62- 3.72 (1H, m), 3.90-4.00 (1H, m), 4.14-4.26 (2H, m), 4.63 (1H, t). Example 6 6) (5R,6S,8R,2’S,4’S)-3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(l-hydroxyethyl)-1-azabicyclo[ Synthesis of 3.2.0]-hept-2-en-7-one-2-carboxylate (I)21.3 g (0.152 mol) of dimethylcyclohexanedione in Example 5 was replaced with 45.1 g (0.155 mol) of tributyltin hydride, and 0.125 g (0.108 mmol) of tetrakistriphenylphosphine palladium was added dropwise, and the other amount was added. And the same method, the obtained 16.2g (0.0426mol, 82.5%) (5R,6S,8R,2’S,4’S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl Sulfur]-6-(l-hydroxyethyl)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (1) ~ meropenem. Example 7 7) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-hydroxyethyl)-1- Synthesis of azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) of (5R, 6S, 8R, 2, S, 4’S)-[(R)-l-(hydroxy)ethyl]-3-[4-(1-allyl was added) Oxycarbonyl-1-ylaminocarbonylcarbonylpyrrolidinothio]-6-(1-allyloxycarbonylethoxy)azaabicyclo[3. 2.]-hept-2-ene-7- Ketone-2-carboxylate 01), 6.0 g (0.0387 mol) of N, N-dimethylbarbituric acid and 500 ml of dichloromethane, and 6.0 g (5.2 mmol) of tetratriphenylphosphine was added dropwise thereto. A solution of palladium in 100 ml of dichloromethane was reacted at room temperature for 5 h under nitrogen. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, and the aqueous layer was washed with ethyl acetate. THF was evaporated and evaporated, and the crystals were evaporated, and crystals were collected, and the crystals were dried in vacuo to give 15.7 g (0.0413 mol, yield: 80.1%). 5R, 6S, 8R, 2,S,4,S) – 3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl)-1-azabicyclo [3. 2. 0] -Hept-2-ene-7-keto-2-carboxylic acid trihydrate (I)-Meropectin.
In a 500 ml reaction flask, 40; 7 g (0.0622 mol) of (3R, 4S)-3-[(R)-l-(tert-butyldimethylsilyloxy) was added. Ethyl]-4-[(2,S,4,R)-1-(indolyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinylsulfate]small (sweetoxypropanoyl)-2-nitrogen Heterocyclic butanone (III) and 150 ml of toluene, 22 ml of trimethyl phosphite (furrowing lg of hydroquinone) were added under nitrogen. After reacting at 60 ° C for 16 hours, the solvent was evaporated under reduced pressure. It was recrystallized by adding 300 ml of ethyl acetate, and the solid was collected, and vacuum-dried at 40 ° C to obtain 32.8 g (0.0528 mol, yield: 85.0%) (5R, 6S, 8R, 2’S, 4,S)-[(R)- 1-(tert-Butyldimethylsilyloxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-ene Propoxycarbonyl ethoxy) small azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (oxime).1H-NMR (300 MHz, CDC 13):0.82(9H, s), 1.24(6H, d), 1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.69(1 H, m), 2.97-3.11(6H, m ), 3.15-3.74(4H, m), 4.35(2H,m), 4.37-4.67(5H, m), 5.24-5.28(4H, m), 5.84(1H, m). Example 44) (5R, 6S, 8R, 2, S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyrrolidinyl Synthesis of thio]-6-(1-allyloxycarbonylethoxy)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (Vffl) at room temperature , in a 2000ml reaction flask, add 32.8g (0.0528mol) (5R,6S,8R,2’S,4,S)-[(R)-1-(tert-butyldimethylsilyloxy)ethyl] 3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-indolyloxycarbonylethoxy)-1-azabicyclo[3.2.0 -Hept-2-ene-7-one-2-carboxylate (W), 27.4 ml of acetic acid, 41.3 g of fluorohydrogenamine and 1000 ml of dichloromethane, stirred at room temperature for 48 h. After completion of the reaction, 500 ml of a saturated aqueous solution of sodium hydrogencarbonate was added to the reaction mixture, and the mixture was stirred for 10 minutes, and the methylene chloride layer was separated and dried over anhydrous magnesium sulfate to give a white solid (26.2 g (0.0517 mol, yield 98.0). %) (5R, 6S, 8R, 2’S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyr Rhodium