VX- ?
CAS 2446817-72-5
HYDRATE 2446818-26-2
Acetic acid, 1-methylethyl ester 2446818-27-3
C21 H20 F N3 O3, 381.4
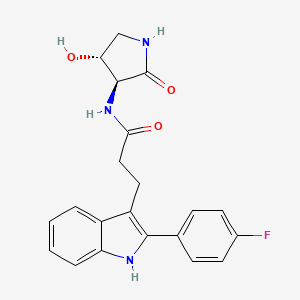
1H-Indole-3-propanamide, 2-(4-fluorophenyl)-N-[(3S,4R)-4-hydroxy-2-oxo-3-pyrrolidinyl]-
3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide
use in treating focal segmental glomerulosclerosis (FSGS) and/or non-diabetic kidney disease (NDKD).

PATENT
SOLID FORMS OF APOL1 INHIBITOR AND METHODS OF USING SAME
Compound I is disclosed as Compound 87 in U.S. Provisional Application No.62/780,667 filed on December 17, 2018, U.S. Application No. 16/717,099 filed onDecember 17, 2019, and PCT International Application No. PCT/US2019/066746 filed on December 17, 2019, the entire contents of each of which are incorporated herein by reference.
Compound I, which can be employed in the treatment of diseases mediated by APOLl, such as FSGS and NDKD
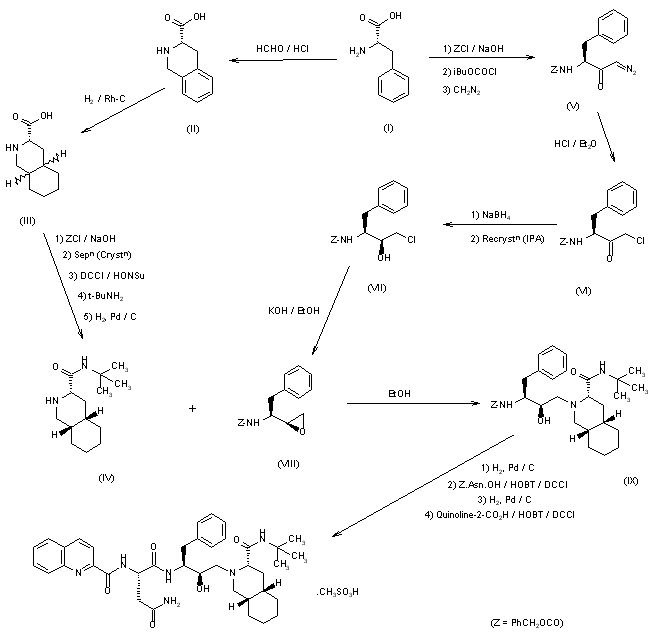
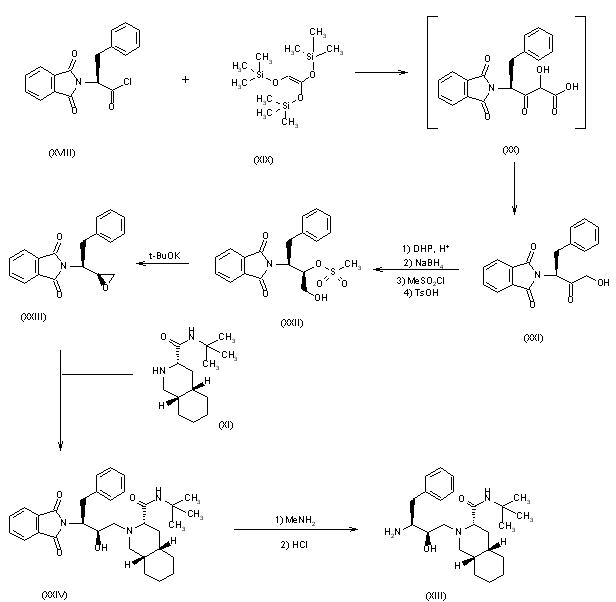
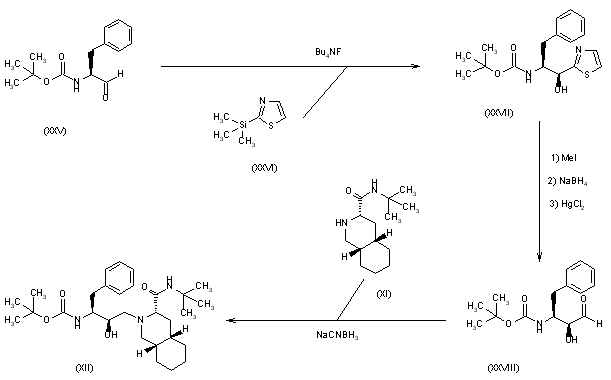
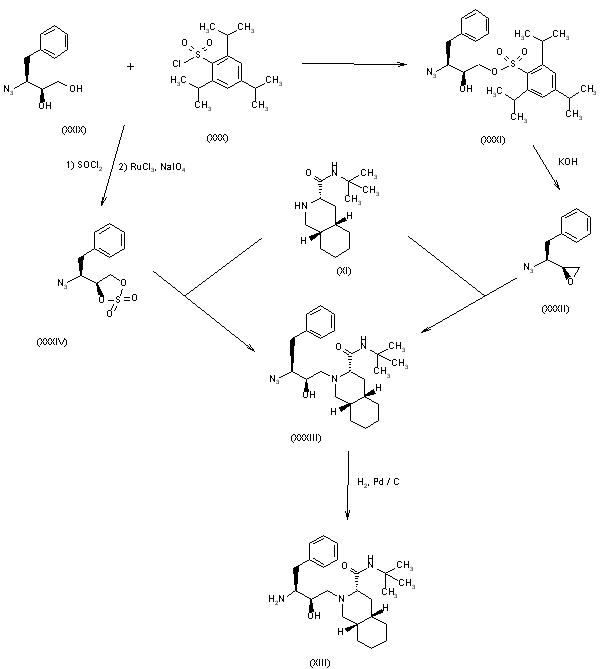
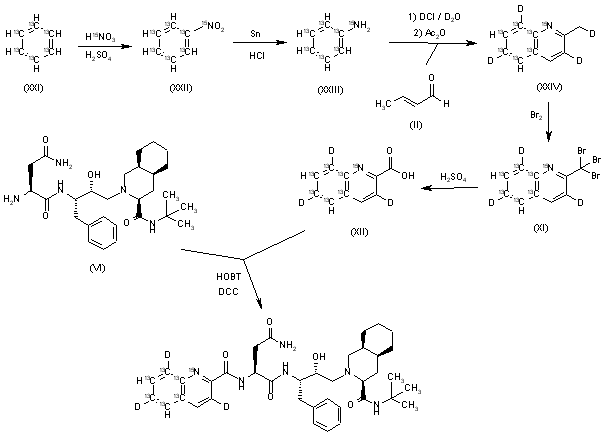
Example 1. Synthesis of Compound
Preparation of Compound I and Forms Thereof
Compound I Compound I /‘– PrOAc solvate Form A
n-pentanol/
n-heptane
Compound I
Form B
Step 1. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00156] To a mixture of C104 (100.0 g, 1.0 equiv) and phenyl hydrazine hydrochloride (72.2 g, 1.05 eqiv) was charged AcOH (800 mL, 8 vol). The mixture was agitated and heated to 85 °C for 16 hours. The batch was cooled to 22 °C. A vacuum was applied and the batch distill at <70 °C to ~3 total volumes. The batch was cooled to 19- 25 °C. The reactor was charged with iPrOAc (800 mL, 8 vol) and then charged with water (800 mL, 8 vol). The internal temperature was adjusted to 20 – 25 °C and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and the phases allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. 1 N HC1 (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the
biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The reactor was charged with 1 N HC1 (500 mL, 5 vol). The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The organic phase was distilled under vacuum at <75 °C to 3 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The resulting slurry was heated to an internal temperature of 85 °C until complete dissolution of solids was achieved. The mixture was allowed to stir for 0.5 h at 85 °C and then cooled to an internal temperature of 19 – 25 °C over 5 h. The mixture was allowed to stir at 25 °C for no less than 2 h. The slurry was filtered. The filter cake was washed with toluene (1 x 2 vol (200 mL) and 1 x 1.5 vol (150 mL)). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford product C101 (95.03 g, 70%).
