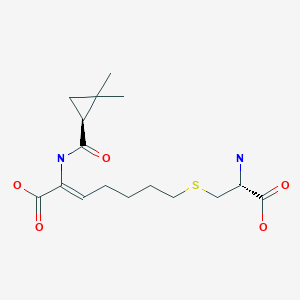
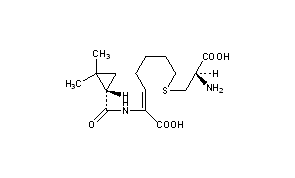

Cilastatin
シラスタチン
UNII141A6AMN38
CAS number 82009-34-5
WeightAverage: 358.453
Monoisotopic: 358.156242642
Chemical FormulaC16H26N2O5S
- (L)-7-(2-Amino-2-carboxy-ethylsulfanyl)-2-[(2,2-dimethyl-cyclopropanecarbonyl)-amino]-hept-2-enoic acid
- (Z)-(S)-6-carboxy-6-[(S)-2,2-dimethylcyclopropanecarboxamido]hex-5-enyl-L-cysteine
- (Z)-7-((R)-2-Amino-2-carboxy-ethylsulfanyl)-2-[((S)-2,2-dimethyl-cyclopropanecarbonyl)-amino]-hept-2-enoic acid
- (2Z)-7-{[(2R)-2-amino-2-carboxyethyl]sulfanyl}-2-{[(1S)-2,2-dimethylcyclopropyl]formamido}hept-2-enoic acid
FDA 2019 APPROVED 2019/7/16, Imipenem, cilastatin and relebactam, Recarbrio
|
Antibacterial
|
|
| Disease |
Uncomplicated urinary tract infection
|
|---|
Cilastatin inhibits the human enzyme dehydropeptidase.[1]
Yatendra Kumar, “Process for the preparation of amorphous cilastatin sodium.” U.S. Patent US20040152780, issued August 05, 2004.US20040152780
Cilastatin is an inhibitor of renal dehydropeptidase, an enzyme responsible for both the metabolism of thienamycin beta-lactam antibiotics as well as conversion of leukotriene D4 to leukotriene E4. Since the antibiotic, imipenem, is one such antibiotic that is hydrolyzed by dehydropeptidase, cilastatin is used in combination with imipenem to prevent its metabolism. The first combination product containing both drugs was approved by the FDA in November of 1985 under the trade name Primaxin, marketed by Merck & Co.9 A newer triple-drug product was approved in July 2019 under the trade name Recarbrio which also contains relebactam.8
Cilastatin is indicated, in combination with imipenem with or without relebactam, for the treatment of bacterial infections including respiratory, skin, bone, gynecologic, urinary tract, and intra-abdominal as well as septicemia and endocarditis.6,5

Uses
Dehydropeptidase is an enzyme found in the kidney and is responsible for degrading the antibiotic imipenem. Cilastatin can therefore be combined intravenously with imipenem in order to protect it from degradation, prolonging its antibacterial effect.
Imipenem alone is an effective antibiotic and can be given without cilastatin. Cilastatin itself does not have antibiotic activity, although it has been proved to be active against a zinc-dependent beta-lactamase that usually confers antibiotic resistance to certain bacteria, more precisely, the carbapenem family of antibiotics. This property is due to the physicochemical similarities between membrane dipeptidase (MDP), the compound it is usually set to target, and the bacterial metallo-beta-lactamase carried by the CphA gene.[1] The combination allows the antibiotic to be more effective by changing the pharmacokinetics involved. Thus imipenem/cilastatin, like amoxicillin/clavulanic acid, is a commonly used combination product.
PATENT
https://patents.google.com/patent/EP2402312A1
Cilastatin sodium is the sodium salt of a derivatized heptenoic acid. Its chemical name is [R-[R*,S*-(Z)]]-7-[(2-amino-2-carboxyethyl)thio]-2-[[(2,2-dimethylcyclopropyl)carbonyl]amino]-2-heptenoic acid, monosodium salt. It is an off-white to yellowish-white, hygroscopic, amorphous compound. PRIMAXIN (Imipenem and Cilastatin) is a formulation of Imipenem (a thienamycin antibiotic) and Cilastatin sodium.
Imipenem with Cilastatin acts as an effective antibiotic for the treatment of infections of various body systems. PRIMAXIN is a potent broad-spectrum antibacterial agent for intramuscular administration. Imipenem can be further described as a semi-synthetic thienamycin that is administered intravenously or intramuscularly in combination with Cilastatin to reduce toxicity. Cilastatin, a renal dipeptidase inhibitor, inhibits the enzymatic breakdown of Imipenem and increases urinary excretion of the active drug.
Originally Cilastatin was disclosed in US patent number 5,147,868 . This patent also discloses various processes for the preparation of Cilastatin, particularly example 19 A of this patent disclose a process for the preparation of Cilastatin. According to this example the condensation of 7-chloro-2-oxoheptanoic acid ethyl ester (I) with (S)-2,2-dimethylcyclopropanecarboxamide (II) by means of p-toluene sulphonic acid in refluxing toluene gives (S)-7-chloro-2-(2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid ethyl ester (III), which is hydrolyzed in aq. NaOH to yield the corresponding carboxylic acid (IV). Finally, this compound is condensed with (R)-cysteine (V) by means of NaOH in water to afford the target Cilastatin, followed by isomerisation to at 3.0 pH. The process followed in this example is depicted as below:


