![Elexacaftor.png]()
![str1]()
![]()
VX-445, Elexacaftor, エレクサカフトル
597.658 g/mol, C26H34F3N7O4S
3-Pyridinecarboxamide, N-((1,3-dimethyl-1H-pyrazol-4-yl)sulfonyl)-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)-2-((4S)-2,2,4-trimethyl-1-pyrrolidinyl)-
N-[(1,3-Dimethyl-1H-pyrazol-4-yl)sulfonyl]-6-[3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl]-2-[(4S)-2,2,4-trimethyl-1-pyrrolidinyl]-3-pyridinecarboxamide
3-Pyridinecarboxamide, N-((1,3-dimethyl-1H-pyrazol-4-yl)sulfonyl)-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)-2-((4S)-2,2,4-trimethyl-1-pyrrolidinyl)-
UNII-RRN67GMB0V
RRN67GMB0V
VX-445
WHO 11180
Cas 2216712-66-0
WHO 11180
Treatment of cystic fibrosis, CFTR modulator
Elexacaftor is under investigation in clinical trial NCT03525548 (A Study of VX-445 Combination Therapy in CF Subjects Homozygous for F508del (F/F)).
Cystic fibrosis transmembrane conductance regulator (CFTR) corrector designed to restore Phe508del CFTR protein function in patients with cystic fibrosis when administered with tezacaftor and ivacaftor.
VX-445 (elexacaftor), tezacaftor, and ivacaftor triple-drug combo
Vertex Pharmaceuticals (NASDAQ: VRTX) already claims a virtual monopoly in treating the underlying cause of cystic fibrosis (CF). The biotech’s current three CF drugs should generate combined sales of close to $3.5 billion this year. Another blockbuster is likely to join those three drugs on the market in 2020 — Vertex’s triple-drug CF combo featuring VX-445 (elexacaftor), tezacaftor, and ivacaftor.
EvaluatePharma projects that this triple-drug combo will rake in close to $4.3 billion by 2024. The market researcher pegs the net present value of the drug at nearly $20 billion, making it the most valuable pipeline asset in the biopharmaceutical industry right now.
PATENT
WO 2018107100
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018107100&tab=PCTDESCRIPTION&queryString=novozymes&recNum=152&maxRec=27502
Also disclosed herein is Compound 1:
![]()
[0013] N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide.
Synthesis of Compound 1
[00256] Part A: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
![]()
[00257] Step 1: methyl-2,4-dimethyl-4-nitro-pentanoate
![]()
[00258] Tetrahydrofuran (THF, 4.5 L) was added to a 20 L glass reactor and stirred under N2 at room temperature.2-Nitropropane (1.5 kg, 16.83 mol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.282 kg, 8.42 mol) were then charged to the reactor, and the jacket temperature was increased to 50 °C. Once the reactor contents were close to 50 °C, methyl methacrylate (1.854 kg, 18.52 mol) was added slowly over 100 minutes. The reaction temperature was maintained at or close to 50 °C for 21 hours. The reaction mixture was concentrated in vacuo then transferred back to the reactor and diluted with methyl tert-butyl ether (MTBE) (14 L).2 M HCl (7.5 L) was added, and this mixture was stirred for 5 minutes then allowed to settle. Two clear layers were visible– a lower yellow aqueous phase and an upper green organic phase. The aqueous layer was removed, and the organic layer was stirred again with 2 M HCl (3 L). After separation, the HCl washes were recombined and stirred with MTBE (3 L) for 5 minutes. The aqueous layer was removed, and all of the organic layers were combined in the reactor and stirred with water (3 L) for 5 minutes. After separation, the organic layers were concentrated in vacuo to afford a cloudy green oil. Crude product was treated with MgSO4 and filtered to afford methyl-2,4-dimethyl-4-nitro-pentanoate as a clear green oil (3.16 kg, 99% yield).
[00259] 1H NMR (400 MHz, Chloroform-d) δ 3.68 (s, 3H), 2.56– 2.35 (m, 2H), 2.11 – 2.00 (m, 1H), 1.57 (s, 3H), 1.55 (s, 3H), 1.19 (d, J = 6.8 Hz, 3H).
[00260] Step 2: Synthesis of methyl (2S)-2,4-dimethyl-4-nitro-pentanoate
![]()
[00261] A reactor was charged with purified water (2090 L; 10 vol) and then potassium phosphate monobasic (27 kg, 198.4 moles; 13 g/L for water charge). The pH of the reactor contents was adjusted to pH 6.5 (± 0.2) with 20% (w/v) potassium carbonate solution. The reactor was charged with racemic methyl-2,4-dimethyl-4-nitro-pentanoate (209 kg; 1104.6 moles), and Palatase 20000L lipase (13 L, 15.8 kg; 0.06 vol).
[00262] The reaction mixture was adjusted to 32 ± 2 °C and stirred for 15-21 hours, and pH 6.5 was maintained using a pH stat with the automatic addition of 20% potassium carbonate solution. When the racemic starting material was converted to >98% ee of the S-enantiomer, as determined by chiral GC, external heating was switched off. The reactor was then charged with MTBE (35 L; 5 vol), and the aqueous layer was extracted with MTBE (3 times, 400-1000L). The combined organic extracts were washed with aqueous Na2CO3 (4 times, 522 L, 18 % w/w 2.5 vol), water (523 L; 2.5 vol), and 10% aqueous NaCl (314 L, 1.5 vol). The organic layer was concentrated in vacuo to afford methyl (2S)-2,4-dimethyl-4-nitro-pentanoate as a mobile yellow oil (>98% ee, 94.4 kg; 45 % yield).
[00263] Step 3: Synthesis of (3S)-3,5,5-trimethylpyrrolidin-2-one
![]()
[00264] A 20 L reactor was purged with N2. The vessel was charged sequentially with DI water-rinsed, damp Raney® Ni (2800 grade, 250 g), methyl (2S)-2,4-dimethyl-4-nitro-pentanoate (1741g, 9.2 mol), and ethanol (13.9 L, 8 vol). The reaction was stirred at 900 rpm, and the reactor was flushed with H2 and maintained at ~2.5 bar. The reaction mixture was then warmed to 60 °C for 5 hours. The reaction mixture was cooled and filtered to remove Raney nickel, and the solid cake was rinsed with ethanol (3.5 L, 2 vol). The ethanolic solution of the product was combined with a second equal sized batch and concentrated in vacuo to reduce to a minimum volume of ethanol (~1.5 volumes). Heptane (2.5 L) was added, and the suspension was concentrated again to ~1.5 volumes. This was repeated 3 times; the resulting suspension was cooled to 0-5 °C, filtered under suction, and washed with heptane (2.5 L). The product was dried under vacuum for 20 minutes then transferred to drying trays and dried in a vacuum oven at 40 °C overnight to afford (3S)-3,5,5-trimethylpyrrolidin-2-one as a white crystalline solid (2.042 kg, 16.1 mol, 87 %).1H NMR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (dd, J = 12.4, 8.6 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
[00265] Step 4: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
![]()
[00266] A glass lined 120 L reactor was charged with lithium aluminum hydride pellets (2.5 kg, 66 mol) and dry THF (60 L) and warmed to 30 °C. The resulting suspension was charged with (S)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C, then cautiously quenched with the addition of ethyl acetate (EtOAc) (1.0 L, 10 moles), followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq), and then a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 equiv water with 1.4 equiv sodium hydroxide relative to aluminum), followed by 7.5 L water. After the addition was complete, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HCl (1.05 equiv.) while maintaining the temperature below 30°C. The resultant solution was concentrated by
vacuum distillation to a slurry. Isopropanol (8 L) was added and the solution was concentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added, and the product was slurried by warming to about 50 °C. MTBE (6 L) was added, and the slurry was cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L MTBE and dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford (4S)-2,2,4-trimethylpyrrolidine•HCl as a white, crystalline solid (6.21 kg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (br d, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H).
[00267] Part B: Preparation of N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (Compound 1)
![]()
![]()
[00268] Preparation of starting materials:
[00269] 3,3,3-Trifluoro-2,2-dimethyl-propan-1-ol
![]()
[00270] A 1 L 3 neck round bottom flask was fitted with a mechanical stirrer, a cooling bath, an addition funnel, and a J-Kem temperature probe. The vessel was charged with lithium aluminum hydride (LAH) pellets (6.3 g, 0.1665 mol) under a nitrogen atmosphere. The vessel was then charged with tetrahydrofuran (200 mL) under a nitrogen atmosphere. The mixture was allowed to stir at room temperature for 0.5 hours to allow the pellets to dissolve. The cooling bath was then charged with crushed ice in water and the reaction temperature was lowered to 0 oC. The addition funnel was charged with a solution of 3,3,3-trifluoro-2,2-dimethyl-propanoic acid (20 g, 0.1281 mol) in tetrahydrofuran (60 mL) and the clear pale yellow solution was added drop wise over 1 hour. After the addition was complete the mixture was allowed to slowly warm to room temperature and stirring was continued for 24 hours. The suspension was cooled to 0 oC with a crushed ice-water in the cooling bath and then quenched by the very slow and drop wise addition of water (6.3 ml), followed by sodium hydroxide solution (15 weight %; 6.3 mL) and then finally with water (18.9 mL). The reaction temperature of the resulting white suspension was recorded at 5 oC. The suspension was stirred at ~5 oC for 30 minutes and then filtered through a 20 mm layer of Celite. The filter cake was washed with tetrahydrofuran (2 x 100 mL). The filtrate was dried over sodium sulfate (150 g) and then filtered. The filtrate was concentrated under reduced pressure to provide a clear colorless oil (15 g) containing a mixture of the product 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol in THF (73 % weight of product ~10.95g, and 27 wt.% THF as determined by 1H-NMR). The distillate from the rotary evaporation was distilled at atmospheric pressure using a 30 cm Vigreux column to provide 8.75 g of a residue containing 60 % weight of THF and 40 % weight of product (~3.5 g). The estimated total amount of product is 14.45 g (79% yield).1H NMR (400 MHz, DMSO-d6) δ 4.99 (t, J = 5.7 Hz, 1H), 3.38 (dd, J = 5.8, 0.9 Hz, 2H), 1.04 (d, J = 0.9 Hz, 6H).
[00271] tert-Butyl 3-oxo-2,3-dihydro-1H-pyrazole-1-carboxylate
![]()
[00272] A 50L Syrris controlled reactor was started and jacket set to 20 °C, stirring at 150 rpm, reflux condenser (10 °C) and nitrogen purge. MeOH (2.860 L) and methyl (E)-3-methoxyprop-2-enoate (2.643 kg, 22.76 mol) were added and the reactor was capped. The reaction was heated to an internal temperature of 40 °C and the system was set to hold jacket temp at 40 °C. Hydrazine hydrate (1300 g of 55 %w/w, 22.31 mol) was added portion wise via addition funnel over 30 min. The reaction was heated to 60 ^C for 1 h. The reaction mixture was cooled to 20 ^C and triethyamine (2.483 kg, 3.420 L, 24.54 mol) was added portion wise (exothermic), maintaining reaction temp <30 °C.
A solution of Boc anhydride (di-tert-butyl dicarbonate) (4.967 kg, 5.228 L, 22.76 mol) in MeOH (2.860 L) was added portion wise maintaining temperature <45 °C. The reaction mixture was stirred at 20 ^C for 16 h. The reaction solution was partially concentrated to remove MeOH, resulting in a clear light amber oil. The resulting oil was transferred to the 50L reactor, stirred and added water (7.150 L) and heptane (7.150 L). The additions caused a small amount of the product to precipitate. The aqueous layer was drained into a clean container and the interface and heptane layer were filtered to separate the solid (product). The aqueous layer was transferred back to the reactor, and the collected solid was placed back into the reactor and mixed with the aqueous layer. A dropping funnel was added to the reactor and loaded with acetic acid (1.474 kg, 1.396 L, 24.54 mol), then began dropwise addition of acid. The jacket was set to 0 °C to absorb the quench exotherm. After addition (pH=5), the reaction mixture was stirred for 1 h. The solid was collected by filtration and washed with water (7.150 L), and washed a second time with water (3.575 L) and pulled dry. The crystalline solid was scooped out of the filter into a 20L rotovap bulb and heptane (7.150 L) was added. The mixture was slurried at 45 °C for 30 mins, and then distilled off 1-2 volumes of solvent. The slurry in the rotovap flask was filtered and the solids washed with heptane (3.575 L) and pulled dry. The solid was further dried in vacuo (50 °C , 15 mbar) to give tert-butyl 5-oxo-1H-pyrazole-2-carboxylate (2921 g, 71%) as coarse, crystalline solid.1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 7.98 (d, J = 2.9 Hz, 1H), 5.90 (d, J = 2.9 Hz, 1H), 1.54 (s, 9H).