Step 2. Synthesis of Compound I
[00157] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (50 g, 1.0 equiv), S2 hydrochloride (28.3 g, 1.05 equiv), and CDMT (34.1 g, 1.1 equiv) was charged with 2-MeTHF (200 mL, 4 vol) and DMF (50 mL, 1 vol) and the mixture was agitated. The internal temperature adjusted to <13 °C. The reactor was charged with NMM (64.5 g, 3.5 equiv) over 1 h, while maintaining internal temperature <20 °C. The internal temperature was adjusted to 25 °C and the batch was stirred at that temperature for 14 h. The batch was cooled to 10 °C and charged with water (250 mL, 5 vol) while keeping the internal temperature <20 °C. The batch was then warmed to 20 – 25 °C. Stirring was stopped, and the phases allowed to separate for 10 min. The lower aqueous phase was removed. The aqueous layer was back extracted with 2-MeTHF (2 x 200 mL, 2 x 4 vol) at
20 – 25 °C. The combined organic phases were washed with 1 N HC1 (500 mL, 10 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The lower aqueous phase was removed. The organic phases were washed with 0.25 N HC1 (2 x 250 mL, 2 x 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min for each wash. Lower aqueous phases were removed after each wash. The organic phase was washed with water (250 mL, 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The reactor was charged with 20 wt % Nuchar RGC® and stirred for 4 h. The reaction mixture was filtered through a pad of celite®. The reactor and celite® pad were rinsed with 2-MeTHF. The combined organics were distilled under vacuum at <50 °C to 5 total volumes. The reactor was charged with iPrOAc (500 mL, 10 vol). The organic phase was distilled under vacuum at <50 °C to 5 total volumes. The mixture was charged with additional iPrOAc (400 mL, 8 vol) and distillation under vacuum was repeated. The mixture was charged with additional iPrOAc (250 mL, 5 vol), heated to an internal temperature of 75 °C and stirred for 5 h. The slurry was cooled to 25 °C, over 5 h and stirred for no less than 12 h. The slurry was filtered and the filter cake washed with iPrOAc (2 x 50 mL, 2 x 1 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C to afford Compound I as an iPrOAc solvate (60.38 g including 9.9% w/w iPrOAc, 80.8% yield).
Recrystallization to Form A of Compound I
[00158] Compound I as an iPrOAc solvate (17.16 g after correction for iPrOAc content, 1.0 equiv) was charged to a reactor. A mixture of IP A (77 mL, 4.5 vol) and water (137 mL, 8 vol) were charged to the reactor. The slurry was heated to an internal temperature of 75 °C. The batch was cooled to an internal temperature of 25 °C over 10 h and then stirred at 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 52 mL, 2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford Compound I as a neat, crystalline form (Form A, 15.35 g, 89%).
[00159] The X-ray powder diffractogram of Compound I Form A (FIG. 50) was acquired at room temperature using a PANalytical Empyrean diffractometer equipped with PIXcel ID detector. The peaks are listed in Table A below.
Table A. XRPD of Form A of Compound I
|
I
PATENT
- WO2020131807
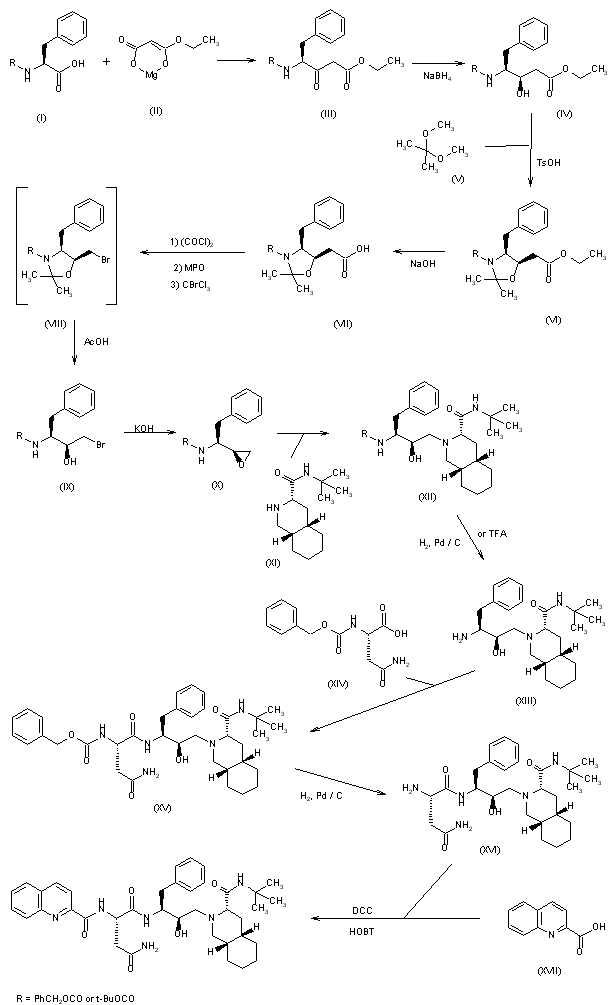
Alternative Preparation I of Compound 87 (Indole preparation route C)
Step 1. Synthesis of 2-(4-fluorophenyl)-lH-indole (C98)
[00401] To a stirred suspension of indole (5 g, 42.7 mmol) and (4- fluorophenyl)boronic acid (8.96 g, 64.0 mmol) in AcOH (200 mL) was
added Pd(OAc)2.Trimer (1.44 g, 6.4 mmol) and the mixture stirred at room temperature for 16 h under 02-balloon pressure. Then the reaction mixture was filtered through a Celite® pad, washed with EtOAc (500 mL). The filtrates were washed with water, sat. NaHC03 solution, brine solution, then dried over Na2S04 and concentrated under reduced pressure. Purification by silica gel chromatography (Gradient: 0-10 % EtOAc in heptane) yielded the product afforded 2-(4-fluorophenyl)-lH-indole (5.5 g, 61 %). ‘H NMR (300 MHz, DMSO-de) 5 11.51 (s, 1H), 7.9 (t, J = 5.4 Hz, 2H), 7.52 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.30 (t, J = 8.7 Hz, 2H), 7.09 (t, J = 12 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.86 (s, 1H). LCMS m/z 212.4 [M+H]+.