Example 1Preparation of 7-chloro-2-[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid (II) (starting material):
- [0032]
To the solution of S-2, 2-dimethylcylopropyl carboxamide (100gm) in toluene (500) was added Ethyl-7-chloro-2-oxo-heptanoate (270gm) and p-toluene sulphonic acid (1.5gm). The resulted solution was refluxed for 20hrs azeotropically. The resulted mass was cooled to 5-10°C and added the solution of sodium hydroxide (140gm) in water 500 ml and the resulted two-layered solution was stirred for 8hrs at 25-30°C up to the complete disappearance of ester. The toluene layer was separated and the aqueous layer was washed with toluene. The pH of the aqueous layer was adjusted to 4.0 to 4.5 and extracted with toluene (1 lt). The toluene layer containing 7-chloro-2-[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid was washed with water and used as such for the next step. The ratio of Z and E isomer 90:10% was obtained.
Example 2Isomerisation of 7-chloro-2-[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid (II):
- [0033]
To the toluene layer, obtained from example -1, was added hydrochloric acid (11t) and stirred for 4hrs at 25-30°C till the disappearance of E isomer. The toluene layer was separated and washed with water and followed by brine. The toluene layer was distilled out under vacuum up to 50% of the original volume. To the reaction mass hexane/IPE was added at 50°C and cooled to 0-5°C. The precipitated mass was filtered and washed with hexane (200ml) and dried under vacuum to obtained 99% pure Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid (150gm) as white solid.
Example 3Preparation of Cilastatin Acid (I):
- [0034]
To the solution of sodium hydroxide (90gm) in water (11t) was added L-Cysteine hydrochloride monohydrate (96gm) and Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid and stirred at 25-30°C till the disappearance of Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid. After completion of reaction, the reaction mass was washed with dichloromethane (500ml). To the aqueous layer was added carbon (10 gm) and stirred and filtered. To the filtrate was added water (11t) and the pH of the solution was adjusted to 3.0 and stirred for 24 hrs. The precipitated mass was filtered, washed with water (200ml) and with acetone (500ml) and dried to obtain 110gm white solid with 97% purity. The solid was dissolved in water (700ml) and added MDC (700ml) and ethyl acetate (100ml) and stirred for 10hrs. The precipitated mass was filtered and washed with water (100ml) and acetone (200ml) and dried to obtain 100gm white Cilastatin acid with 99.5% purity.
Example 4Preparation of Cilastatin Sodium:
- [0035]
The Cilastatin acid (100gm, 99.5%) was dissolved in the mixture of ethanol (2.5lt) and triethylamine (30gm) at 25 to 30°C. To the resulted clear solution was added carbon (10gm) and stirred and filtered. The filtrated was filtered again through sterile micron (0.2 µ) filter. To the resulted clear solution was added solution of sodium ethyl hexanoate (70gm) in ethanol (70ml) and stirred for 3hrs at 25 to 30°C.The precipitated Cilastatin sodium was filtered and washed with ethanol (80ml) and followed by acetone (200ml) and dried under vacuum to obtained 95gm Cilastatin sodium as amorphous white solid with 99.5% purity.
Example 5Preparation of Cilastatin Acid:
- [0036]
To the solution of sodium hydroxide (88gm) in methanol (1500ml) was added Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid and stirred to dissolve. To the resulted clear solution was added L-Cysteine hydrochloride monohydrate (97gm) and stirred the resulted suspension at 60 to 65°C till the disappearance of Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid. After completion of reaction, the pH insoluble salts were filtered. The filtrate was distilled out under vacuum. The residue was dissolved in water (500ml) and washed with dichloromethane (500ml). The pH of aqueous layer was adjusted to 3 to 4 from the original pH in the range of 5.5, and with n-butanol (500ml). The butanol layer was washed with water and distilled. The residue was dissolved in water (100ml) and added acetonitrile (1500ml) at 50°C and further refluxed at 80°C for one hr. The precipitated cilastatin acid was filtered and washed with acetonitrile (100ml). The crude wet cake (60gm) was refluxed with acetonitrile water mixture (9:1,1500ml), and cooled to yield 60gm pure cilastatin acid with 99.5% purity.
Example 6Preparation of Cilastatin Acid:
- [0037]
To the solution of sodium hydroxide (88gm) in methanol (1500ml) was added Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid and stirred to dissolve. To the resulted clear solution was added L-Cysteine hydrochloride monohydrate (97gm) and stirred the resulted suspension at 60 to 65°C till the disappearance of Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid. The pH of the reaction mass was adjusted to 7.0 with conc.HCl and filterd the insoluble salts. The filtrated was distilled out under vacuum. The residue was dissolved in water (500ml) and washed with dichloromethane (500ml). The pH of aqueous layer was adjusted to 3 to 4 from the original pH in the range of 5.5, and with n-butanol (500ml). The butanol layer was washed with water and distilled up to 50% of original volume and stirred at 25°C. The precipitated cilastatin acid was filtered and washed with n-butanol (100ml) followed by acetone to yield 60gm pure cilastatin acid with 99.7% purity.
Example 7Preparation of Cilastatin, Sodium:
- [0038]
The Cilastatin acid (100gm, 99.5%) was dissolved in the mixture of n-butanol (2.5lt) and triethylamine (30gm) at 25 to 30°C. To the resulted clear solution was added carbon (10gm) and stirred and filtered. The filtrated was filtered again through sterile micron (0.2 µ) filter. To the resulted clear solution was added solution of sodium ethyl hexanoate (70gm) in n-butanol (70ml) and stirred for 3hrs at 25 to 30°C. The precipitated Cilastatin sodium was filtered and washed with n-butanol (80ml) and followed by acetone (200ml) and dried under vacuum to obtained 80gm Cilastatin sodium as amorphous white solid with 99.78% purity.
Abbreviations;
- [0039]
- DBU: diazabicyclo[5,4,0]undec-7-en
- DBN : 1,5-diazabicyclo[4,3,0]-non-5-ene
- TMG: 1,1,3,3-tetramethylguanidine
- DABCO: 1,4-diazabicyclo-[2,2,2]-octane
PATENT
https://patents.google.com/patent/WO2006022511A1/en
Cilasatin sodium salt i.e., [R-[R*, S*-(Z)]] –
7-[(2-amino-2-carboxyethylthio)-2-[[(2,2-dimethylcyclopropyl)carbonyl]amino-2-hepa tenoic acid monosodium salt represented by following chemical formulae (1)1 has been used with imipenem in order to prevent its renal metabolism. Imipenem/cilastatin sodium is used as a potent broad spectrum antibacterial agent. [3] There have been several reports on the method for preparing a cilastatin sodium until now: for example, EP 48301 Bl discloses a method for the preparation of a cilastatin sodium salt using by Grignard reaction started from l-bromo-5-chloropentane (2′) explained by following Reaction Scheme 1; Donald W.
Graham et al discloses a preparation method using ethyl- 1, 3-dithian-2-carboxylate as a starting material (Donald W. Graham et al, J. Med. Chem., 30, pplO74, 1987) etc. [4] [Reaction Scheme 1]
[5]