[00273] Step A: tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate
![]()
[00274] A mixture of 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol (10 g, 70.36 mmol) and tert-butyl 3-hydroxypyrazole-1-carboxylate (12.96 g, 70.36 mmol) in toluene (130 mL) was treated with triphenyl phosphine (20.30 g, 77.40 mmol) followed by isopropyl N-isopropoxycarbonyliminocarbamate (14.99 mL, 77.40 mmol) and the mixture was stirred at 110 °C for 16 hours. The yellow solution was concentrated under reduced
pressure, diluted with heptane (100mL) and the precipitated triphenylphosphine oxide was removed by filtration and washed with heptane/toluene 4:1 (100mL). The yellow filtrate was evaporated and the residue purified by silica gel chromatography with a linear gradient of ethyl acetate in hexane (0-40%) to give tert-butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (12.3 g, 57%) as an off white solid. ESI-MS m/z calc.308.13477, found 309.0 (M+1) +; Retention time: 1.84 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.10 (d, J = 3.0 Hz, 1H), 6.15 (d, J = 3.0 Hz, 1H), 4.18 (s, 2H), 1.55 (s, 9H), 1.21 (s, 6H).
[00275] Step B: 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole
![]()
[00276] tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (13.5 g, 43.79 mmol) was treated with 4 M hydrogen chloride in dioxane (54.75 mL, 219.0 mmol) and the mixture was stirred at 45 °C for 1 hour. The reaction mixture was evaporated to dryness and the residue was extracted with 1 M aqueous NaOH (100ml) and methyl tert-butyl ether (100ml), washed with brine (50ml) and extracted with methyl tert-butyl ether (50ml). The combined organic phases were dried, filtered and evaporated to give 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 96%) as an off white waxy solid. ESI-MS m/z calc.208.08235, found 209.0 (M+1) +;
Retention time: 1.22 minutes.1H NMR (400 MHz, DMSO-d6) δ 11.91 (s, 1H), 7.52 (d, J = 2.2 Hz, 1H), 5.69 (t, J = 2.3 Hz, 1H), 4.06 (s, 2H), 1.19 (s, 6H).
[00277] Step C: tert-Butyl 2,6-dichloropyridine-3-carboxylate
![]()
[00278] A solution of 2,6-dichloropyridine-3-carboxylic acid (10 g, 52.08 mmol) in THF (210 mL) was treated successively with di-tert-butyl dicarbonate (17 g, 77.89 mmol) and 4-(dimethylamino)pyridine (3.2 g, 26.19 mmol) and left to stir overnight at room temperature. At this point, HCl 1N (400 mL) was added and the mixture was stirred vigorously for about 10 minutes. The product was extracted with ethyl acetate (2x300mL) and the combined organics layers were washed with water (300 mL) and brine (150 mL) and dried over sodium sulfate and concentrated under reduced pressure to give 12.94 g (96% yield) of tert-butyl 2,6-dichloropyridine-3-carboxylate as a colorless oil. ESI-MS m/z calc.247.01668, found 248.1 (M+1) +; Retention time: 2.27 minutes.1H NMR (300 MHz, CDCl3) ppm 1.60 (s, 9H), 7.30 (d, J=7.9 Hz, 1H), 8.05 (d, J=8.2 Hz, 1H).
[00279] Step D: tert-Butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate
![]()
[00280] To a solution of tert-butyl 2,6-dichloropyridine-3-carboxylate (10.4 g, 41.9 mmol) and 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 41.93 mmol) in DMF (110 mL) were added potassium carbonate (7.53 g, 54.5 mmol) and 1,4-diazabicyclo[2.2.2]octane (706 mg, 6.29 mmol) and he mixture was stirred at room temperature for 16 hours. The cream suspension was cooled in a cold water bath and cold water (130 mL) was slowly added. The thick suspension was stirred at room temperature for 1 hour, filtered and washed with plenty of water to give tert-butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 99%) as an off white solid. ESI-MS m/z calc.419.12234, found 420.0 (M+1) +; Retention time: 2.36 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 2.9 Hz, 1H), 8.31 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 6.26 (d, J = 2.9 Hz, 1H), 4.27 (s, 2H), 1.57 (s, 9H), 1.24 (s, 6H).
[00281] Step E: 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid
![]()
[00282] tert-butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 40.25 mmol) was suspended in isopropanol (85 mL) treated with hydrochloric acid (34 mL of 6 M, 201 mmol) and heated to reflux for 3 hours (went almost complete into solution at reflux and started to precipitate again). The suspension was diluted with water (51 mL) at reflux and left to cool to room
temperature under stirring for 2.5 h. The solid was collected by filtration, washed with isopropanol/water 1:1 (50mL), plenty of water and dried in a drying cabinet under vacuum at 45-50 °C with a nitrogen bleed overnight to give 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (13.7 g, 91%) as an off white solid. ESI-MS m/z calc.363.05975, found 364.0 (M+1) +; Retention time: 1.79 minutes. 1H NMR (400 MHz, DMSO-d6) δ 13.61 (s, 1H), 8.44 (d, J = 2.9 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 6.25 (d, J = 2.9 Hz, 1H), 4.28 (s, 2H), 1.24 (s, 6H).
[00283] Step F: 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide
![]()
[00284] 2-Chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (100 mg, 0.2667 mmol) and CDI (512 mg, 3.158 mmol) were combined in THF (582.0 µL) and the mixture was stirred at room temperature. Meanwhile, 1,3-dimethylpyrazole-4-sulfonyl chloride (62 mg, 0.3185 mmol) was combined with ammonia (in methanol) in a separate vial, instantly forming a white solid. After stirring for an additional 20 min, the volatiles were removed by evaporation, and 1 mL of dichloromethane was added to the solid residue, and was also evaporated. DBU (100 µL, 0.6687 mmol) was then added and the mixture stirred at 60 °C for 5 minutes, followed by addition of THF (1 mL) which was subsequently evaporated. The contents of the vial containing the CDI activated carboxylic acid in THF were then added to the vial containing the newly formed sulfonamide and DBU, and the reaction mixture was stirred for 4 hours at room temperature. The reaction mixture was diluted with 10 mL of ethyl acetate, and washed with 10 mL solution of citric acid (1 M). The aqueous layer was extracted with ethyl acetate (2x 10 mL) and the combined organics were washed with brine, dried over sodium sulfate, and concentrated to give the product as white solid (137 mg, 99%) that was used in the next step without further purification. ESI-MS m/z calc.520.09076, found 521.1 (M+1) +; Retention time: 0.68 minutes.
[00285] Step G: N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide
![]()
[00286] 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (137 mg, 0.2630 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (118 mg, 0.7884 mmol) , and potassium carbonate (219 mg, 1.585 mmol) were combined in DMSO (685.0 µL) and the mixture was heated at 130 ^C for 16 hours. The reaction was cooled to room temperature, and 1 mL of water was added. After stirring for 15 minutes, the contents of the vial were allowed to settle, and the liquid portion was removed via pipet and the remaining solids were dissolved with 20 mL of ethyl acetate and were washed with 1 M citric acid (15 mL). The layers were separated and the aqueous layer was extracted two additional times with 15 mL of ethyl acetate. The organics were combined, washed with brine, dried over sodium sulfate and concentrated. The resulting solid was further purified by silica gel chromatography eluting with a gradient of methanol in dichloromethane (0-10%) to give N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (72 mg, 41%) as a white solid. ESI-MS m/z calc.597.2345, found 598.3 (M+1) +; Retention time: 2.1 minutes.1H NMR (400 MHz, DMSO) δ 12.36 (s, 1H), 8.37 (s, 1H), 8.22 (d, J = 2.8 Hz, 1H), 7.74 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 6.17 (d, J = 2.8 Hz, 1H), 4.23 (s, 2H), 3.81 (s, 3H), 2.56 (d, J = 10.4 Hz, 1H), 2.41 (t, J = 8.7 Hz, 1H), 2.32 (s, 3H), 2.18 (dd, J = 12.4, 6.1 Hz, 1H), 1.87 (dd, J = 11.7, 5.5 Hz, 1H), 1.55 (d, J = 11.2 Hz, 6H), 1.42 (t, J = 12.0 Hz, 1H), 1.23 (s, 6H), 0.81 (d, J = 6.2 Hz, 3H).
[00287] Alternative Steps F and G:
[00288] Alternative Step F: 2-chloro-N-((1,3-dimethyl-1H-pyrazol-4-yl)sulfonyl)-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinamide
[00289]
[ ![]()
[00291] To a suspension of 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (20.0 g, 53.89 mmol) in THF (78.40 mL) was added solid carbonyldiimidazole (approximately 10.49 g, 64.67 mmol) portion wise and the resulting solution was stirred at room temperature (slight exotherm from 18-21 °C was observed). After 1 h, solid 1,3-dimethylpyrazole-4-sulfonamide
(approximately 11.33 g, 64.67 mmol) was added, followed by DBU (approximately 9.845 g, 9.671 mL, 64.67 mmol) in two equal portions over 1 min (exotherm from 19 to 35 °C). The reaction mixture was stirred at room temperature for 16 h. The reaction mixture was diluted with ethyl acetate (118 mL) and then HCl (approximately 107.8 mL of 2 M, 215.6 mmol). The phases were separated and the aqueous phase was extracted
with ethyl aceate (78 mL). The combined organics were washed with water (39.2 mL), then brine (40 mL), dried over sodium sulfate and concentrated. The resulting foam was crystallized from a 1:1 isopropanol:heptane mixture (80 mL) to afford 2-chloro-N-((1,3-dimethyl-1H-pyrazol-4-yl)sulfonyl)-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinamide (26.1 g, 93%) as a white solid. ESI-MS m/z calc.520.0, found 520.9 (M+1) +; Retention time: 1.83 minutes.
[00292] Alternative Step G: N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide
[ ![]()
[00294] 2-chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (20.0 g, 38.39 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (approximately 14.36 g, 95.98 mmol), and K2CO3 (approximately 26.54 g, 192.0 mmol) were combined in DMSO (80.00 mL) and 1,2-diethoxyethane (20.00 mL) in a 500-mL flask with reflux condenser. The reaction mixture was heated at 120 °C for 16 h then cooled to room temperature. The reaction was diluted with DCM (200.0 mL) and HCl (approximately 172.8 mL of 2 M, 345.5 mmol); aqueous pH ~1. The phases were separated, and the aqueous phase was extracted with DCM (100.0 mL). The organic phases were combined, washed with water (100.0 mL) (3 x), and dried (Na2SO4) to afford an amber solution. The solution was filtered through a DCM-packed silica gel bed (80 g; 4 g/g) and washed with 20% EtOAc/DCM (5 x 200 mL). The combined filtrate/washes were concentrated to afford 22.2 g of an off-white powder. The powder was slurried in MTBE (140 mL) for 30 min. The solid was collected by filtration (paper/sintered-glass) to afford 24 g after air-drying. The solid was transferred to a drying dish and vacuum-dried (40 °C/200 torr/N2 bleed) overnight to afford 20.70 g (90%) of a white powder. ESI-MS m/z calc.
597.2345, found 598.0 (M+1)+; Retention time: 2.18 minutes.
[00295] 1H NMR (400 MHz, Chloroform-d) δ 13.85 (s, 1H), 8.30 (d, J = 8.6 Hz, 1H), 8.23 (d, J = 2.8 Hz, 1H), 8.08 (s, 1H), 7.55 (d, J = 8.5 Hz, 1H), 5.98 (d, J = 2.8 Hz, 1H), 4.24 (s, 2H), 3.86 (s, 3H), 3.44 (dd, J = 10.3, 8.4 Hz, 1H), 3.09 (dd, J = 10.3, 7.8 Hz, 1H), 2.67– 2.52 (m, 1H), 2.47 (s, 3H), 2.12 (dd, J = 12.3, 7.8 Hz, 1H), 1.70 (dd, J = 12.4, 9.6 Hz, 1H), 1.37 (s, 3H), 1.33 (s, 3H), 1.27 (s, 6H), 1.20 (d, 3H).
[00296] Alternative Synthesis of 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole
![]()
Step 1: Preparation of 3,3,3-trifluoro-2,2-dimethylpropan-1-ol
![]()
A reactor was loaded with toluene (300 mL) and 3,3,3-trifluoro-2,2-dimethylpropanoic acid (30 g, 192.2 mmol), capped, purged under nitrogen. The reaction was set to control the internal temperature to 40 °C. A solution of Vitride (65% in toluene. approximately 119.6 g of 65 %w/w, 115.4 mL of 65 %w/w, 384.4 mmol) was set up for addition via syringe, and addition was begun at 40 °C, with the target addition temperature between 40 and 50 °C. The reaction was stirred at 40 °C for 90 min. The reaction was cooled to 10 °C then the remaining Vitride was quenched with slow addition of water (6 mL). A solution of 15 % aq NaOH (30 mL) was added in portions, and solids precipitated half way through the base addition. Water (60.00 mL) was added. The mixture was warmed to 30 °C and held for at least 15 mins. The mixture was then cooled to 20 °C. The
aqueous layer was removed. The organic layer was washed with water (60 mL x 3), and then washed with brine (60 mL). The washed organic layer was dried under Na2SO4, followed with MgSO4. The mix was filtered through Celite, and the cake washed with toluene (60.00 mL) and pulled dry. The product 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol (22.5 g, 82%) was obtained as clear colorless solution.