Step 2. Synthesis of methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (C99)
[00402] 2-(4-fluorophenyl)-lH-indole (1.0 g, 4.76 mmol) and methyl 3,3-dimethoxypropanoate (0.81 mL, 5.7 mmol) were suspended in dichloromethane (15 mL). Trifluoroacetic acid (2.00 mL, 26 mmol) was added rapidly via syringe, resulting in a clear brown solution. The reaction mixture was heated to 40 °C for three hours. The reaction was diluted with dichloromethane (15 mL) to give an amber solution which was washed with saturated aqueous NaHCCh (25 mL) to yield a bright yellow/light amber biphasic mixture. The phases were separated and the organic layer was washed with saturated NaHCCh (30 mL), then dried (MgSCh) and filtered. The mixture was concentrated under a nitrogen stream overnight. The crude product was obtained as a yellow powder. The product was dissolved in minimum 2-MeTHF and pentane added until the suspension became lightly cloudy. The suspension was allowed to stand overnight, and the precipitate was filtered off. The filter cake was washed with heptane (2 x 15 mL), and dried in vacuo at 40 °C to afford the product as a yellow powder. Methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (1.30 g, 86 %). ¾ NMR (300 MHz, Chloroform -if) d 8.41 (s, 1H), 8.01 – 7.95 (m, 1H), 7.92 (d, J = 16.0 Hz,
1H), 7.58 – 7.50 (m, 2H), 7.46 – 7.41 (m, 1H), 7.33 – 7.27 (m, 2H), 7.22 (t, J = 8.6 Hz, 2H), 6.59 (d, J = 16.0 Hz, 1H), 3.79 (s, 3H). LCMS m/z 295.97 [M+H]+.
Step 3. Synthesis of methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (CIOO)
[00403] To a solution of methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (7 g, 0.02 mol) in EtOAc (350 mL) was added Palladium on carbon (4 g, 10 %w/w, 0.004 mol) and stirred at room temperature for 2 h under an atmosphere of H2 (bladder pressure). The reaction mixture was filtered through a pad of Celite® and washed with EtOAc (400 mL). The filtrates was concentrated to afford methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (7.1 g, 100 %). 1H MR (300 MHz, DMSO-<fc) 5 11.2 (s, 1H), 7.65 (q, J = 5.4 Hz, 2H), 7.54 (d, J = 8.1 Hz, 1H), 7.36 (t, J = 9.0 Hz, 3H), 7.10 (t, J = 8.1 Hz, 1H), 7.02 (t, J = 7.8 Hz, 1H), 3.53 (s, 3H), 3.10 (t, J = 15.9 Hz, 2H), 2.63 (t, J = 15.9 Hz, 2H). LCMS m/z 298.21 [M+H]+. The product was used directly in the subsequent step without further purification.
Step 4. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00404] To stirred solution of methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (14.4 g, 0.05mol) in THF (300 mL), MeOH (300 mL) and H2O (250 mL) was cooled to -10°C. LiOH.H20 (10.1 g, 0.24 mol) was slowly added in a portion-wise manner. The reaction mixture was allowed to stir at room temperature for 16 h. The mixture was
evaporated and ice cold water (200 mL) was added, pH was adjusted to pH- 2 with 1M HC1 (400 mL, Cold solution). The mixture was stirred for 10 minutes, filtered and dried to afford 3-[2-(4-fhiorophenyl)-lH-indol-3-yl]propanoic acid (12.9 g, 94 %). ‘H NMR (400 MHz, DMSCMJ) 5 12.11 (s, 1H), 11.18 (s, 1H), 7.65 (q, J = 5.2 Hz, 2H), 7.56 (d, J = 7.6 Hz, 1H), 7.36 (t, J = 8.8 Hz, 3H), 7.10 (t, J = 8 Hz, 1H), 7.01 (t, J = 8 Hz, 1H), 3.06 (t, J = 16.4 Hz, 2H), 2.55 (t, J = 16 Hz, 2H). LCMS m/z 284.21 [M+H]+.