[6]

[7] [8] As shown in the above Reaction Scheme 1, l-bromo-5-chloropentane (2′) is reacted with diethyl oxalate through Grignard reaction to afford ethyl 7-chloro-2-oxo-hepanoate (3′)at the 1st step; ethyl 7-chloro-2-oxo-heptanoate (3′) is reacted with (S)-2, 2-dimethylcyclopropanecarboxamide to obtain ethyl (Z )-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate (4′) at the 2n step.
[9] However, present inventors has confirmed that considerable amount (about 10 to 13%) of (E)-form isomer thereof (7′)was produced during the 2nd step as a reaction impurity by gas chromatography. The (E)-form isomer is further subjected to hydrolysis resulting in (E)-7-chloro-2-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (8′)as shown in following Reaction Scheme 2.
[10] [H] However, present inventors has confirmed that considerable amount (about 10 to 13%) of (E)-form isomer thereof (7′) was produced during the 2nd step as an reaction impurities by gas chromatography as shown in following Reaction Scheme 2. The (E )-form isomer is further subjected to hydrolysis resulting in (E)-7-chloro-2-((S)-2, 2-dimethylcyclopropylcarboxamide)-2-heptanoic acid (8′).
[12] [Reaction Scheme 2] [13]


[14] [15] There have been tried to solve the problems for example, the isomer impurity was removed by the acidification followed by recrystallization step or by adding cysteine to the reaction solution obtained in the 3r step at the above described 4 step, reacting with together to form (E)-7-(L-amino-2-carboxyethylthio)-2-((S )-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid and finally removing the reacted impurity by acidifying and heating step in the known preparation till now. However, the present inventors found that there remained unsolved problem such that the recrystallization yield of the product, i.e., (Z)-7-chloro-2-((S )-2,2-dimethylcyclopropylcarboxamide)-2-heptanoic acid was very poor because of the formed byproduct, i.e., (E
)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid in 3rdstep and further the unknown impurity (10′) and (S)-2,2-dimethylcyclopropanecarboxamide (H’) were produced by acidifying and heating reaction solution at the above described the 4 step as shown in following Reaction Scheme 3 confirmed by HPLC analysis, which give rise to another difficulty in the purification of final products. [16] [17] [Reaction Scheme 3] [18]

NH
(Z) and (E) m ix ture (91)