Step 2: Preparation of 1-(tert-butyl) 4-ethyl 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-1,4-dicarboxylate
![]()
A reactor was charged with 3,3,3-trifluoro-2,2-dimethylpropan-1-ol (17.48 g, 123.0 mmol) solution in toluene (250g), 1-(tert-butyl) 4-ethyl 3-hydroxy-1H-pyrazole-1,4-dicarboxylate (30.0 g, 117.1 mmol), and PPh3 (35.33 g, 134.7 mmol). The reaction was heated to 40 °C. DIAD (26.09 mL, 134.7 mmol) was weighed and placed into a syringe and added over 10 minutes while maintaining an internal temperature ranging between 40 and 50 °C. The reaction was then heated to 100 °C over 30 minutes. After holding at 100 °C for 30 minutes, the reaction was complete, and the mixture was cooled to 70 °C over 15 minutes. Heptane (180.0 mL) was added, and the jacket was cooled to 15 °C over 1 hour. (TPPO began crystallizing at ~35 °C). The mixture stirring at 15 °C was filtered (fast), the cake was washed with a pre-mixed solution of toluene (60 mL) and heptane (60 mL) and then pulled dry. The clear solution was concentrated to a waxy solid (45 °C, vacuum, rotovap). Crude 1-(tert-butyl) 4-ethyl 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-1,4-dicarboxylate (53.49g) was obtained as a waxy solid, (~120% of theoretical mass recovered).
Step 3: Preparation of 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid
![]()
A solution of 1-(tert-butyl) 4-ethyl 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-1,4-dicarboxylate (50.0 g, 131 mmol) in 2-methyltetrahydrofuran (500 mL) was prepared in a reactor and stirred at 40 °C. Portions of KOt-Bu (80.85 g, 720.5 mmol) were then added over 30 minutes. Addition was exothermic. After 2053.49g UPLC-MS showed complete removal of the Boc group, so water (3.53 g, 3.53 mL, 196 mmol) was added drop-wise addition via syringe over 20 min to keep the reaction temperature between 40-50 °C. The mixture was then stirred for 17 hours to complete the reaction. The mixture was then cooled to 20 °C and water (400 mL) was added. The stirring was stopped and the layers were separated. The desired product in the aqueous layer was returned to the reactor and the organic layer was discarded. The aqueous layer was washed with 2-Me-THF (200 mL). Isopropanol (50. mL) was added followed by dropwise addition of aqueous HCl (131 mL of 6.0 M, 786.0 mmol) to adjust the pH to ![❤]() while maintaining the temperature below 30 °C. The resulting solid was then isolated by filtration and the filter cake washer with water (100 mL) then pulled dry until a sticky cake was obtained. The solids were then dried under vacuum at 55 °C to afford 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid (23.25 g) as an off-white fine solid.
while maintaining the temperature below 30 °C. The resulting solid was then isolated by filtration and the filter cake washer with water (100 mL) then pulled dry until a sticky cake was obtained. The solids were then dried under vacuum at 55 °C to afford 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid (23.25 g) as an off-white fine solid.
[00297] Step 4: Preparation of 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole
3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid (1.0 equiv) was added to a reactor followed by DMF (6.0 vol, 2.6 wt equiv). The mixture was stirred at 18– 22 °C. DBU (0.2 equiv.) was charged to the reaction mixture at a rate of approximately 45 mL/min. The reaction temperature was then raised to 98– 102 °C over 45 minutes. The reaction mixture was stirred at 98– 102 °C for no less than 10 h. The reaction mixture was then cooled to -2°C to 2 °C over approximately 1 hour and was used without isolation to make ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate.
[00298] Alternate procedure for the preparation of 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid
![]()
[00299] Step 1. Ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate
![]()
[00300] A solution of ethyl 2,6-dichloronicotinate (256 g, 1.16 mol) and 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (242 g, 1.16 mol) in DMF (1.53 L) was treated with potassium carbonate (209 g, 1.51 mol) and DABCO (19.6 g, 174 mmol). The resultant suspension was stirred allowed to exotherm from 14 to 25 °C and then maintained at 20– 25 °C with external cooling for 3 days. The suspension was cooled to below 10 °C when water (2.0 L) was added in a thin stream while maintaining the temperature below 25 °C. After the addition was complete, the suspension was stirred for an additional 1 h. The solid was collected by filtration (sintered-glass/polypad) and the filter-cake was washed with water (2 x 500-mL) and dried with suction for 2 h to afford water-damp ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate (512 g; 113% yield) as white powder which was used without further steps in the subsequent reaction.
[00301] Step 2.2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1h-pyrazol-1-yl)nicotinic acid
![]()
[00302] The water-damp ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate (455 g, 1.16 mol; assumed 100% yield from previous step) in EtOH (1.14 L) and THF (455 mL) was stirred at ambient temperature (17 °C) when 1 M NaOH (1.16 L, 1.16 mol) was added. The reaction mixture exothermed to 30 °C and was further warmed at 40 °C for 2 h. The solution was quenched with 1 M HCl (1.39 L, 1.39 mol) which resulted in an immediate precipitation which became thicker as the acid was added. The creamy suspension was allowed to cool to room temperature and was stirred overnight. The solid was collected by filtration (sintered-glass/poly pad). The filter-cake was washed with water (2 x 500-mL). The filter-cake was dried by suction for 1 h but remained wet. The damp solid was transferred to a 10-L Buchi flask for further drying (50 °C/20 torr), but was not effective. Further effort to dry by chasing with i-PrOH was also ineffective. Successful drying was accomplished after the damp solid was backfilled with i-PrOAc (3 L), the suspension was heated at 60 °C (homogenization), and re-concentrated to dryness (50 °C/20 torr) to afford dry 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1h-pyrazol-1-yl)nicotinic acid (408 g; 97% yield for two steps) as a fine, white powder. The product was further dried in a vacuum oven (50 °C/10 torr/N2 bleed) for 2 h but marginal weight loss was observed. 1H NMR (400 MHz, DMSO-d6) δ 13.64 (s, 1H), 8.49– 8.36 (m, 2H), 7.77 (d, J = 8.4 Hz, 1H), 6.26 (d, J = 2.8 Hz, 1H), 4.28 (s, 2H), 1.24 (s, 6H).19F NMR (376 MHz, DMSO-d6) δ -75.2. KF analysis: 0.04% water.
2. Preparation of Form A of Compound 1
[00303] The crystalline Form A of Compound 1 was obtained as a result of the following synthesis. Combined 2-chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide(108 g, 207.3 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (77.55 g, 518.2 mmol), was combined with K2CO3 (143.2 g, 1.036 mol) in DMSO (432.0 mL) and 1,2-
diethoxyethane (108.0 mL) in a 1-L RB flask with a reflux condenser. The resulting suspension was heated at 120°C and was stirred at temperature overnight. Then the reaction was diluted with DCM (1.080 L) and HCl (933.0 mL of 2 M, 1.866 mol) was slowly added. The liquid phases were separated, and the aqueous phase was extracted with DCM (540.0 mL).The organic phases were combined, washed with water (540.0 mL) (3 x), then dried with (Na2SO4) to afford an amber solution. Silica gel (25 g) was added and then the drying agent/silica gel was filtered off. The filter-bed was washed with DCM (3 x 50-mL). The organic phases were combined and concentrated (40 °C/40 torr) to afford crude N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (198.6 g, 160% theory) as an off-white solid. The solid was diluted with MTBE (750 mL), warmed at 60 °C (external temperature), and mixed to a homogenous suspension. The suspension was cooled to 30 °C with stirring and the solid was collected by filtration, air-dried, and vacuum-dried to afford Compound 1 (111.1 g; 90 %) as a fine, white powder.
[00304] The crystalline Form A of Compound 1 was also obtained through the following procedure. A suspension of Compound 1 (150.0 g, 228.1 mmol) in iPrOH (480 mL) and water (120 mL) was heated at 82 °C to obtain a solution. The solution was cooled with a J-Kem controller at a cooling rate of 10 °C/h. Once the temperature reached 74 °C, the solution was seeded with a sample of Compound 1 in crystalline Form A. Crystallization occurred immediately. The suspension was cooled to 20 °C. The solid was collected by filtration, washed with i-PrOH (2 x 75 mL), air-dried with suction, and vacuum-dried (55 °C/300 torr/N2 bleed) to afford Compound 1, Form A (103.3 g) as a white powder.. The sample was cooled to ~5 °C, let stir for 1 h, and then the solid was collected by filtration (sintered glass/paper). the filter-cake was washed with i-PrOH (75 mL) (2 x), air-dried with suction, air-dried in a drying dish (120.6 g mostly dried), vacuum-dried (55 °C/300 torr/N2 bleed) for 4 h, and then RT overnight. Overnight drying afforded 118.3 g (87% yield) of a white powder.
PATENT
WO-2019113476
Example 1: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
![]()
Step 1: methyl-2,4-dimethyl-4-nitro-pentanoate
![]()
[00110] Tetrahydrofuran (THF, 4.5 L) was added to a 20 L glass reactor and stirred under N2 at room temperature. 2-Nitropropane (1.5 kg, 16.83 mol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.282 kg, 8.42 mol) were then charged to the reactor, and the jacket temperature was increased to 50 °C. Once the reactor contents were close to 50 °C, methyl methacrylate (1.854 kg, 18.52 mol) was added slowly over 100 minutes. The reaction temperature was maintained at or close to 50 °C for 21 hours. The reaction mixture was concentrated in vacuo then transferred back to the reactor and diluted with methyl tert-butyl ether (MTBE) (14 L). 2 M HCl (7.5 L) was added, and this mixture was stirred for 5 minutes then allowed to settle. Two clear layers were visible– a lower yellow aqueous phase and an upper green organic phase. The aqueous layer was removed, and the organic layer was stirred again with 2 M HCl (3 L). After separation, the HCl washes were recombined and stirred with MTBE (3 L) for 5 minutes. The aqueous layer was removed, and all of the organic layers were combined in the reactor and stirred with water (3 L) for 5 minutes. After separation, the organic layers were concentrated in vacuo to afford a cloudy green oil. Crude product was treated with MgSO4 and filtered to afford methyl-2,4-dimethyl-4-nitro-pentanoate as a clear green oil (3.16 kg, 99% yield).
[00111] 1H NMR (400 MHz, Chloroform-d) δ 3.68 (s, 3H), 2.56– 2.35 (m, 2H), 2.11 – 2.00 (m, 1H), 1.57 (s, 3H), 1.55 (s, 3H), 1.19 (d, J = 6.8 Hz, 3H).
Step 2: Synthesis of methyl (2S)-2,4-dimethyl-4-nitro-pentanoate
![]()
[00112] A reactor was charged with purified water (2090 L; 10 vol) and then potassium phosphate monobasic (27 kg, 198.4 moles; 13 g/L for water charge). The pH of the reactor contents was adjusted to pH 6.5 (± 0.2) with 20% (w/v) potassium carbonate solution. The reactor was charged with racemic methyl-2,4-dimethyl-4-nitro-pentanoate (209 kg; 1104.6 moles), and Palatase 20000L lipase (13 L, 15.8 kg; 0.06 vol).
[00113] The reaction mixture was adjusted to 32 ± 2 °C and stirred for 15-21 hours, and pH 6.5 was maintained using a pH stat with the automatic addition of 20% potassium carbonate solution. When the racemic starting material was converted to >98% ee of the S-enantiomer, as determined by chiral GC, external heating was switched off. The reactor was then charged with MTBE (35 L; 5 vol), and the aqueous layer was extracted with MTBE (3 times, 400-1000L). The combined organic extracts were washed with aqueous Na2CO3 (4 times, 522 L, 18 % w/w 2.5 vol), water (523 L; 2.5 vol), and 10% aqueous NaCl (314 L, 1.5 vol). The organic layer was concentrated in vacuo to afford methyl (2S)-2,4-dimethyl-4-nitro-pentanoate as a mobile yellow oil (>98% ee, 94.4 kg; 45 % yield).
