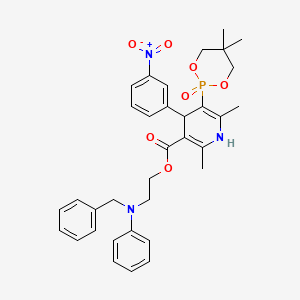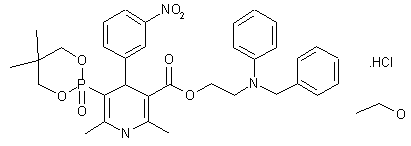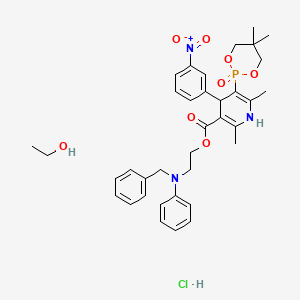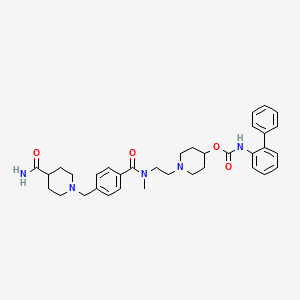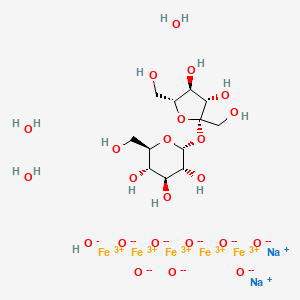

Sucroferric oxyhydroxide
Iron sucrose (USP);
Ferric oxide, saccharated;
Sucroferric oxyhydroxide;
Venofer (TN)
含糖酸化鉄;
スクロオキシ水酸化鉄
| Molecular Formula: | C12H29Fe5Na2O23 |
|---|---|
| Molecular Weight: | 866.546 g/mol |
|
CAS REGISTRY NUMBER 12134-57-5, 8047-67-4
disodium;(2R,3R,4S,5S,6R)-2-[(2S,3S,4S,5R)-3,4-dihydroxy-2,5-bis(hydroxymethyl)oxolan-2-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol;iron(3+);oxygen(2-);hydroxide;trihydrate
Iron sugar; Saccharated iron; Sucroferric oxyhydroxide; Saccharated iron oxide; Saccharated ferric oxide; Ferrivenin
Ferric oxyhydroxide; Ferrihydrite; Iron oxyhydroxide; P-TOL; PA-21; PA21-1; Phosphate binder – Vifor Pharma; suroferric oxyhydroxide tablets; Velphoro
NDC 49230-645-51
Iron saccharate (Sucroferric oxyhydroxide or Iron Sucrose) is used as a source of iron in patients with iron deficiency anemia with chronic kidney disease (CKD), including those who are undergoing dialysis (hemodialysis or peritoneal) and those who do not require dialysis. Due to less side effects than iron dextran, iron saccharate is more preferred in chronic kidney disease patients.
Mixture of polynuclear iron(III)-oxyhydroxide, starch and sucrose
VIFOR FRESENIUS MEDICAL CARE RENAL PHARMA FRANCE
Approved in US
Indicated for the control of serum phosphorus levels in patients with chronic kidney disease on dialysis.
THERAPEUTIC CLAIM Oral phosphate binder, treatement of elevated
phosphate levels in patients undergoing dialysis
CHEMICAL DESCRIPTIONS
1. Ferric hydroxide oxide
2. Mixture of iron(III) oxyhydroxide, sucrose, starches
3. Polynuclear iron(III) oxyhydroxide stabilized with sucrose and starches
structure
O =Fe -OH
MOLECULAR FORMULA FeHO2•xC12H22O11•y(C6H10O5)n
SPONSOR Vifor (International) Inc.