C ondition
(105 0 15
[19] In addition to above described problems, present inventors have found that the cilastatin isolated through the above described 4th step consisting of eluting the cation exchange resin with ammonia solution, concentrating the eluate and solidifying with ethanol and diethyl ether exists in the form of its ammonium salt not free acid form as disclosed in the patent. Using an acid such as hydrochloric acid in order to obtain free acid accompany with unwanted formation of inorganic ammonium salt such as ammonium chloride, which could not afford high purity of cilastatin sodium salt in the end.
[20] [21] Therefore, there have been tried to solve the above-described problems: for example, PCTAVO 0318544 (Al) discloses the isolation method using by neutral HP 20 resin column instead of cationic resin disclosed in EP 48301 Bl; PCTAVO 02094742 (Al) discloses the method for preparing cilastatin sodium salt (Ia) from cilastatin (6′), the disclosure of which cited documents are incorporated herein by reference.
[22]
[23] However, the above-described methods for preparing cilastatin using column chro¬ matographic process are not suitable for commercial mass production.
[24]
[25] The present inventors have made extensive researches to discover novel method for preparing cilastatin sodium salt with high yield and mass production and finally completed the invention by founding novel preparation for obtaining purposed cilastatin sodium salt; i.e., selectively hydrolyzing (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate, isolating (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt from the reaction mixture, adopting the cilastatin amine salt instead of free acid form disclosed in cited references and the use of sodium hydroxide and cationic exchange resin with pH control in order to obtain cilastatin sodium salt with high purity and high yield.
Example 1: Preparation of ethyl (Z)-7-chloro-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate (4)
[67]
[68] l-bromo-5-chloropentane (29 Ig, 1.57 mol) was reacted with diethyl oxalate
(206.5g) through Grignard reaction to obtain ethyl 7-chloro-2-oxo-heptanoate (3) and the compound (3) was reacted with (S)-2,2-dimethylcyclopropanecarboxamide to obtain ethyl (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptanoate (237g, 0.79 mol). The above-described step was performed by the procedure according to the procedure disclosed in EP 48301 (Bl).
[69]
[70] Example 1: Preparation of ethyl (Z)-7-chloro-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12)
[71]
[72] 1-1. ( Z V7-chloro-(YSV2. 2-dimethylcyclopropanecarboxamidoV2-heptanoic acid sodium salt
[73] The ethyl (Z)-7-chloro-2-((S
)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoate (237g, 0.79 mol) obtained in Comparative Example 1 was dissolved in 877ml of methanol and 1.8 L of sodium hydroxide solution (0.48 M) was added with stirring at room temperature. The reaction was finished when the area ratio of (Z) isomer and (E) isomer becomes 20: 1 by HPLC analysis and the un-reacted organic reagent was extracted with 490 ml of dichloromethane. The pH of the solution was adjusted to 7-8 with 3N HCl and the un- reacted organic reagent was extracted with 490 ml of dichloromethane again. The water layer was concentrated under reduced pressure and 650ml of ethanol was added and stirred until the solid had been dissolved at 50°C, for 30 minute to 1 hour. The un- dissolved solid was removed with filtration and the filtrate was concentrated under reduced pressure. 2.4 L of acetonitrile is added thereto and stirred to obtain 140.8g of ( Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12 ; 55% yield).
[74]
[75]
[76] m.p.: 219°C;
[77] 1H-NMR (D2O, 300MHz) δppm: 0.87 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.61 (m, 2H), 1.68 (dd, IH), 1.78 (m, 2H), 2.12 (m, 2H), 3.62 (t, 2H), 6.47 (t, IH);
[78]
13
[79] 13C ( -NMR (D2O, 300MHz) δppm: 19.47, 19.99, 22.55, 25.74, 26.75, 27.53, 29.44,
32.27, 46.11, 131.41, 136.52, 172.74, 174.62.
[80] [81] [82]
[83] 1-2. ( Z V7-chloro-(YSV2. 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid
(12-D