Step 3: Synthesis of (3S)-3,5,5-trimethylpyrrolidin-2-one
![]()
[00114] A 20 L reactor was purged with N2. The vessel was charged sequentially with DI water-rinsed, damp Raney® Ni (2800 grade, 250 g), methyl (2S)-2,4-dimethyl-4-nitro-pentanoate (1741g, 9.2 mol), and ethanol (13.9 L, 8 vol). The reaction was stirred at 900 rpm, and the reactor was flushed with H2 and maintained at ~2.5 bar. The reaction mixture was then warmed to 60 °C for 5 hours. The reaction mixture was cooled and filtered to remove Raney nickel, and the solid cake was rinsed with ethanol (3.5 L, 2 vol). The ethanolic solution of the product was combined with a second equal sized batch and concentrated in vacuo to reduce to a minimum volume of ethanol (~1.5 volumes). Heptane (2.5 L) was added, and the suspension was concentrated again to ~1.5 volumes. This was repeated 3 times; the resulting suspension was cooled to 0-5 °C, filtered under suction, and washed with heptane (2.5 L). The product was dried under vacuum for 20 minutes then transferred to drying trays and dried in a vacuum oven at 40 °C overnight to afford (3S)-3,5,5-trimethylpyrrolidin-2-one as a white solid (2.042 kg, 16.1 mol, 87 %). 1H NMR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (dd, J = 12.4, 8.6 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
Step 4: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
![]()
[00115] A glass lined 120 L reactor was charged with lithium aluminum hydride pellets (2.5 kg, 66 mol) and dry THF (60 L) and warmed to 30 °C. The resulting suspension was charged with (S)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C, then cautiously quenched with the addition of ethyl acetate (EtOAc) (1.0 L, 10 moles), followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq), and then a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 equiv water with 1.4 equiv sodium hydroxide relative to aluminum), followed by 7.5 L water. After the addition was complete, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HCl (1.05 equiv.) while maintaining the temperature below 30°C. The resultant solution was concentrated by vacuum distillation to a slurry. Isopropanol (8 L) was added and the solution was concentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added, and the product was slurried by warming to about 50 °C. MTBE (6 L) was added, and the slurry was cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L MTBE and dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford (4S)-2,2,4-trimethylpyrrolidine•HCl as a white solid (6.21 kg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (br d, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H).
Example 2: Synthesis of 5,5-dimethyl-3-methylenepyrrolidin-2-one
![]()
Example 2A
[00116] 2,2,6,6-tetramethylpiperidin-4-one (50.00 g, 305.983 mmol, 1.000 equiv), tributylmethyl ammonium chloride (2.89 g, 3.0 mL, 9.179 mmol, 0.030 equiv), chloroform (63.92 g, 43.2 mL, 535.470 mmol, 1.750 equiv), and DCM
(dichloromethane) (100.0 mL, 2.00 vol) were charged to a 1000 mL three-neck round bottom flask equipped with an overhead stirrer. The reaction mixture was stirred at 300 rpm, and 50 wt% NaOH (195.81 g, 133.2 mL, 2,447.863 mmol, 8.000 equiv) was added dropwise (via addition funnel) over 1.5 h while maintaining the temperature below 25 °C with intermittent ice/acetone bath. The reaction mixture was stirred at 500 rpm for 18 h, and monitored by GC (3% unreacted piperidinone after 18 h). The suspension was diluted with DCM (100.0 mL, 2.00 vol) and H2O (300.0 mL, 6.00 vol), and the phases were separated. The aqueous phase was extracted with DCM (100.0 mL, 2.00 vol). The organic phases were combined and 3 M hydrochloric acid (16.73 g, 153.0 mL, 458.974 mmol, 1.500 equiv) was added. The mixture was stirred at 500 rpm for 2 h. The conversion was complete after approximately 1 h. The aqueous phase was saturated with NaCl, H2O (100.0 mL, 2.00 vol) was added to help reduce the emulsion, and the phases were separated. The aqueous phase was extracted with DCM (100.0 mL, 2.00 vol) twice. H2O (100.0 mL, 2.00 vol) was added to help with emulsion separation. The organic phases were combined, dried (MgSO4), and concentrated to afford 32.6 g (85%) of crude 5,5-dimethyl-3-methylenepyrrolidin-2-one (19) as a pale orange clumpy solid. The crude was recrystallized from hot (90°C) iPrOAc (71.7 mL, 2.2 vol. of crude), cooled to 80 °C, and ~50 mg of crystalline 5,5-dimethyl-3-methylenepyrrolidin-2-one (19) was added for seeding. Crystallization started at 77 °C, the mixture was slowly cooled to ambient temperature, and aged for 2 h. The solid was collected by filtration, washed with 50/50 iPrOAc/heptane (20.0 mL, 0.40 vol) twice, and dried overnight in the vacuum oven at 40 °C to afford the desired product (23.70 g, 189.345 mmol, 62% yield) as a white sand colored crystalline solid.
1H NMR (400 MHz, CDCl3, 7.26 ppm) δ 7.33 (bs, 1H), 5.96– 5.95 (m, 1H), 5.31-5.30 (m, 1H), 2.6 (t, J = 2.5 Hz, 2H), 1.29 (s, 6H).
Example 2B
[00117] Step 1: Under a nitrogen atmosphere, 2,2,6,6-tetramethylpiperidin-4-one (257.4 kg, 1658.0 mol, 1.00 eq.), tri-butyl methyl ammonium chloride (14.86 kg, 63.0 mol, 0.038 eq.), chloroform (346.5 kg, 2901.5 mol, 1.75 eq.) and DCM (683.3 kg) were added to a 500 L enamel reactor. The reaction was stirred at 85 rpm and cooled to 15~17°C. The solution of 50wt% sodium hydroxide (1061.4 kg, 13264.0 mol, 8.00 eq.) was added dropwise over 40 h while maintaining the temperature between 15~25°C. The reaction mixture was stirred and monitored by GC.
[00118] Step 2: The suspension was diluted with DCM (683.3 kg) and water (1544.4 kg). The organic phase was separated. The aqueous phase was extracted with DCM (683.3 kg). The organic phases were combined, cooled to 10°C and then 3 M
hydrochloric acid (867.8 kg, 2559.0 mol, 1.5 eq.) was added. The mixture was stirred at 10~15 °C for 2 h. The organic phase was separated. The aqueous phase was extracted with DCM (683.3 kg x 2). The organic phases were combined, dried over Na2SO4 (145.0 kg) for 6 h. The solid was filtered off and washed with DCM (120.0 kg). The filtrate was stirred with active charcoal (55 kg) for 6 h. The resulting mixture was filtered and the filtrate was concentrated under reduced pressure (30~40°C, -0.1MPa). Then isopropyl acetate (338 kg) was added and the mixture was heated to 87~91°C, stirred for 1 h. Then the solution was cooled to 15 °C in 18 h and stirred for 1 h at 15 °C. The solid was collected by filtration, washed with 50% isopropyl acetate/hexane (80.0 kg x 2) and dried overnight in the vacuum oven at 50 °C to afford 5,5-dimethyl-3-methylenepyrrolidin-2-one as an off white solid, 55% yield.
Example 3: Synthesis of (S)-3,5,5-trimethyl-pyrrolidin-2-one from 5,5-dimethyl-3- methylenepyrrolidin-2-one
![]()
Example 3A – Use of Rh Catalyst
Step 1 – Preparation of Rh Catalyst Formation:
[00119] In a 3 L Schlenk flask, 1.0 l of tetrahydrofurn (THF) was degassed with an argon stream. Mandyphos Ligand SL-M004-1 (1.89 g) and [Rh(nbd)Cl]2 (98%, 0.35 g) (chloronorbornadiene rhodium(I) dimer) were added. The resulting orange catalyst solution was stirred for 30 min at room temperature to form a catalyst solution.
Step 2:
[00120] A 50 L stainless steel autoclave was charged with 5,5-dimethyl-3-methylenepyrrolidin-2-one (6.0 kg) and THF (29 L). The autoclave was sealed and the resulting suspension was flushed with nitrogen (3 cycles at 10 bar), and then released of pressure. Next the catalyst solution from Step 1 was added. The autoclave was flushed with nitrogen without stirring (3 cycles at 5 bar) and hydrogen (3 cycles at 5 bar). The pressure was set to 5 bar and a 50 L reservoir was connected. After 1.5 h with stirring at 1000 rpm and no hydrogen uptake the reactor was flushed again with nitrogen (3 cycles at 10 bar) with stirring and additional catalyst solution was added. The autoclave was again flushed to hydrogen with the above described procedure (3 x 5 bar N2, 3 x 5 bar H2) and adjusted to 5 bar. After 2 h, the pressure was released, the autoclave was flushed with nitrogen (3 cycles at 5 bar) and the product solution was discharged into a 60 L inline barrel. The autoclave was charged again with THF (5 L) and stirred with 1200 rpm for 5 min. The wash solution was added to the reaction mixture.
Step 3:
[00121] The combined solutions were transferred into a 60 L reactor. The inline barrel was washed with 1 L THF which was also added into the reactor. 20 L THF were removed by evaporation at 170 mbar and 40°C.15 L heptane were added. The distillation was continued and the removed solvent was continuously replaced by heptane until the THF content in the residue was 1% w/w (determined by NMR). The reaction mixture was heated to 89°C (turbid solution) and slowly cooled down again (ramp: 14°C/h). Several heating and cooling cycles around 55 to 65°C were made. The off-white suspension was transferred to a stirred pressure filter and filtered (ECTFE-pad, d = 414 mm, 60 my, Filtration time = 5 min). 10 L of the mother liquor was transferred back into the reactor to wash the crystals from the reactor walls and the obtained slurry was also added to the filter. The collected solid was washed with 2 x 2.5 l heptane, discharged and let dry on the rotovap at 40°C and 4 mbar to obtain the product, (S)-3,5,5-trimethyl-pyrrolidin-2-one; 5.48Kg (91%), 98.0% ee.
Example 3B – Use of Ru Catalyst
[00122] The reaction was performed in a similar manner as described above in Example 3A except the use of a Ru catalyst instead of a Rh catalyst.
[00123] Compound (15) (300 g) was dissolved in THF (2640 g, 10 Vol) in a vessel. In a separate vessel, a solution of [RuCl(p-cymene){(R)-segphos}]Cl (0.439g, 0.0002 eq) in THF (660 g, 2.5 Vol) was prepared. The solutions were premixed in situ and passed through a Plug-flow reactor (PFR). The flow rate for the Compound (15) solution was at 1.555 mL/min and the Ru catalyst solution was at 0.287 mL/min. Residence time in the PFR was 4 hours at 30 °C, with hydrogen pressure of 4.5 MPa. After completion of reaction, the THF solvent was distilled off to give a crude residue. Heptane (1026 g, 5 vol) was added and the resulting mixture was heated to 90 °C. The mixture was seeded with 0.001 eq. of Compound 16S seeds. The mixture was cooled to -15 °C at 20 °C/h. After cooling, heptane (410 g, 2 vol) was added and the solid product was recovered by filtration. The resulting product was dried in a vacuum oven at 35 °C to give (S)-3,5,5-trimethyl-pyrrolidin-2-one (281.77 g, 98.2 % ee, 92 % yield).
Example 3C – Analytical Measurements
[00124] Analytical chiral HPLC method for the determination of the conversion, chemoselectivity, and enantiomeric excess of the products from Example 3A and 3B was made under the following conditions
Instrument: Agilent Chemstation 1100
Column: Phenomenex Lux 5u Cellulose-2, 4.6 mm x 250 mm x 5 um, LHS6247 Solvent: Heptane/iPrOH (90:10)
Flow: 1.0 ml/min
Detection: UV (210 nm)
Temperature: 25°C
Sample concentration: 30 μl of reaction solution evaporated, dissolved in 1 mL heptane/iPrOH (80/20)
Injection volume: 10.0 μL, Run time 20 min
Retention times:
5,5–‐dimethyl–3–methylenepyrrolidin–‐2–‐one: 13.8 min (S)-3,5,5-trimethyl-pyrrolidin-2-one: 10.6 min
(R)-3,5,5-trimethyl-pyrrolidin-2-one: 12.4 min
Example 4: Synthesis of (S)-3,5,5-trimethyl-pyrrolidin-2-one from 5,5-dimethyl-3- methylenepyrrolidin-2-one
![]()
[00125] Mandyphos (0.00479 mmol, 0.12 eq) was weighed into a GC vial. In a separate vial Ru(Me-allyl)2(COD) (16.87 mg, 0.0528 mmol) was weighed and dissolved in DCM (1328 µL). In another vial HBF4•Et2O (6.6 µL) and BF3 ^Et2O (2.0 µL) were dissolved in DCM (240 µL). To the GC vial containing the ligand was added, under a flow of argon, the Ru(Me-allyl)2(COD) solution (100 µL; 0.00399 mmol, 0.1eq) and the HBF4•Et2O / BF3 ^Et2O solution (20 µL; 1 eq HBF4 ^Et2O and catalytic BF3 ^Et2O). The resulting mixtures were stirred under a flow of argon for 30 minutes.
[00126] 5,5-dimethyl-3-methylenepyrrolidin-2-one (5 mg, 0.0399 mmol) in EtOH (1 mL) was added. The vials were placed in the hydrogenation apparatus. The apparatus was flushed with H2 (3×) and charged with 5 bar H2. After standing for 45 minutes, the apparatus was placed in an oil bath at temperature of 45°C. The reaction mixtures were stirred overnight under H2.200 µL of the reaction mixture was diluted with MeOH (800 µL) and analyzed for conversion and ee.