Step 5. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide (87)
[00405] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (40 g, 120.0 mmol) and (3S,4R)-3-amino-4-hydroxy-pyrrolidin-2-one (Hydrochloride salt) S2 (23.8 g, 156.0 mmol) in DMF (270 mL) was stirred at room temperature for 5 minutes. CDMT (27.2 g, 154.9 mmol) and NMM (53 mL, 482.1 mmol) were added and the mixture was stirred at room temperature for 2 h. The mixture was poured into water (140 mL) and then stirred for 1 h at room temperature, then filtered and washing the solids with water (50 mL). The solids were dissolved in 1 : 1 IP A/water (-400 mL, until all solids dissolved) with heating (reflux) and stirring. The mixture was allowed to cool slowly to room temperature overnight. The mixture was cooled to 0 oC and stirred to break up crystals for filtration. The crystals were then filtered off, rinsed with cold 1 : 1 IP A/water to afford a tan solid (45 g). The solid was dissolved in IPA (200 mL) and heated to 80 °C to dissolve the solid. Activated charcoal (10 g) was added and the mixture was heated with stirring for 30 minutes. The mixture was filtered through Celite ® and solvent removed under reduced pressure. A mixture of 40:60 IP A/water (350 mL) was added to the solid and the mixture was heated until all solids dissolved. The mixture was cooled to room temperature over 5 h. Solids precipitated within the mixture. The mixture was then cooled to 0 °C and stirred for 1 h. The solids were filtered off and air dried on funnel for 1 h, then in a vacuum at 55 °C overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (36.6 g, 79 %). ¾ NMR (300 MHz, Methanol-i¾) d 7.63 (ddt, J= 8.6, 5.1, 2.7 Hz, 3H), 7.35 (dt, J= 8.1, 1.0 Hz, 1H), 7.25 – 7.16 (m, 2H), 7.11 (ddd, J= 8.1, 7.0, 1.3 Hz, 1H), 7.03 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H), 4.34 (td, J= 7.6, 6.8 Hz, 1H), 4.22 (d, J= 7.7 Hz, 1H), 3.55 (dd, J= 9.9, 7.5 Hz, 1H), 3.26 – 3.18 (m, 2H), 3.10 (dd, J= 9.9, 6.8 Hz, 1H), 2.69 – 2.59 (m, 2H). LCMS m/z 382.05 [M+H]+. The
product contained 0.23 % IPA by weight by NMR (1439 ppm IPA by residual solvent analysis). Purity is 99.5 % by (qNMR).
Alternative Preparation II of Compound 87 ( Indole Preparation route D)
Step 1. Synthesis of 5-(4-fluorophenyl)-5-oxo-pentanoic acid (Cl 04)
[00406] To a stirred suspension of AlCb(13.9 g, 0.10 mol) in dichloromethane (50 mL) was added a solution of tetrahydropyran-2,6-dione (5.93 g, 0.05
mol) in dichloromethane (100 mL) at 0 °C over a period of 15 minutes and stirred for 30 min. Then to the reaction mixture was added fluorobenzene (5 g, 0.05 mol) at 0 °C over a period of 15 min, gradually allowed to room temperature and stirred for 16 h. Then the reaction mixture was added to ice water (50 mL) under stirring. The resulting solid was filtered to afford a light yellow solid. The solid was diluted with 3 % NaOH solution (50 mL) and dichloromethane (50 mL). The aqueous layer was separated and acidified with IN HC1 at 0 °C. The mixture was then extracted with EtOAc (100 mL), dried over Na2SC>4, and concentrated under reduced pressure. The solid was then washed with pentane and dried to afford 5-(4-fluorophenyl)-5-oxo-pentanoic acid as an off white solid. (6 g, 53 %). ¾ NMR (300 MHz, DMSO-^) d 12.07 (s, 1H), 8.06 (d, J = 6 Hz, 1H), 8.02 (d, J = 5.4 Hz, 1H), 7.36 (t, J = 8.7 Hz, 2H), 3.06 (t, J = 12 Hz,
2H), 2.31 (t, J = 7.2 Hz, 2H), 1.86-1.78 (m, 2H). LCMS m/z 211.18 [M+H]+.