[84] 140.8g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12) obtained from Example 1-1 was dissolved in 422 ml of distilled water. The pH of the solution was adjusted to 2.0-3.0 with 3N HCl, extracted with 592 ml of isopropylether two times and 59.2g of anhydrous magnesium sulfate was added to isopropylether layer, stirred and subjected to filtration. The filtrate was concentrated to afford 127.7g of (Z)-7-chloro-2-((S )-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (12-1, 98% yield).
[85]
[86] 1H-NMR (CDCl3, 300MHz) δppm: 0.83 (dd, IH), 1.19 (s, 7H), 1.44 (dd, IH), 1.19
(s, 3H), 1.64 (m, 2H), 1.81 (m, 2H), 2.21 (m, 2H), 3.54 (t, 2H), 6.78 (t, IH), 7.04 (br, IH);
[87]
13
[88] 13C ( -NMR (CDCl3, 300MHz) δppm: 18.69, 20.82, 22.86, 25.36, 27.03, 28.53,
29.27, 32.17, 44.60, 124.88, 139.49, 168.96, 170.15.
[89]
[90]
[91] 1-3. ( Z V7-chloro-((SV2. 2-dimethylcyclopropanecarboxamidoV2-heptenoic acid ammonium salt (12-2)
[92] 127.7g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (12-1) obtained from Example 1-2 was dissolved in 422 ml of EtOH. 100 ml of 25% ammonia water solution was added thereto, stirred and concentrated to obtain 135.6g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid ammonium salt (12-2, 100% yield).
[93]
[94] Example 2: Preparation of cilastatin ammonium salt (13-1)
[95] 4Og of (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12, 0.14 mol) obtained in Example 1-1 was dissolved in 120 ml of 0.48 M sodium hydroxide solution and 240 ml of ethanol and the mixture of 1.4g of NaBr (0.013 mol) and 25.3g of L-cysteineDHClDH) was added thereto, stirred at 55°C, for 8 hours.
[96] The pH of the reaction solution was adjusted to 5.5-5.0 with 3N HCl, concentrated and 800ml of methanol was added, stirred at 55°C for 1 hour and un-dissolved salt was filtered out. The filtrate was concentrated to the extent that the volume of total solution was reduced to about 1/2. The concentrate was adsorbed with cationic exchange resin (PK208 model, Samyang Co.), washed with distilled water to the extent that the con¬ ductivity of the solution became less than lθμs(microsiemens), eluted with 2N ammonia water and the eluate was concentrated under the reduced pressure to give brown solid compound. The compound was dissolved in 40 ml of distilled water. 0.8 L of 2-propanol was added thereto and the solution was subjected to salting out method with reflux for 2 hours. The resulting solid was cooled and filtered to obtain 45.66g of cilastatin ammonium salt (13-1. 90% yield).
[97]
[98] m. p.: 161°C;
[99] Element Analysis: C16H29N3O5S (MW: 375.183): CaI. Q51.18; 7.78; N:11.19; Est.
C:51.01; H: 7.97; N: 11.04;
[100] MS m/z : 375 (M+, 49), 312(36), 97 (84.2), 69 (100);
[101] 1H-NMR (D2O, 300MHz) δppm: 0.87 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.62 (m, 5H), 2.1 l(q, 2H), 2.62 (t, 2H), 3.06 (m, 4H), 3.91 (dd, IH), 6.47 (t, IH);
13
[102] ” (C-NMR (D2O, 300MHz) δppm: 19.49, 19.97, 22.53, 26.74, 27.44, 27.86, 29.09,
29.43, 31.94, 32.85, 54.44, 131.23, 136.83, 172.70, 173.71, 174.64.
[103]
[104] Example 3: Preparation of cilastatin ethylamine salt (13-2)
[105] 4Og of (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12-1, 0.15 mol) obtained in Example 1-2 was dissolved in 165 ml of 0.66 M sodium hydroxide solution and 330 ml of ethanol and the mixture of 1.5g of NaBr (0.015 mol) and 27.6g of L-cysteineDHClDH) was added thereto, stirred at 55°C, for 8 hours.
[106] The pH of the reaction solution was adjusted to 5.5-5.0 with 3N HCl, concentrated and 800ml of methanol was added, stirred at 55°C for 1 hour and un-dissolved salt was filtered out.. The filtrate was concentrated to the extent that the volume of total solution was reduced to about 1/2. The concentrate was adsorbed with cationic exchange resin (PK208 model, Samyang Co.), washed with distilled water to the extent that the conductivity of the solution became less than 10μs(microsiemens), eluted with 2N ethylamine water and the eluate was concentrated under the reduced pressure to give brown solid compound. The compound was dissolved in 40 ml of distilled water. 0.8 L of 2-propanol was added thereto and the solution was subjected to salting out method with reflux for 2 hours. The resulting solid was cooled and purified with filtration to obtain 49.38g of cilastatin ethylamine salt (13-2. 90% yield).