1H NMR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (ddd, J = 12.4, 8.6, 0.8 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
![]()
Table 1: IPC method for Asymmetric Hydrogenation
![]()
Example 5. Synthesis of (S)-2,2,4-trimethylpyrrolidine hydrochloride from (S)- 3,5,5-trimethyl-pyrrolidin-2-one
![]()
Example 5A
[00127] Anhydrous THF (100ml) was charged to a dry 750ml reactor and the jacket temperature was set to 50°C. Once the vessel contents were at 50°C LiAlH4pellets (10g, 263mmol, 1.34 eq.) were added. The mixture was stirred for 10 minutes, then a solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) (25g, 197mmol) in anhydrous THF (100ml) was added dropwise over 45 minutes, maintaining the temperature between 50-60°C. Once the addition was complete the jacket temperature was increased to 68°C and the reaction stirred for 18.5hrs. The reaction mixture was cooled to 30°C then saturated sodium sulfate solution (20.9ml) was added dropwise over 30 minutes, keeping the temperature below 40°C. Vigorous evolution of hydrogen was observed and the reaction mixture thickened but remained mixable. The mixture thinned towards the end of the addition. The mixture was cooled to 20°C, diluted with iPrOAc (100ml) and stirred for an additional 10 minutes. The suspension was then drained and collected through the lower outlet valve, washing through with additional iPrOAc (50ml). The collected suspension was filtered through a celite pad on a sintered glass funnel under suction and washed with iPrOAc (2x50ml).
[00128] The filtrate was transferred back to the cleaned reactor and cooled to 0°C under nitrogen. 4M HCl in dioxane (49.1ml, 197mmol, 1eq.) was then added dropwise over 15 minutes, maintaining the temperature below 20°C. A white precipitate formed. The reactor was then reconfigured for distillation, the jacket temperature was increased to 100 °C, and distillation of solvent was carried out. Additional i-PrOAc (100 mL) was added during concentration, after >100 mL distillate had been collected. Distillation was continued until ~250 mL total distillate was collected, then a Dean-Stark trap was attached and reflux continued for 1 hour. No water was observed to collect. The reaction mixture was cooled to 20 °C and filtered under suction under nitrogen. The filtered solid was washed with i-PrOAc (100 mL), dried under suction in nitrogen, then transferred to a glass dish and dried in a vacuum oven at 40 °C with a nitrogen bleed. (S)-2,2,4-Trimethylpyrrolidine hydrochloride (17S•HCl) was obtained as a white solid (24.2g, 82%).
GC analysis (purity): >99.5%
GC chiral purity: 99.5%
Water content (by KF): 0.074%
Residual solvent (by 1H-NMR): 0.41%
Example 5B
[00129] To a glass lined 120 L reactor was charged LiAlH4 pellets (2.5 kg 66 mol, 1.2 equiv.) and dry THF (60 L) and warmed to 30 °C. To the resulting suspension was
charged (S)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C and sampled to check for completion, then cautiously quenched with the addition of EtOAc (1.0 L, 10 moles, 0.16 eq) followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq) then followed by a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 eq water with 1.4 eq sodium hydroxide relative to aluminum), followed by 7.5 L water (6 eq“Fieser” quench). After the addition was completed, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HCl (1.05 equiv.) while maintaining the temperature below 30°C.
[00130] The resultant solution was concentrated by vacuum distillation to a slurry in two equal part lots on the 20 L Buchi evaporator. Isopropanol (8 L) was charged and the solution reconcentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added and the product slurried by warming to about 50 °C. Distillation from Isopropanol continued until water content by KF is≤ 0.1 %. Methyl tertbutyl ether (6 L) was added and the slurry cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L methyl tert-butyl ether and pulled dry with a strong nitrogen flow and further dried in a vacuum oven (55 °C/300 torr/N2bleed) to afford (S)-2,2,4-trimethylpyrrolidine•HCl as a white, crystalline solid (6.21 kg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, , 3H).
Example 5C
[00131] With efficient mechanical stirring, a suspension of LiAlH4 pellets (100 g 2.65 mol; 1.35 eq.) in THF (1 L; 4 vol. eq.) warmed at a temperature from 20 °C– 36 °C (heat of mixing). A solution of (S)-3,5,5-trimethylpyrrolidin-2-one (250 g; 1.97 mol) in THF (1 L; 4 vol. eq.) was added to the suspension over 30 min. while allowing the reaction temperature to rise to ~60 °C. The reaction temperature was increased to near reflux (~68 °C) and maintained for about 16 h. The reaction mixture was cooled to below 40 °C and cautiously quenched with drop-wise addition of a saturated aqueous solution of Na2SO4 (209 mL) over 2 h. After the addition was completed, the reaction mixture was cooled to ambient temperature, diluted with i-PrOAc (1 L), and mixed thoroughly. The solid was removed by filtration (Celite pad) and washed with i-PrOAc (2 x 500 mL). With external cooling and N2 blanket, the filtrate and washings were combined and treated with drop-wise addition of anhydrous 4 M HCl in dioxane (492 mL; 2.95 mol; 1 equiv.) while maintaining the temperature below 20 °C. After the addition was completed (20 min), the resultant suspension was concentrated by heating at reflux (74– 85 °C) and removing the distillate. The suspension was backfilled with i-PrOAc (1 L) during concentration. After about 2.5 L of distillate was collected, a Dean-Stark trap was attached and any residual water was azeotropically removed. The suspension was cooled to below 30 °C when the solid was collected by filtration under a N2 blanket. The solid is dried under N2 suction and further dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford 261 g (89% yield) of (S)-2,2,4-trimethylpyrrolidine•HCl as a white, crystalline solid. 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H). 1H NMR (400 MHz, CDCl3) δ 9.55 (d, J = 44.9 Hz, 2H), 3.52 (ddt, J = 12.1, 8.7, 4.3 Hz, 1H), 2.94 (dq, J = 11.9, 5.9 Hz, 1H), 2.70– 2.51 (m, 1H), 2.02 (dd, J = 13.0, 7.5 Hz, 1H), 1.62 (s, 3H), 1.58– 1.47 (m, 4H), 1.15 (d, J = 6.7 Hz, 3H).
Example 5D
[00132] A 1L four-neck round bottom flask was degassed three times. A 2M solution of LiAlH4 in THF (100 mL) was charged via cannula transfer. (S)-3,5,5-trimethylpyrrolidin-2-one (19.0 g) in THF (150 mL) was added dropwise via an addition funnel over 1.5 hours at 50-60 °C, washing in with THF (19 mL). Upon completion of the addition, the reaction was stirred at 60 °C for 8 hours and allowed to cool to room temperature overnight. GC analysis showed <1% starting material remained.
[00133] Deionized water (7.6 mL) was added slowly to the reaction flask at 10-15 °C, followed by 15% potassium hydroxide (7.6 mL). Isopropyl acetate (76 mL) was added, the mixture was stirred for 15 minutes and filtered, washing through with isopropyl acetate (76 mL).
[00134] The filtrate was charged to a clean and dry 500 mL four neck round bottom flask and cooled to 0-5 °C. 36% Hydrochloric acid (15.1 g, 1.0 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (190 mL), was carried out to leave a residual volume of ~85 mL. Karl Fischer analysis = 0.11% w/w H2O. MTBE (methyl tertiary butyl ether) (19 mL) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (25 mL) and drying under vacuum at 40-45 °C to give crude (S)-2,2,4-trimethylpyrrolidine hydrochloride as a white crystalline solid (17.4 g, 78% yield). GC purity = 99.5%. Water content = 0.20% w/w. Chiral GC gave an ee of 99.0% (S).
Ruthenium content = 0.004 ppm. Lithium content = 0.07 ppm.
[00135] A portion of the dried crude (S)-2,2,4-trimethylpyrrolidine hydrochloride (14.3g) was charged to a clean and dry 250 mL four-neck round bottom flask with isopropanol (14.3 mL) and the mixture held at 80-85 °C (reflux) for 1 hour to give a clear solution. The solution was allowed to cool to 50 °C (solids precipitated on cooling) then MTBE (43 mL) was added and the suspension held at 50-55 °C (reflux) for 3 hours. The solids were filtered off at 10 °C, washing with MTBE (14 mL) and dried under vacuum at 40 °C to give recrystallised (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystallised solid (13.5 g, 94% yield on recrystallisation, 73% yield). GC purity = 99.9%. Water content = 0.11% w/w. Chiral GC gave an ee of 99.6 (S). Ruthenium content = 0.001 ppm. Lithium content = 0.02 ppm.
Example 5E:
[00136] A reactor was charged with lithium aluminum hydride (LAH) (1.20 equiv.) and 2-MeTHF (2-methyltetrahydrofuran) (4.0 vol), and heated to internal temperature of 60 °C while stirring to disperse the LAH. A solution of (S)-3,5,5-trimethylpyrrolidin-2-one (1.0 equiv) in 2-MeTHF (6.0 vol) was prepared and stirred at 25 °C to fully dissolve the (S)-3,5,5-trimethylpyrrolidin-2-one. The (S)-3,5,5-trimethylpyrrolidin-2-one solution was added slowly to the reactor while keeping the off-gassing manageable, followed by rinsing the addition funnel with 2-MeTHF (1.0 vol) and adding it to the reactor. The reaction was stirred at an internal temperature of 60 ± 5 °C for no longer than 6 h. The internal temperature was set to 5 ± 5 °C and the agitation rate was increased. A solution of water (1.35 equiv.) in 2-MeTHF (4.0v) was prepared and added slowly to the reactor while the internal temperature was maintained at or below 25 °C. Additional water (1.35 equiv.) was charged slowly to the reactor while the internal temperature was maintained at or below 25 °C. Potassium hydroxide (0.16 equiv.) in water (0.40 vol) was added to the reactor over no less than 20 min while the temperature was maintained at or below 25 °C. The resulting solids were removed by filtration, and the reactor and cake were washed with 2-MeTHF (2 x 2.5 vol). The filtrate was transferred back to a jacketed
vessel, agitated, and the temperature was adjusted to 15 ± 5 °C. Concentrated aqueous HCl (35-37%, 1.05 equiv.) was added slowly to the filtrate while maintaining the temperature at or below 25 °C and was stirred no less than 30 min. Vacuum was applied and the solution was distilled down to a total of 4.0 volumes while maintaining the internal temperature at or below 55 °C, then 2-MeTHF (6.00 vol) was added to the vessel. The distillation was repeated until Karl Fischer analysis (KF) < 0.20% w/w H2O. Isopropanol was added (3.00 vol), and the temperature was adjusted to 70 °C (65– 75 °C) to achieve a homogenous solution, and stirred for no less than 30 minutes at 70 °C. The solution was cooled to 50 °C (47– 53 °C) over 1 hour and stirred for no less than 1 h, while the temperature was maintained at 50°C (47– 53 °C). The resulting slurry was cooled to -10 °C (-15 to -5°C) linearly over no less than 12 h. The slurry was stirred at -10 °C for no less than 2 h. The solids were isolated via filtration or centrifugation and were washed with a solution of 2-MeTHF (2.25 vol) and IPA (isopropanol) (0.75 vol). The solids were dried under vacuum at 45 ± 5 °C for not less than 6 h to yield (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl).
Example 6: Phase Transfer Catalyst (PTC) Screens for the Synthesis of 5,5- dimethyl-3-methylenepyrrolidin-2-one
![]()
[00137] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), PTC (0.05 eq.), and chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added dropwise over 2 min. The reaction mixture was stirred until completion as assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion and assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the
organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC. Reaction results are summarized in Table 2.
Table 2
![]()
Example 7: Solvent Screens for the Synthesis of 5,5-dimethyl-3- methylenepyrrolidin-2-one
![]()
[00138] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), tetrabutylammonium hydroxide (0.12 g, 0.153 mmol, 0.050 eq), chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.), and solvent (2 vol. or 4 vol., as shown in Table 3 below) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion and assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL,
2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC.
Reaction results are summarized in Table 3.
Table 3
![]()
![]()
Example 8: Base Screens for the Synthesis of 5,5-dimethyl-3-methylenepyrrolidin- 2-one
![]()
[00139] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), tetrabutylammonium hydroxide (0.12 g, 0.153 mmol, 0.050 eq), and chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath, and a solution of an amount wt% sodium hydroxide as shown in Table 4 below in water (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion and assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase is extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as
an internal HPLC standard. Solution yield was assessed by HPLC. Reaction results are summarized in Table 4.
Table 4
![]()
Example 9: Various Amounts of Phase Transfer Catalyst (PTC) for the Synthesis of 5,5-dimethyl-3-methylenepyrrolidin-2-one
![]()
[00140] In this experiment, various amounts of PTCs were tested as described below: Tetrabutylammonium hydroxide (0.01 eq.), TBAB (0.01 eq.), Tributylmethylammonium chloride (0.01 eq.), Tetrabutylammonium hydroxide (0.02 eq.), TBAB (0.02 eq.), Tributylmethylammonium chloride (0.02 eq.), Tetrabutylammonium hydroxide (0.03 eq.), TBAB (0.03 eq.), Tributylmethylammonium chloride (0.03 eq.).