Step 2. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (Cl 01) [00407] Phenylhydrazine (Hydrochloride salt) (375.7 g, 2.6 mol) was combined with the 5-(4-fluorophenyl)-5-oxo-pentanoic acid (507.7 g, 2.4 mol) in a 12 L three-necked round-bottomed flask equipped with an overhead stirrer, temperature probe, and reflux condenser. AcOH (5 L) was added. The stirring was initiated and ZnCk (605 g, 4.44 mol) was added. The white suspension rapidly thickened after a few minutes (due to formation of the hydrazine intermediate). Approx. 500 mL of extra AcOH was added to aid stirring. The reaction was then heated to 100 °C for three hours. The reaction was cooled to room temperature and poured into water (approx. 6 L). The mixture was extracted with EtOAc (approx 8 L). The extract was washed with water, dried
(MgS04), filtered, and evaporated in vacuo to afford a golden yellow solid. The solid was triturated with approx. 4 L of 10 % EtOAc/DCM and filtered. The filter cake was washed with 50 % dichloromethane/heptane (approx 1 L). The filter cake was dissolved in 40 % EtOAc/dichloromethane (approx. 2L) and filtered over a plug of silica gel. The plug was eluted with 40 % EtOAc/ dichloromethane until the product had been eluted (checked by TLC (25 % EtOAc/ dichloromethane)). The filtrate was evaporated in vacuo to afford 382.6 g of an off-white solid (Crop 1). All filtrates were combined and evaporated in vacuo. The remaining solid was dissolved in 10 %
EtOAc/dichloromethane (approx. 1 L) and chromatographed on a 3 kg silica gel cartridge on the ISCO Torrent (isocratic gradient of 10 % EtOAc/dichloromethane). Product fractions were combined and evaporated in vacuo to afford a yellow solid that was slurried with dichloromethane, cooled under a stream of nitrogen, and filtered. The filter cake was washed with 50 % dichloromethane/heptane and dried in vacuo to afford 244.2 g of product (Crop 2). Altogether, both crops afforded 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (626.8 g, 93 %). ¾ NMR (300 MHz, DMSO-i/e) d 12.15 (s, 1H), 11.20 (s, 1H), 7.74 – 7.62 (m, 2H), 7.57 (d, J = 7.8 Hz, 1H), 7.47 – 7.28 (m, 3H), 7.11 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.02 (ddd, J = 7.9, 7.0, 1.1 Hz, 1H), 3.17 – 2.85 (m, 2H), 2.61 – 2.52 (m, 2H) ppm. 19F NMR (282 MHz, DMSO-i/e) d -114.53 ppm. LCMS m/z 284.15 [M+H]+.
Step 3. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide (87)
[00408] A 3-L three neck RBF under nitrogen was equipped with a 150 mL addition funnel and thermocouple, then loaded with 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (77.2 g, 228.6 mmol), (3S,4R)-3-amino-4-hydroxy-pyrrolidin-2-one
(Hydrochloride salt) (36.6 g, 239.9 mmol) and CDMT (44.2 g, 251.7 mmol). DMF (320 mL) was added and the orange slurry was cooled to -5 °C (acetone/brine/dry ice). NMM (88 mL, 800.4 mmol) was added via a funnel over 75 minutes to keep the internal temp <0 °C. The slurry was stirred at between -10 and 0 °C for 1 hour, then allowed to warm to ambient temperature progressively over 2 hours. Additional reagents were added (10 % of the initial quantities), and the mixture was stirred overnight at ambient temperature. Water (850 mL) was added over 60 minutes, maintaining the internal temperature at <25 °C (ice bath). This slow water addition allows for complete dissolution of any visible salt before precipitation of the product. The resulting thick slurry was stirred at ambient temperature overnight. The solid was recovered by filtration and washed with water (3 x 500 mL). The solid was dried under a stream of air at ambient temperature, then purified by crystallization.
Crystallization of 3- [2-( 4-fluorophenyl)-lH-indol-3-yl ]-N-[ ( 3S, 4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl ] propanamide (87)
[00409] Under nitrogen atmosphere, a 2-L, 3 -neck flask equipped with addition funnel and thermocouple was charged with a light brown suspension of the crude 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yljpropanamide (89.5 g) in IPA (225 mL, 2.5 vol). The slurry was heated to 50 °C and water (675 mL, 7.5 vol) was added until near-complete dissolution of solid was observed. The temperature was adjusted to 70 °C-to achieve full dissolution, yielding a clear amber solution. After 30 minutes, the heat source was removed and the mixture was cooled to ambient temperature over the weekend, stirring gently while maintaining the nitrogen atmosphere. The solid was recovered by filtration, washed with IPA:H20 = 1 :2 (2 x 300 mL, 2 x 3.3 vol) dried under a stream of air overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (84.8 g, 92 %). ¾ NMR (300 MHz, DMSO-^) d 11.19 (s, 1H), 8.23 (d, J= 7.5 Hz, 1H), 7.77 (s, 1H), 7.72 – 7.63 (m, 2H), 7.60 (d, J= 7.8 Hz, 1H), 7.41 -7.31 (m, 3H), 7.12 (ddd, J= 8.1, 7.0, 1.2 Hz, 1H), 7.03 (ddd, J= 8.0, 7.0, 1.1 Hz, 1H), 5.49 (d, J= 5.0 Hz, 1H), 4.20 – 4.06 (m, 2H), 3.38 (s, 1H), 3.11 – 3.00 (m, 2H), 2.92 (dd, J= 9.4, 6.6 Hz, 1H). LCMS m/z 382.15 [M+H]+.