[107]
[108] 1H-NMR (D2O, 300MHz) δppm: 0.86 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.27 (t, 3H), 1.60 (m, 5H), 2.1 l(q, 2H), 2.62 (t, 2H), 3.06 (m, 4H), 3.91 (dd, IH), 6.47 (t, IH); [109] 13C-NMR (D2O, 300MHz) δppm: 14.7, 21.57, 22.04, 24.63. 28.82, 29.52, 29.94,
31.16, 31.49, 34.00, 34.91, 37.78, 56.50, 133.27, 138.96, 174.75, 175.81, 176.74.
[HO]
[111] Example 4 : Purification of cilastatin ammonium salt
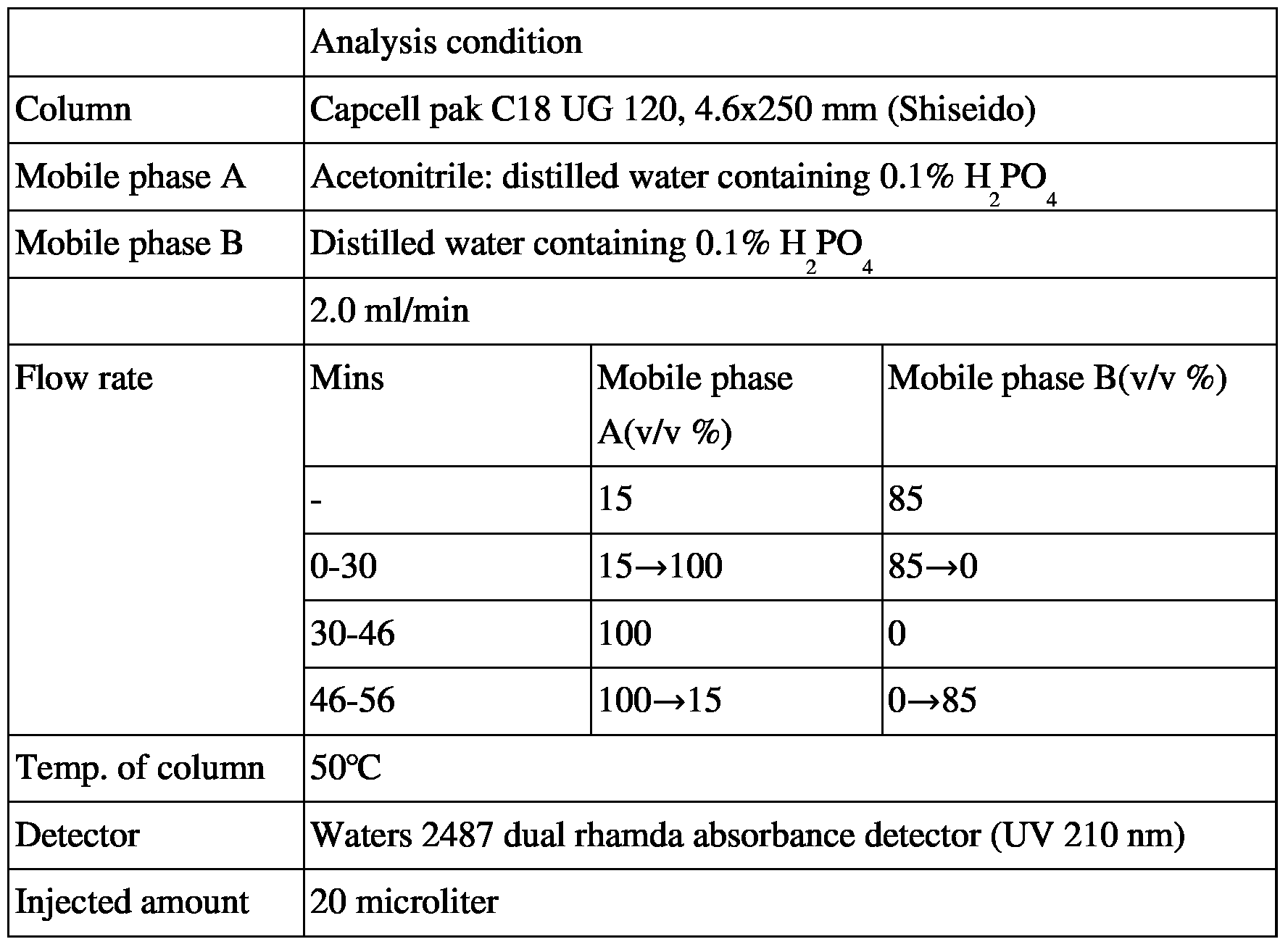
[112] 4-1. Purification using by water and ethanol
[113] 45.66g of cilastatin ammonium salt (13-1,0.12 mol) obtained in Example 2 was dissolved in 45.66 ml of distilled water and 1.3L of anhydrous ethanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 38.81g of cilastatin ammonium salt (Yield: 85%, Purity: 99.8%).
[114]
[115] 4-2. Purification using by ammonia water and propanol Q)
[116] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 50 ml of 25% ammonia water and 1.5L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 41.2g of cilastatin ammonium salt (Yield: 82.4%, Purity: 99.3%).
[117]
[118] 4-3. Purification using by ammonia water and propanol (1)
[119] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in
100 ml of 25% ammonia water and 2.0L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was purified with filtration to obtain 35.4g of cilastatin ammonium salt (Yield: 70.8%, Purity: 99.3%)
[120]
[121] 4-4. Purification using by ammonia water and ethanol
[122] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 50 ml of 25% ammonia water and 1.5 L of anhydrous ethanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 35.6g of cilastatin ammonium salt (Yield: 71.2%, Purity: 99.8%).
[123]
[124] 4-5. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (1)
[125] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 50ml of 4N ammonia water, and 2.0 L of 1 -propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 89.3g of cilastatin ammonium salt (Yield: 89.3%, Purity: 99.6%).
[126]
[127] 4-6. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (1) [128] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 50ml of 2N ammonia water, and 2.0 L of 1-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 95.0g of cilastatin ammonium salt (Yield: 95.0%, Purity: 99.5%).
[129]
[130] 4-7. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (3)
[131] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 100 ml of distilled water and 50ml of 25% ammonia water, and 2.0 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 89.7g of cilastatin ammonium salt (Yield: 89.7%, Purity: 99.8%).
[132]
[133] 4-8. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (4)
[134] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 100ml of 25% ammonia water, and 3.0 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 80.Og of cilastatin ammonium salt (Yield: 80.0%, Purity: 99.7%).
[135]
[136] 4-9. Purification using by water and propanol
[137] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 100 ml of distilled water and 1.5 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 87.2g of cilastatin ammonium salt (Yield: 87.2%, Purity: 99.6%).
[138]
[139] Example 5: Preparation of cilastatin sodium salt
[140] 4.28g of sodium hydroxide (0.107 mol) was dissolved in 38.3 ml of distilled water and 191.5 ml of ethanol. 38.81g of cilastatin ammonium salt (0.1 mol) obtained in Example 4-1 was added thereto and stirred for 30 minutes. The solution was con¬ centrated under reduced pressure at 60°C and 153 ml of distilled water was added to the concentrate. The solution was stirred to dissolve the concentrate and the pH of the solution was adjusted to 7.0 using by cationic exchange resin and filtered. The filtrate was lyophilized to obtain high purity (99.4%) of cilastatin sodium salt.
[141]
[142] Experimental Example 1: Purity Determination [143] The purity of cilastatin ammonium salt obtained in Example 4 was determined by
HPLC on condition as shown in Table 1 and the determined result was shown in Table 2.
[144] Table 1
[145] Table 2