[00141] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), PTC (0.12 g, 0.153 mmol, 0.050 eq), and chloroform (1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath, and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion, assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by
HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC. The reaction results are summarized in Table 5.
Table 5
![]()
Example 10: Preparation of 2,2,6,6-tetramethylpiperidin-4-one hydrochloride (14•HCl)
![]()
[00142] 2,2,6,6-tetramethyl-4-piperidinone (14) (30 g, 193.2 mmol, 1.0 eq) was charged to a 500 mL nitrogen purged three necked round bottomed flask equipped with condenser. IPA (300 mL, 10 vol) was added to the flask and the mixture heated to 60 °C until dissolved.
[00143] To the solution at 60 °C was added 5-6 M HCl in IPA (40 mL, 214.7 mmol, 1.1 eq) over 10 min and the resulting suspension stirred at 60 °C for 30 min then allowed to cool to ambient temperature. The suspension was stirred at ambient temperature overnight, then filtered under vacuum and washed with IPA (3 x 60 mL, 3 x 2 vol). The cream colored solid was dried on the filter under vacuum for 10 min.
[00144] The wet cake was charged to a 1 L nitrogen purged three necked round bottomed flask equipped with condenser. IPA (450 mL, 15 vol) was added to the flask and the suspension heated to 80 °C until dissolved. The mixture was allowed to cool slowly to ambient temperature over 3 h and the resulting suspension stirred overnight at ambient temperature.
[00145] The suspension was filtered under vacuum, washed with IPA (60 mL, 2 vol) and dried on the filter under vacuum for 30 min. The resulting product was dried in a vacuum oven at 40 °C over the weekend to give 2,2,6,6-tetramethylpiperidin-4-one hydrochloride (14•HCl) a white crystalline solid, 21.4 g, 64% yield.
Example 11: Synthesis of (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) from (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S)
![]()
[00146] Each reactor was charged with (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF, H2, and the catalyst shown in the below table. The reactor was heated to 200 °C and pressurized to 60 bar, and allowed to react for 12 hours. GC analysis showed that (S)-2,2,4-trimethylpyrrolidine was produced in the columns denoted by“+.”
![]()
[00147] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 2% Pt-0.5%Sn/SiO2 catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 130 °C under 80 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h-1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (74.8% yield, 96.1% ee).
Alternate synthesis
[00148] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 4% Pt-2%Sn/TiO2catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 200 °C under 50 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h-1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (88.5% yield, 29.6% ee).
Alternate synthesis
[00149] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 2% Pt-0.5%Sn/TiO2 catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 150 °C under 50 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h-1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (90.9% yield, 98.0% ee).
Alternate synthesis
[00150] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.03 mL/min into a packed bed reactor prepacked with 2% Pt-8%Sn/TiO2catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 40 mL/min. The reaction was carried out at 180 °C under 55 bar pressure with a residence time of 6 min. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (90.4% yield, 96.8% ee).
Example 12: Preparation of N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3- trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4- trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (Compound 1)
![]()
![]()
Compound 1
![]()
I. Preparation of Starting Materials:
A. Synthesis of 3,3,3-Trifluoro-2,2-dimethylpropionic acid (31), morpholine salt:
![]()
Step 1: tert-Butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane (28)
![]()
[00151] A 2L 3-necked round-bottom flask, equipped with a J-Kem thermocouple and an overhead stirrer, was purged with nitrogen for >20 minutes. Hexyllithium solution (2.3 M in hexanes; 1.05 equiv; 0.260 L, 597 mmol) was transferred into the flask via cannula. The flask was then cooled to–65°C in a dry ice/isopropyl alcohol bath and diisopropylamine (1.05 equiv; 0.842 L; 597mmol) was added via an addition funnel, and the internal temperature was maintained at–40 ±5 °C. Once the diisopropylamine addition was complete, tetrahydrofuran (THF) (0.423 L; 6.4 vol) was added to the reactor and the reaction was warmed to room temperature and stirred for 15 minutes. The solution was then cooled to–60 °C and ethyl isobutyrate (1.0 equiv; 0.754 L; 568 mmol) was added dropwise maintaining the temperature below–45 °C. 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU) (0.9 equiv; 0.616 L; 511 mmol) was then added dropwise to the reaction flask and the temperature was maintained below–45 °C. In a separate flask, tert-butyldimethylsilyl chloride (TBSCl) (1.05 equiv; 89.9 g; 597 mmol) was dissolved in THF (2.2 vol w.r.t. TBSCl) and then added to the 2L reactor. The internal temperature was maintained at≤–30°C during the addition of the TBSCl solution. The resulting reaction mixture was allowed to warm to room
temperature and stirred overnight under inert atmosphere. The reaction solution was transferred to a 2L one-neck round-bottom flask. Additional THF (50 mL, x 2) was used to rinse and transfer. The solution was concentrated in vacuo to remove most of the THF. Hexanes were added to the concentrated tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane (500 mL). The organic phase was washed with three times with water (500 mL x 3), to remove salts. The organic layer was dried over Na2SO4 (100 g). The solution was filtered and the waste cake washed with additional hexanes (100 mL). The resulting hexanes solution of tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane was concentrated in vacuo. A quantitative 1H-NMR assay was performed with benzyl benzoate as an internal standard. The quantitative NMR assay indicated that 108.6 grams of tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane (83% yield) was present, and that 1.2 mol% of ethyl isobutyrate relative to tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane was also present. The resulting tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane solution was used without further purification for the photochemical reaction of Step 2. Step 2: 3,3,3-Trifluoro-2,2-dimethylpropionic acid (31), morpholine salt
![]()
[00152] Stock solution A: The concentrated tert-butyl((1-ethoxy-2-methylprop-1-en-1- yl)oxy)dimethylsilane (198 g; 0.86 mol) was dissolved in acetonitrile (895 g; 1.14 L; 5.8 vol) to give a cloudy, yellow solution that was then filtered. The density of the clear, filtered solution was measured to be 0.81 g/mL and the molar concentration was calculated to be 0.6 M. This is referred to as stock solution A (substrate).
[00153] Stock solution B: The catalyst and reagent solution was prepared by dissolving Ru(bpy)3Cl2 hexahydrate in acetonitrile, followed by adding ethanol and pyrrolidine to give a red-colored solution (density measured: 0.810 g/mL). The molar concentration of the catalyst was calculated to be 0.00172 M. The molar concentration of the solution with respect to EtOH/pyrrolidine was calculated to be ~2.3 M. See Table 6.
Table 6
![]()
(i) Photochemical Trifluoromethylation
[00154] CF3I gas was delivered to the reactor directly from the lecture bottle using a regulator and mass flow controller. Stock solutions A and B were pumped at 6.7 g/min and 2.07 g/min, respectively, to mix in a static mixer. The resulting solution was then combined with CF3I in a static mixer. The CF3I was metered into the reactor via a mass flow controller at 2.00 g/min (2 equiv). Liquid chromatography (LC) assay indicated that 1.0% of the tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane was left unreacted. Details of the reaction parameters are shown in the table below. The reaction stream was passed through the 52 mL photoreactor while being irradiated with the 800 W 440-445 LED light source. The first 5 minutes of eluent was discarded. Thereafter the eluent was collected for a total of 3.05 hours. A total of ~2.3 L of solution was collected during the reaction (~1.06 mol). See Table 7.
Table 7
![]()
(ii) Saponification & Salt Formation
[00155] The saponication of the crude solution (4.1 L, from 1.60 mol tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane) was carried out in a 5L 4-necked round-bottomed flask in 2 roughly equal size batches using 15 wt% NaOH (aq) (total ~320g NaOH) at 50 °C for 2-4 h. Upon completion of the reaction determined by gas chromatography (GC) analysis, the re-combined batches were cooled to room
temperature and hexanes (500 mL) and toluene (500 mL) were added to give a clear phase separation. The top organic layer was washed with half-brine (1 L) and combined with the first portion of the product-containing aqueous solution (4.5 L). The combined aqueous stream was washed with hexanes (500 mL) and concentrated to 2-3 L to remove a majority of volatile acetonitrile. To the aqueous phase was added concentrated HCl (1 L, 12 N) and the resulting mixture was extracted with hexanes (4 x 1 L). The combined hexanes extracts were washed with half brine (2 x 500 mL) and concentrated to give an oil (216 g). The oil was dissolved in THF (580 mL), and morpholine (120 mL, 1.0 equiv) was added slowly via an addition funnel. Upon completion of addition, the batch was seeded (0.5-1 g) with morpholine salt, and the seeds were held and allowed to thicken over 30 min. Hexanes (1660 mL) were added over ~ 2 h, and the mixture was aged for another 3 h. The batch was filtered, washed with hexanes (~500 mL) in portions and dried under vacuum/dry air flush to give 3,3,3-trifluoro-2,2-dimethylpropionic acid, morpholine salt as a white solid (283 g, 73%).1H NMR (400 MHz, CD3OD) δ 3.84-3.86 (m, 4H), 3.15-3.18 (m, 4H), 1.33 (s, 6H); 19F NMR (376 MHz, CD3OD): δ -75.90 (s, 3F).
B. Synthesis of 3,3,3-Trifluoro-2,2-dimethyl-propan-1-ol (5)
![]()
[00156] A 1 L 3 neck round bottom flask was fitted with a mechanical stirrer, a cooling bath, an addition funnel, and a J-Kem temperature probe. The vessel was charged with lithium aluminum hydride (LAH) pellets (6.3 g, 0.1665 mol) under a nitrogen atmosphere. The vessel was then charged with tetrahydrofuran (200 mL) under a nitrogen atmosphere. The mixture was allowed to stir at room temperature for 0.5 hours to allow the pellets to dissolve. The cooling bath was then charged with crushed ice in water and the reaction temperature was lowered to 0 oC. The addition funnel was charged with a solution of 3,3,3-trifluoro-2,2-dimethyl-propanoic acid (20 g, 0.1281 mol) in tetrahydrofuran (60 mL) and the clear pale yellow solution was added drop wise over 1 hour. After the addition was complete the mixture was allowed to slowly warm to room temperature and stirring was continued for 24 hours. The suspension was cooled to 0 oC with a crushed ice-water in the cooling bath and then quenched by the very slow and drop wise addition of water (6.3 ml), followed by sodium hydroxide solution (15 weight %; 6.3 mL) and then finally with water (18.9 mL). The reaction temperature of the resulting white suspension was recorded at 5 oC. The suspension was stirred at ~5 oC for 30 minutes and then filtered through a 20 mm layer of Celite. The filter cake was washed with tetrahydrofuran (2 x 100 mL). The filtrate was dried over sodium sulfate (150 g) and then filtered. The filtrate was concentrated under reduced pressure to provide a clear colorless oil (15 g) containing a mixture of the product 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol in THF (73 % weight of product ~10.95g, and 27 wt.% THF as determined by 1H-NMR). The distillate from the rotary evaporation was distilled at atmospheric pressure using a 30 cm Vigreux column to provide 8.75 g of a residue containing 60 % weight of THF and 40 % weight of product (~3.5 g), which corresponds to 14.45 g (79% yield).1H NMR (400 MHz, DMSO-d6) δ 4.99 (t, J = 5.7 Hz, 1H), 3.38 (dd, J = 5.8, 0.9 Hz, 2H), 1.04 (d, J = 0.9 Hz, 6H).
C. Synthesis of tert-Butyl 3-oxo-2,3-dihydro-1H-pyrazole-1-carboxylate (22)
![]()
[00157] A 50L Syrris controlled reactor was started and the jacket was set to 20 °C, stirring at 150 rpm, reflux condenser (10 °C) and nitrogen purge. MeOH (2.860 L) and methyl (E)-3-methoxyprop-2-enoate (2.643 kg, 22.76 mol) were added and the reactor was capped. The reaction was heated to an internal temperature of 40 °C and the system was set to hold jacket temp at 40 °C. Hydrazine hydrate (1300 g of 55 %w/w, 22.31 mol) was added portion wise via addition funnel over 30 min. The reaction was heated to 60 ^C for 1 h. The reaction mixture was cooled to 20 ^C and triethylamine (2.483 kg, 3.420 L, 24.54 mol) was added portion wise, maintaining reaction temp <30 °C. A solution of Boc anhydride (di-tert-butyl dicarbonate) (4.967 kg, 5.228 L, 22.76 mol) in MeOH (2.860 L) was added portion wise maintaining temperature <45 °C. The reaction mixture was stirred at 20 ^C for 16 h. The reaction solution was partially concentrated to remove MeOH, resulting in a clear light amber oil. The resulting oil was transferred to the 50L reactor, stirred and added water (7.150 L) and heptane (7.150 L). The additions caused a small amount of the product to precipitate. The aqueous layer was drained into a clean container and the interface and heptane layer were filtered to separate the solid (product). The aqueous layer was transferred back to the reactor, and the collected solid was placed back into the reactor and mixed with the aqueous layer. A dropping funnel was added to the reactor and loaded with acetic acid (1.474 kg, 1.396 L, 24.54 mol), then began dropwise addition of acid. The jacket was set to 0 °C to absorb the quench exotherm. After addition (pH=5), the reaction mixture was stirred for 1 h. The solid was collected by filtration and washed with water (7.150 L) and washed a second time with water (3.575 L) and pulled dry. The crystalline solid was scooped out of the filter into a 20L rotovap bulb and heptane (7.150 L) was added. The mixture was slurried at 45 °C for 30 mins, and then 1-2 volumes of solvent was distilled off. The slurry in the rotovap flask was filtered and the solids washed with heptane (3.575 L) and pulled dry. The solid was further dried in vacuo (50 °C, 15 mbar) to give tert-butyl 5-oxo-1H-pyrazole-2-carboxylate (2921 g, 71%) as coarse solid.1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 7.98 (d, J = 2.9 Hz, 1H), 5.90 (d, J = 2.9 Hz, 1H), 1.54 (s, 9H).