Crystallization of 3- [2-( 4-fluorophenyl)-lH-indol-3-yl J-N-[ ( 3S, 4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl ] propanamide (87)
[00410] A 2-L, 3-neck flask equipped with addition funnel and thermocouple was charged with a light brown suspension of the crude 3-[2-(4-fluorophenyl)-lH-indol-3- yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide in IPA (225 mL, 1 vol). The slurry was heated to 50 °C and water (675 mL, 3 vol) was added until near- complete dissolution of solid observed (mL). Temperature was increased to 70 °C under nitrogen (full dissolution, yielding a clear amber solution). After 30 minutes, the heat was removed and the mixture cooled to ambient temperature over the weekend, stirring gently under nitrogen atmosphere. The solid was recovered by filtration and washed with IPAiLLO = 1 :2 (2 x 300 mL).The solid was dried under a stream of air overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl]propanamide (84.8 g, 92 %). ¾ NMR (300 MHz, DMSO-i/e) d 11.19 (s, 1H), 8.23 (d, J= 7.5 Hz, 1H), 7.77 (s, 1H), 7.72 – 7.63 (m, 2H), 7.60 (d, J= 7.8 Hz,
1H), 7.41 – 7.31 (m, 3H), 7.12 (ddd, J= 8.1, 7.0, 1.2 Hz, 1H), 7.03 (ddd, 7= 8.0, 7.0,
1.1 Hz, 1H), 5.49 (d, J= 5.0 Hz, 1H), 4.20 – 4.06 (m, 2H), 3.38 (s, 1H), 3.11 – 3.00 (m, 2H), 2.92 (dd, J= 9.4, 6.6 Hz, 1H). LCMS m/z 382.15 [M+H]+.
Large Scale Preparation of Compound 87
/- PrOAc solvate Form A
Step 1. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00411] To a mixture of C104 (100.0 g, 1.0 equiv) and phenyl hydrazine hydrochloride (72.2 g, 1.05 eqiv) was charged AcOH (800 mL, 8 vol). The mixture was agitated and heated to 85 °C for 16 hours. The batch was cooled to 22 °C. A vacuum was applied and the batch distill at <70°C to ~3 total volumes. The batch was cooled to 19- 25 °C. The reactor was charged with iPrOAc (800 mL, 8 vol) and then charged with water (800 mL, 8 vol). The internal temperature was adjusted to 20 – 25 °C and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and the phases allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. 1 N HC1 (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The reactor was charged with 1 N HC1 (500 mL, 5 vol). The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h.
Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor.
The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The organic phase was distilled under vacuum at <75 °C to 3 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The resulting slurry was heated to an internal temperature of 85 °C until complete dissolution of solids was achieved. The mixture was allowed to stir for 0.5 h at 85 °C and then cooled to an internal temperature of 19 – 25 °C over 5 h. The mixture was allowed to stir at 25 °C for no less than 2 h. The slurry was filtered. The filter cake was washed with toluene (1 x 2 vol (200 mL) and 1 x 1.5 vol (150 mL)). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford product C101 (95.03 g, 70%).
Purification of Compound 87 by Recrystallization to Form A
[00412] Compound 87 as an iPrOAc solvate (17.16 g after correction for iPrOAc content, 1.0 equiv) was charged to a reactor. A mixture of IP A (77 mL, 4.5 vol) and water (137 mL, 8 vol) were charged to the reactor. The slurry was heated to an internal temperature of 75 °C. The batch was cooled to an internal temperature of 25 °C over 10 h and then stirred at 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 52 mL, 2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford Compound 87 as a neat, crystalline form (Form A, 15.35 g, 89%).