thio] -6-(l-allyloxycarbonylethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (ring The product was directly charged to the next step without further purification.1H-NMR (300 MHz, CDC 13):1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.67(1H, m), 2.97-3.11(6H, m), 3.2-3.7(4H, m) ; 4.25(2H, m), 4.47-4.87 (5H, m), 5.15-5.50 (4H, m), 5.94 (2H, m). Example 55) (5R,6S,8R,2,S,4,S)-3-[4-dimethylaminocarbonyl)pyrrolidinyl]-6-(l-hydroxyethyl)-1-aza Synthesis of bicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) (5R, 6S, 8R, 2’S, 4’S) was added. – [(R)-l-(hydroxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-allyloxy Carbonyl ethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (VDI), 21.3 g (0.152 mol) dimethylcyclohexane The ketone and 550 ml of ethyl acetate were heated to 30 ° C, and a solution of 1.0 g (0.865 mmol) of tetratriphenylphosphine palladium in 150 ml of dichloromethane was added dropwise thereto, and the mixture was reacted at room temperature for 3 h under nitrogen atmosphere. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, the aqueous layer was washed with ethyl acetate, and then, 500 ml of tetrahydrofuran was added dropwise with stirring in an ice bath, and the crystals were stirred, and the crystals were collected and dried in vacuo to give pale yellow crystals of 13.4 g (0.0352 md, Yield 68.1%) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl) 1-Azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (I)-Meropectin.IR max KBr cm- 1 : 1755, 1627, 1393, 1252, 1130NMR (D20, 300Hz): 1.25 (3H, d), 1.81-1.96 (1H, m), 2.96 (3H, s), 3.03 (3H, s), 3.14-3.20 (3H, m), 3.31-3.41 (2H, m), 3.62- 3.72 (1H, m), 3.90-4.00 (1H, m), 4.14-4.26 (2H, m), 4.63 (1H, t). Example 6 6) (5R,6S,8R,2’S,4’S)-3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(l-hydroxyethyl)-1-azabicyclo[ Synthesis of 3.2.0]-hept-2-en-7-one-2-carboxylate (I)21.3 g (0.152 mol) of dimethylcyclohexanedione in Example 5 was replaced with 45.1 g (0.155 mol) of tributyltin hydride, and 0.125 g (0.108 mmol) of tetrakistriphenylphosphine palladium was added dropwise, and the other amount was added. And the same method, the obtained 16.2g (0.0426mol, 82.5%) (5R,6S,8R,2’S,4’S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl Sulfur]-6-(l-hydroxyethyl)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (1) ~ meropenem. Example 7 7) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-hydroxyethyl)-1- Synthesis of azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) of (5R, 6S, 8R, 2, S, 4’S)-[(R)-l-(hydroxy)ethyl]-3-[4-(1-allyl was added) Oxycarbonyl-1-ylaminocarbonylcarbonylpyrrolidinothio]-6-(1-allyloxycarbonylethoxy)azaabicyclo[3. 2.]-hept-2-ene-7- Ketone-2-carboxylate 01), 6.0 g (0.0387 mol) of N, N-dimethylbarbituric acid and 500 ml of dichloromethane, and 6.0 g (5.2 mmol) of tetratriphenylphosphine was added dropwise thereto. A solution of palladium in 100 ml of dichloromethane was reacted at room temperature for 5 h under nitrogen. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, and the aqueous layer was washed with ethyl acetate. THF was evaporated and evaporated, and the crystals were evaporated, and crystals were collected, and the crystals were dried in vacuo to give 15.7 g (0.0413 mol, yield: 80.1%). 5R, 6S, 8R, 2,S,4,S) – 3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl)-1-azabicyclo [3. 2. 0] -Hept-2-ene-7-keto-2-carboxylic acid trihydrate (I)-Meropectin. 



































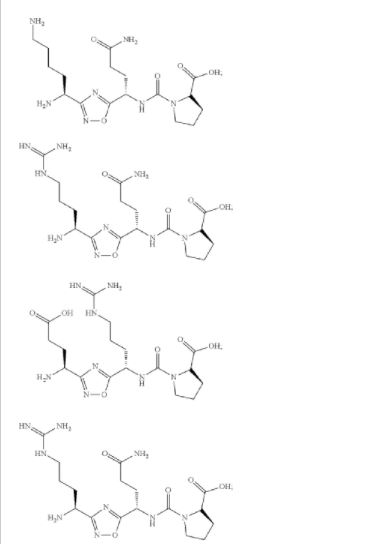


![(2S)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117943691&t=l)



![(2S,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=126843231&t=l)
![(2R,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155529077&t=l)

![(2S,3S)-2-[[(1S)-3-Amino-1-[3-[(1S)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155339943&t=l)