II. Preparation of Compound I
Step A: tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (23)
![]()
[00158] A mixture of 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol (10 g, 70.36 mmol) and tert-butyl 3-hydroxypyrazole-1-carboxylate (12.96 g, 70.36 mmol) in toluene (130 mL) was treated with triphenyl phosphine (20.30 g, 77.40 mmol) followed by isopropyl N-
isopropoxycarbonyliminocarbamate (14.99 mL, 77.40 mmol) and the mixture was stirred at 110 °C for 16 hours. The yellow solution was concentrated under reduced pressure, diluted with heptane (100mL) and the precipitated triphenylphosphine oxide was removed by filtration and washed with heptane/toluene 4:1 (100mL). The yellow filtrate was evaporated and the residue purified by silica gel chromatography with a linear gradient of ethyl acetate in hexane (0-40%) to give tert-butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (12.3 g, 57%) as an off white solid. ESI-MS m/z calc.308.13477, found 309.0 (M+1) +; Retention time: 1.84 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.10 (d, J = 3.0 Hz, 1H), 6.15 (d, J = 3.0 Hz, 1H), 4.18 (s, 2H), 1.55 (s, 9H), 1.21 (s, 6H).
Step B: 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (7)
![]()
[00159] tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (13.5 g, 43.79 mmol) was treated with 4 M hydrogen chloride in dioxane (54.75 mL, 219.0 mmol) and the mixture was stirred at 45 °C for 1 hour. The reaction mixture was evaporated to dryness and the residue was extracted with 1 M aqueous NaOH (100ml) and methyl tert-butyl ether (100ml), washed with brine (50ml) and extracted with methyl tert-butyl ether (50ml). The combined organic phases were dried, filtered and evaporated to give 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 96%) as an off white solid. ESI-MS m/z calc.208.08235, found 209.0 (M+1) +; Retention time: 1.22 minutes.1H NMR (400 MHz, DMSO-d6) δ 11.91 (s, 1H), 7.52 (d, J = 2.2 Hz, 1H), 5.69 (t, J = 2.3 Hz, 1H), 4.06 (s, 2H), 1.19 (s, 6H).
Step C: tert-Butyl 2,6-dichloropyridine-3-carboxylate (25)
![]()
[00160] A solution of 2,6-dichloropyridine-3-carboxylic acid (10 g, 52.08 mmol) in THF (210 mL) was treated successively with di-tert-butyl dicarbonate (17 g, 77.89 mmol) and 4-(dimethylamino)pyridine (3.2 g, 26.19 mmol) and left to stir overnight at room temperature. At this point, HCl 1N (400 mL) was added and the mixture was stirred vigorously for about 10 minutes. The product was extracted with ethyl acetate (2x300mL) and the combined organics layers were washed with water (300 mL) and brine (150 mL) and dried over sodium sulfate and concentrated under reduced pressure to give 12.94 g (96% yield) of tert-butyl 2,6-dichloropyridine-3-carboxylate as a colorless oil. ESI-MS m/z calc.247.01668, found 248.1 (M+1) +; Retention time: 2.27 minutes.1H NMR (300 MHz, CDCl3) ppm 1.60 (s, 9H), 7.30 (d, J=7.9 Hz, 1H), 8.05 (d, J=8.2 Hz, 1H).
Step D: tert-Butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (26)
![]()
[00161] To a solution of tert-butyl 2,6-dichloropyridine-3-carboxylate (10.4 g, 41.9 mmol) and 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 41.93 mmol) in DMF (110 mL) were added potassium carbonate (7.53 g, 54.5 mmol) and 1,4-diazabicyclo[2.2.2]octane (706 mg, 6.29 mmol) and the mixture was stirred at room temperature for 16 hours. The cream suspension was cooled in a cold water bath and cold water (130 mL) was slowly added. The thick suspension was stirred at room temperature for 1 hour, filtered and washed with plenty of water to give tert-butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 99%) as an off white solid. ESI-MS m/z calc.419.12234, found 420.0 (M+1) +; Retention time: 2.36 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 2.9 Hz, 1H), 8.31 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 6.26 (d, J = 2.9 Hz, 1H), 4.27 (s, 2H), 1.57 (s, 9H), 1.24 (s, 6H).
Step E: 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (10)
![]()
[00162] tert-Butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 40.25 mmol) was suspended in isopropanol (85 mL) treated with hydrochloric acid (34 mL of 6 M, 201 mmol) and heated to reflux for 3 hours (went almost complete into solution at reflux and started to precipitate again). The suspension was diluted with water (51 mL) at reflux and left to cool to room temperature under stirring for 2.5 h. The solid was collected by filtration, washed with
isopropanol/water 1:1 (50mL), plenty of water and dried in a drying cabinet under vacuum at 45-50 °C with a nitrogen bleed overnight to give 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (13.7 g, 91%) as an off white solid. ESI-MS m/z calc.363.05975, found 364.0 (M+1) +; Retention time: 1.79 minutes. 1H NMR (400 MHz, DMSO-d6) δ 13.61 (s, 1H), 8.44 (d, J = 2.9 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 6.25 (d, J = 2.9 Hz, 1H), 4.28 (s, 2H), 1.24 (s, 6H).
Step F: 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (13)
![]()
[00163] 2-Chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (100 mg, 0.2667 mmol) and CDI (512 mg, 3.158 mmol) were combined in THF (582.0 µL) and the mixture was stirred at room temperature. Meanwhile, 1,3-dimethylpyrazole-4-sulfonyl chloride (62 mg, 0.3185 mmol) was combined with ammonia (in methanol) in a separate vial, instantly forming a white solid. After stirring for an additional 20 min, the volatiles were removed by evaporation, and 1 mL of dichloromethane was added to the solid residue, and was also evaporated. DBU (100 µL, 0.6687 mmol) was then added and the mixture stirred at 60 °C for 5 minutes, followed by addition of THF (1 mL) which was subsequently evaporated. The contents of the vial containing the CDI activated carboxylic acid in THF were then added to the vial containing the newly formed sulfonamide and DBU, and the reaction mixture was stirred for 4 hours at room temperature. The reaction mixture was diluted with 10 mL of ethyl acetate, and washed with 10 mL solution of citric acid (1 M). The aqueous layer was extracted with ethyl acetate (2x 10 mL) and the combined organics were washed with brine, dried over sodium sulfate, and concentrated to give 2-chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide as white solid (137 mg, 99%) that was used in the next step without further purification. ESI-MS m/z calc.520.09076, found 521.1 (M+1) +;
Retention time: 0.68 minutes.
Step G: N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (Compound 1)
![]()
[00164] 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (137 mg, 0.2630 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (118 mg, 0.7884 mmol), and potassium carbonate (219 mg, 1.585 mmol) were combined in DMSO (685.0 µL) and the mixture was heated at 130 ^C for 16 hours. The reaction was cooled to room temperature, and 1 mL of water was added. After stirring for 15 minutes, the contents of the vial were allowed to settle, and the liquid portion was removed via pipet and the remaining solids were dissolved with 20 mL of ethyl acetate and were washed with 1 M citric acid (15 mL). The layers were separated and the aqueous layer was extracted two additional times with 15 mL of ethyl acetate. The organics were combined, washed with brine, dried over sodium sulfate and concentrated. The resulting solid was further purified by silica gel chromatography eluting with a gradient of methanol in dichloromethane (0-10%) to give N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (72 mg, 41%) as a white solid. ESI-MS m/z calc.597.2345, found 598.3 (M+1) +; Retention time: 2.1 minutes.1H NMR (400 MHz, DMSO) δ 12.36 (s, 1H), 8.37 (s, 1H), 8.22 (d, J = 2.8 Hz, 1H), 7.74 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 6.17 (d, J = 2.8 Hz, 1H), 4.23 (s, 2H), 3.81 (s, 3H), 2.56 (d, J = 10.4 Hz, 1H), 2.41 (t, J = 8.7 Hz, 1H), 2.32 (s, 3H), 2.18 (dd, J = 12.4, 6.1 Hz, 1H), 1.87 (dd, J = 11.7, 5.5 Hz, 1H), 1.55 (d, J = 11.2 Hz, 6H), 1.42 (t, J = 12.0 Hz, 1H), 1.23 (s, 6H), 0.81 (d, J = 6.2 Hz, 3H).