Synthetic Procedure
[00413] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (50 g, 1.0 equiv), S2 hydrochloride (28.3 g, 1.05 equiv), and CDMT (34.1 g, 1.1 equiv) was charged with 2-MeTHF (200 mL, 4 vol) and DMF (50 mL, 1 vol) and the mixture was agitated. The internal temperature adjusted to <13 °C. The reactor was charged with NMM (64.5 g, 3.5 equiv) over 1 h, while maintaining internal temperature <20 °C. The internal temperature was adjusted to 25 °C and the batch was stirred at that temperature for 14 h. The batch was cooled to 10 °C and charged with water (250 mL, 5 vol) while keeping the internal temperature <20 °C. The batch was then warmed to 20 – 25 °C. Stirring was stopped, and the phases allowed to separate for 10 min. The lower aqueous phase was removed. The aqueous layer was back extracted with 2-MeTHF (2 x 200 mL, 2 x 4 vol) at 20 – 25 °C. The combined organic phases were washed with 1 N HC1 (500 mL, 10 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The lower aqueous phase was removed. The organic phases were washed with 0.25 N HC1 (2 x 250 mL, 2 x 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min for each wash. Lower aqueous phases were removed after each wash. The organic phase was washed with water (250 mL, 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The reactor was charged with 20 wt % Nuchar RGC® and stirred for 4 h. The reaction mixture was filtered through a pad of celite®. The reactor and celite® pad were rinsed with 2-MeTHF. The combined organics were distilled under vacuum at <50 °C to 5 total volumes. The reactor was charged with iPrOAc (500 mL, 10 vol). The organic phase was distilled under vacuum at <50 °C to 5 total volumes. The mixture was charged with additional iPrOAc (400 mL, 8 vol) and distillation under vacuum was repeated. The mixture was charged with additional iPrOAc (250 mL, 5 vol), heated to an internal
temperature of 75 °C and stirred for 5 h. The slurry was cooled to 25 °C, over 5 h and stirred for no less than 12 h. The slurry was filtered and the filter cake washed with iPrOAc (2 x 50 mL, 2 x 1 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C to afford Compound 87 as an iPrOAc solvate (60.38 g including 9.9% w/w iPrOAc, 80.8% yield).
Form A of Compound 87
[00414] Compound 87 hydrate form was converted to the dehydrated, neat crystalline form (Form A) after drying.
Hydrate Form A of Compound 87
[00415] A mixture of IP A (4.5 vol) and water (8 vol) was added to compound 87
(iPrOAc solvate containing ~2.5 – 11 wt% iPrOAc, 1.0 equiv). The slurry was heated to an internal temperature of 75 °C and filtered hot. The filtrate was cooled to 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C. The product was isolated as Hydrate form.
IPAC Solvate of Compound 87:
[00416] The large scale synthesis described above provided an iPrOAc solvate containing ~2.5 – 11 wt% iPrOAc after drying.
Amorphous Form of Compound 87
[00417] ~lg of compound 87 was dissolved in 22mL of acetone. The solution was evaporated using a Genevac. The resulted solid was dried at 60C under vacuum overnight. The dried solid was amorphous form.
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2020131807-A1 | Inhibitors of apol1 and methods of using same | 2018-12-17 | |
| US-2020377479-A1 | Inhibitors of apol1 and methods of using same | 2018-12-17 |
///////////
O=C(N[C@@H]1C(=O)NC[C@H]1O)CCc1c2ccccc2[NH]c1c1ccc(F)cc1
SIMILAR

predicted
VX 147
cas 2446816-88-0 predicted
O=C(N[C@@H]1C(=O)NC[C@H]1O)CCc1c2cc(F)cc(F)c2[NH]c1c1ccc(F)cc1
- OriginatorVertex Pharmaceuticals
- ClassSmall molecules; Urologics
- Mechanism of ActionApolipoprotein L1 inhibitors
- Orphan Drug StatusNo
- New Molecular EntityYes
Highest Development Phases
- Phase IIFocal segmental glomerulosclerosis
- Phase IKidney disorders
Most Recent Events
- 14 Apr 2020Phase-II clinical trials in Focal segmental glomerulosclerosis in USA (PO) (EudraCT2020-000185-42) (NCT04340362)
- 31 Dec 2019Vertex Pharmaceuticals completes phase I clinical trial in Focal segmental glomerulosclerosis and Kidney disorders (In volunteers) in USA (PO)
- 05 Aug 2019Vertex Pharmaceuticals plans a phase II proof-of-concept trial for focal segmental glomerulosclerosis in 2020
| NCT Number ICMJE | NCT04340362 |
|---|---|
| Other Study ID Numbers ICMJE | VX19-147-101 2020-000185-42 ( EudraCT Number ) |





























































 technology, is a long-acting prodrug of somatropin that releases the same somatropin used in daily therapies –
technology, is a long-acting prodrug of somatropin that releases the same somatropin used in daily therapies –






























![Synthesis of thiamine, method by Williams and Cline [90].](http://www.researchgate.net/profile/Artur-Ratkiewicz/publication/321626762/figure/fig5/AS:661906792648715@1534822285512/Synthesis-of-thiamine-method-by-Williams-and-Cline-90.png)