[146]
Industrial Applicability
[147] The novel method of the present invention could prevent the formation of (E )-isomer from the preparation of novel intermediate for preparing cilastatin sodium, i.e., (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt and isolate the intermediate in situ providing simpler process with high yield and purity. Furthermore, it can provide with highly purified cilastatin sodium salt by isolating novel cilastatin amine salt and using sodium hydroxide and cationic exchange resin. Accordingly, the method can be very useful in preparing cilastatin sodium salt with high yield and high purity.





References
- ^ Jump up to:a b Keynan S, Hooper NM, Felici A, Amicosante G, Turner AJ (1995). “The renal membrane dipeptidase (dehydropeptidase I) inhibitor, cilastatin, inhibits the bacterial metallo-beta-lactamase enzyme CphA”. Antimicrob. Agents Chemother. 39 (7): 1629–31. doi:10.1128/aac.39.7.1629. PMC 162797. PMID 7492120.
- Keynan S, Hooper NM, Felici A, Amicosante G, Turner AJ: The renal membrane dipeptidase (dehydropeptidase I) inhibitor, cilastatin, inhibits the bacterial metallo-beta-lactamase enzyme CphA. Antimicrob Agents Chemother. 1995 Jul;39(7):1629-31. [PubMed:7492120]
- Buckley MM, Brogden RN, Barradell LB, Goa KL: Imipenem/cilastatin. A reappraisal of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy. Drugs. 1992 Sep;44(3):408-44. [PubMed:1382937]
- Balfour JA, Bryson HM, Brogden RN: Imipenem/cilastatin: an update of its antibacterial activity, pharmacokinetics and therapeutic efficacy in the treatment of serious infections. Drugs. 1996 Jan;51(1):99-136. doi: 10.2165/00003495-199651010-00008. [PubMed:8741235]
- Koller M, Brom J, Raulf M, Konig W: Cilastatin (MK 0791) is a potent and specific inhibitor of the renal leukotriene D4-dipeptidase. Biochem Biophys Res Commun. 1985 Sep 16;131(2):974-9. doi: 10.1016/0006-291x(85)91335-x. [PubMed:3863619]
- FDA: Recarbrio Label [Link]
- FDA: Primaxin Label [Link]
- ChemSpider: Cilastatin [Link]
- FDA Label: Apadaz [Link]
- Drugs@FDA: Primaxin [Link]
Synthesis
By Panchapakesan, Ganapathy et alFrom Indian, 269299, 16 Oct 2015
IN 269299