///////////VX-445, Elexacaftor, VX445, エレクサカフトル , PHASE 3, CYSTIC FIBRIOSIS, VX 445
C[C@@H]1CN(c2nc(ccc2C(=O)NS(=O)(=O)c3cn(C)nc3C)n4ccc(OCC(C)(C)C(F)(F)F)n4)C(C)(C)C1
CC1CC(N(C1)C2=C(C=CC(=N2)N3C=CC(=N3)OCC(C)(C)C(F)(F)F)C(=O)NS(=O)(=O)C4=CN(N=C4C)C)(C)C
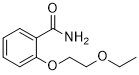



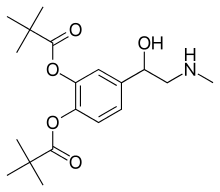













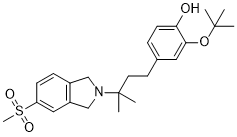
 in Europe
in Europe



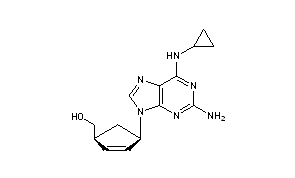
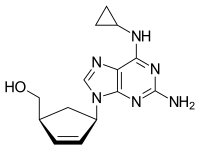












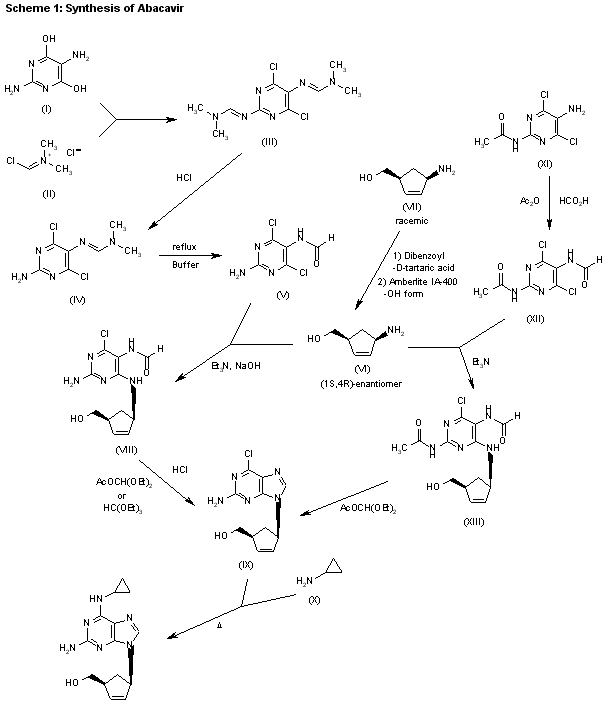
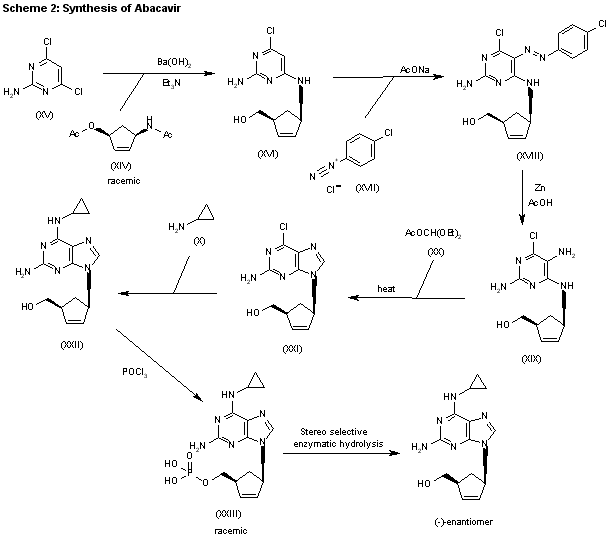
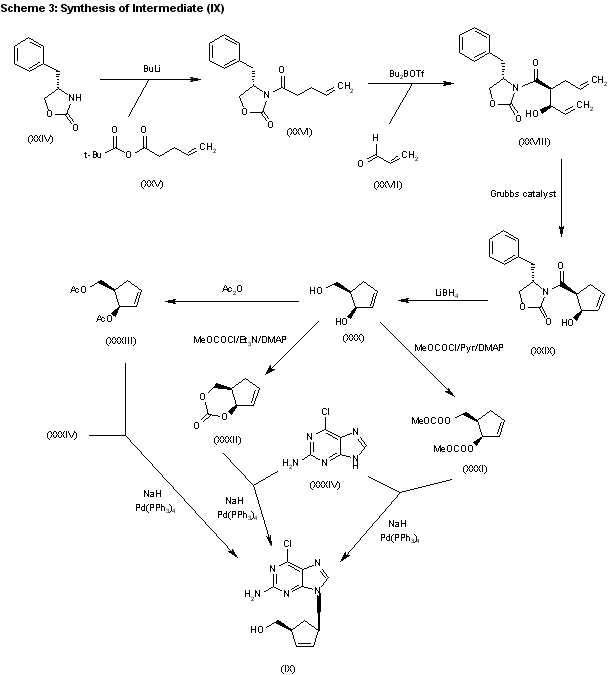
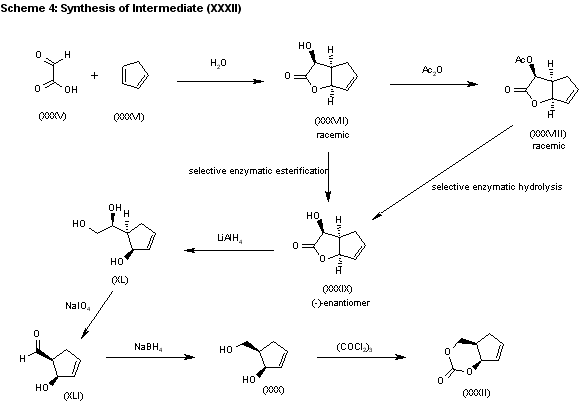
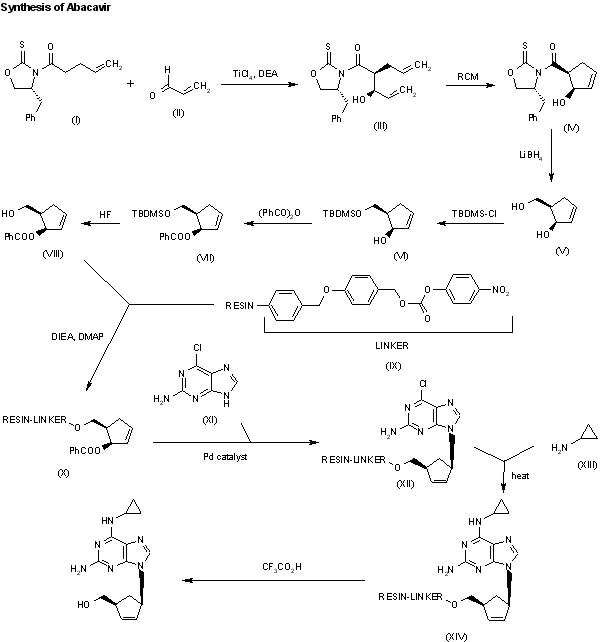
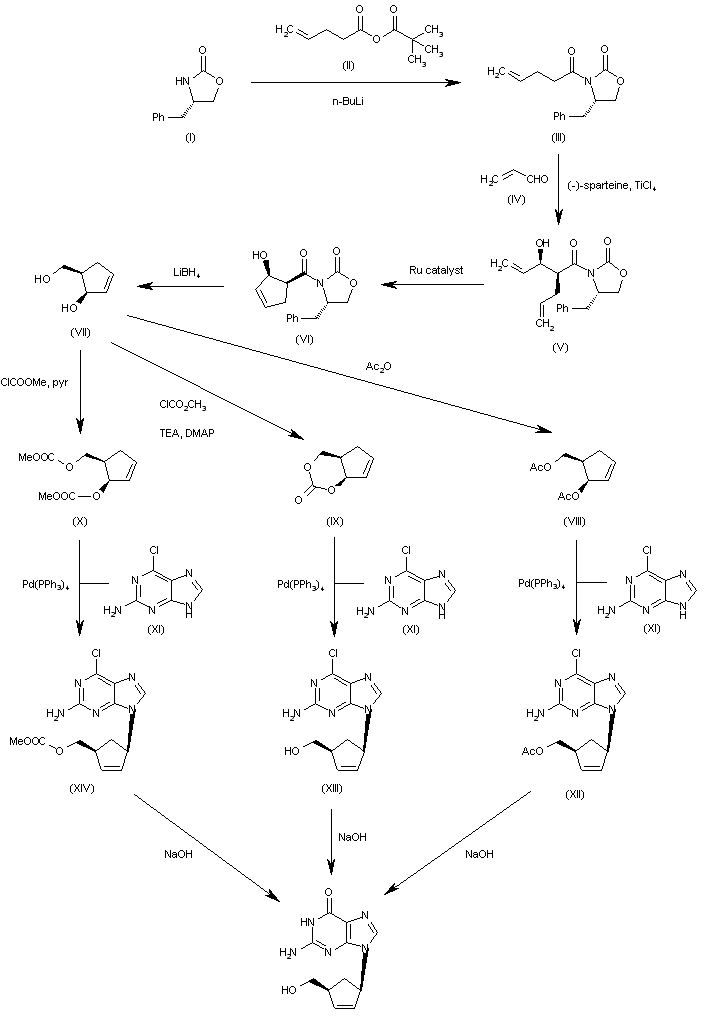 The condensation of the chiral oxazolidinone (I) with the pentenoic anhydride (II) by means of n-BuLi in THF gives the N-pentenoyloxazolidinone (III), which is condensed with acrolein (IV) catalyzed by TiCl4 and (-)-spartein in dichloromethane, yielding the chiral adduct (V). The ring-closing metathesis of (V) by means of a Ru catalyst in dichloromethane affords the chiral cyclopentenol derivative (VI), which is reduced to the (R,R)-5-(hydroxymethyl)-2-cyclopenten-1-ol (VII) by means of LiBH4 in THF. The reaction of diol (VII) with Ac2O; with methyl chloroformate, TEA and DMAP; or with ethyl chloroformate and pyridine gives the diacetate (VIII), the cyclic carbonate (IX) or the dicarbonate (X), respectively. The condensation of (VIII), (IX) or (X) with 2-amino-6-chloropurine (XI) by means of Pd(PPh3)4 yields the carbocyclic purines (XII), (XIII) or (XIV), respectively. Finally, these compounds are hydrolyzed with aqueous NaOH to the target carbocyclic guanine.
The condensation of the chiral oxazolidinone (I) with the pentenoic anhydride (II) by means of n-BuLi in THF gives the N-pentenoyloxazolidinone (III), which is condensed with acrolein (IV) catalyzed by TiCl4 and (-)-spartein in dichloromethane, yielding the chiral adduct (V). The ring-closing metathesis of (V) by means of a Ru catalyst in dichloromethane affords the chiral cyclopentenol derivative (VI), which is reduced to the (R,R)-5-(hydroxymethyl)-2-cyclopenten-1-ol (VII) by means of LiBH4 in THF. The reaction of diol (VII) with Ac2O; with methyl chloroformate, TEA and DMAP; or with ethyl chloroformate and pyridine gives the diacetate (VIII), the cyclic carbonate (IX) or the dicarbonate (X), respectively. The condensation of (VIII), (IX) or (X) with 2-amino-6-chloropurine (XI) by means of Pd(PPh3)4 yields the carbocyclic purines (XII), (XIII) or (XIV), respectively. Finally, these compounds are hydrolyzed with aqueous NaOH to the target carbocyclic guanine.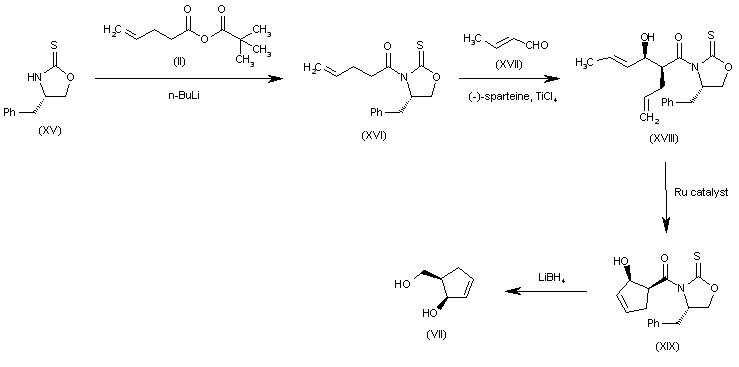
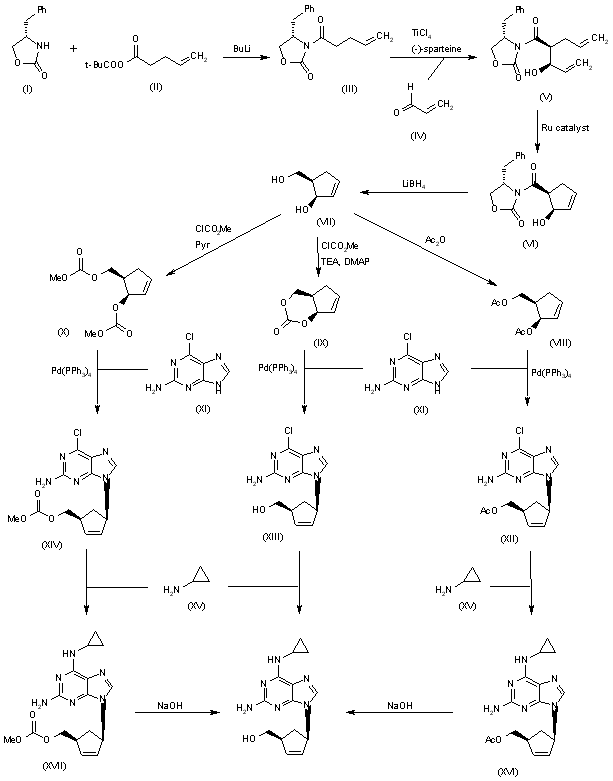
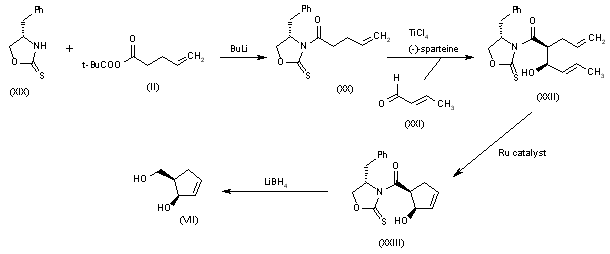


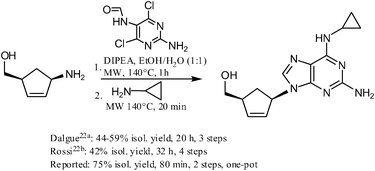

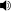








































 while maintaining the temperature below 30 °C. The resulting solid was then isolated by filtration and the filter cake washer with water (100 mL) then pulled dry until a sticky cake was obtained. The solids were then dried under vacuum at 55 °C to afford 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid (23.25 g) as an off-white fine solid.
while maintaining the temperature below 30 °C. The resulting solid was then isolated by filtration and the filter cake washer with water (100 mL) then pulled dry until a sticky cake was obtained. The solids were then dried under vacuum at 55 °C to afford 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid (23.25 g) as an off-white fine solid.


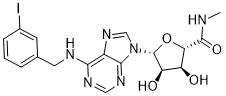

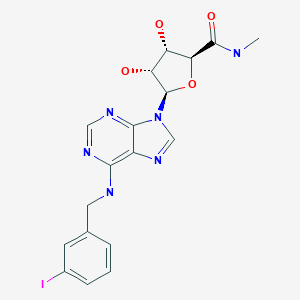




![(E)-3-[6-[2-(6-Methylpyridin-2-yl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl]-[1,2,4]triazolo[1,5-a]pyridin-5-yl]prop-2-enamide.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=132246793&t=l)



![3-[(1R)-1-(2,3-Dichloro-4-pyrazin-2-ylphenyl)-2,2,2-trifluoroethyl]-1-methyl-1-(1-methylpiperidin-4-yl)urea.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=91884613&t=l)