SYN
Patent
Cilastatin
-
- ATC:J01DH51
- Use:dehydropeptidase inhibitor (for combination with imipenem)
- Chemical name:[R-[R*,S*-(Z)]]-7-[(2-amino-2-carboxyethyl)thio]-2-[[(2,2-dimethylcyclopropyl)carbonyl]amino]-2-heptenoic acid
- Formula:C16H26N2O5S
- MW:358.46 g/mol
- CAS-RN:82009-34-5
- InChI Key:DHSUYTOATWAVLW-WFVMDLQDSA-N
- InChI:InChI=1S/C16H26N2O5S/c1-16(2)8-10(16)13(19)18-12(15(22)23)6-4-3-5-7-24-9-11(17)14(20)21/h6,10-11H,3-5,7-9,17H2,1-2H3,(H,18,19)(H,20,21)(H,22,23)/b12-6-/t10-,11+/m1/s1
- EINECS:279-875-8
- LD50:8 g/kg (M, route unreported);
8 g/kg (R, route unreported)
Derivatives
monosodium salt
- Formula:C16H25N2NaO5S
- MW:380.44 g/mol
- CAS-RN:81129-83-1
- EINECS:279-694-4
- LD50:6786 mg/kg (M, i.v.); >10 g/kg (M, p.o.);
5027 mg/kg (R, i.v.); >10 g/kg (R, p.o.)
|
|
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a686013 |
| Routes of administration |
IV |
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.072.592 |
| Chemical and physical data | |
| Formula | C16H26N2O5S |
| Molar mass | 358.454 g/mol g·mol−1 |
| 3D model (JSmol) | |

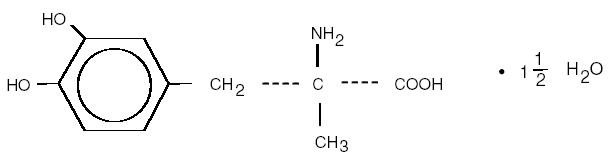
/////////////cilastatin, シラスタチン , FDA 2019, циластатин , سيلاستاتين , 西司他丁 , MK-791, Recarbrio
CC1(C)C[C@@H]1C(=O)N\C(=C/CCCCSC[C@H](N)C(O)=O)C(O)=O


























































 ”
”




















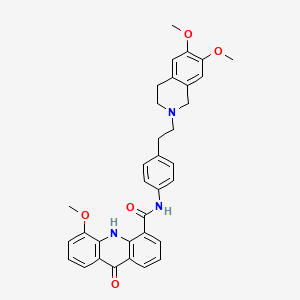

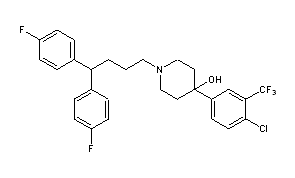
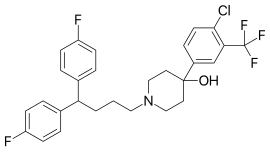








 with 4-chloro-3-trifluoromethyl phenyl magnesium bromide reacted Penfluridol (I).
with 4-chloro-3-trifluoromethyl phenyl magnesium bromide reacted Penfluridol (I).