CODE DESIGNATIONS PA21
CAS REGISTRY NUMBER 12134-57-5
- ClassFerric compounds; Hyperphosphataemia therapies
- Mechanism of ActionPhosphate binding modulators
- MarketedHyperphosphataemia
- 24 Jun 2018Biomarkers information updated
- 19 Jun 2018Kissei Pharmaceutical completes a phase III trial in Hyperphosphataemia (Treatment-experienced) in Japan (PO) (UMIN000023657)
- 09 Jun 2017Phase-II clinical trials in Hyperphosphataemia in Austria (PO) (NCT03010072)
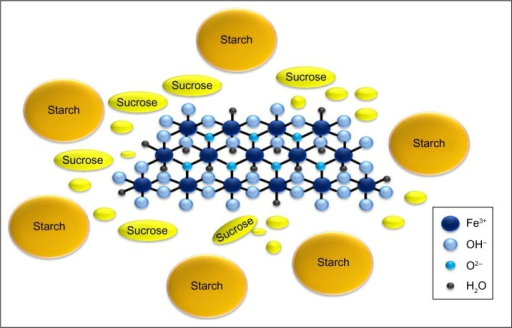
Sucroferric oxyhydroxide is a brown, amorphous powder. The drug substance is relatively poorly defined, so that the manufacturing process is particularly important. Sucroferric oxyhydroxide is prepared by basifying a ferric chloride solution, giving a polynuclear iron(III)-oxyhydroxide suspension which is mixed with potato and maize starches and sucrose. Vifor states that the sucrose stabilises the iron core and thus maintain the high phosphate adsorption capacity while the starches function as processing aids, but they are added simultaneously and the drug substance is probably a complex mixture of species.
The solubility of the active moiety, polynuclear iron oxyhydroxide, is evidently low in the gastrointestinal (GI) tract so that iron absorption is low. Aqueous solubility at different pH has been very poorly quantified. Vifor states that the “sucrose part is soluble in water, iron(III)-oxyhydroxide/starch mixture is practically insoluble in water.” While the iron oxide particle size is important in determining the phosphate binding, it is relatively difficult to directly measure. The sucrose/starch “wrapped” drug substance particle size is established and the process is controlled, but it does not correlate well with phosphate adsorption. Sucroferric oxyhydroxide cannot be controlled in the manner of a well-defined molecular drug and some variability between batches is likely. The drug substance specification includes a phosphate adsorption test. Vifor has tested the adsorption of a range of other in vivo chemical species to sucroferric oxyhydroxide and not identified any likely to be strongly bound, or affect phosphate binding, except for oxalate. Some drugs, however, do interact, for example alendronate is strongly absorbed (and the PI warnings in that context should be generalised to all bisphosphonates, not just identify the single drug in class studied)….https://www.tga.gov.au/sites/default/files/auspar-sucroferric-oxyhydroxide-150219.pdf
EMA
| Name | Active substance | Therapeutic area | Date of authorisation / refusal | Has current safety alert | Status | ||||
|---|---|---|---|---|---|---|---|---|---|
| Velphoro | mixture of polynuclear iron(III)-oxyhydroxide, sucrose and starches | HyperphosphatemiaRenal Dialysis | 26/08/2014 | Authorised |
Product details
| Name | Velphoro |
|---|---|
| Agency product number | EMEA/H/C/002705 |
| Active substance | mixture of polynuclear iron(III)-oxyhydroxide, sucrose and starches |
| International non-proprietary name(INN) or common name | mixture of polynuclear iron(III)-oxyhydroxide, sucrose and starches |
| Therapeutic area | HyperphosphatemiaRenal Dialysis |
| Anatomical therapeutic chemical (ATC) code | V03AE05 |
| Additional monitoring | This medicine is under additional monitoring. This means that it is being monitored even more intensively than other medicines. For more information, see medicines under additional monitoring. |
Publication details
| Marketing-authorisation holder | Vifor Fresenius Medical Care Renal Pharma France |
|---|---|
| Revision | 5 |
| Date of issue of marketing authorisation valid throughout the European Union | 26/08/2014 |
Contact address:
Vifor Fresenius Medical Care Renal Pharma France
100-101 Terrasse Boieldieu
Tour Franklin- La Défense 8
92042 Paris la Défense Cedex
France
Sucroferric oxyhydroxide (INN; trade name Velphoro, by Vifor Fresenius Medical Care Renal Pharma) is a non-calcium, iron-based phosphate binder used for the control of serum phosphorus levels in adult patients with chronic kidney disease (CKD) on haemodialysis(HD) or peritoneal dialysis (PD).[1] It is used in form of chewable tablets.
Hyperphosphatemia
In a healthy person, normal serum phosphate levels are maintained by the regulation of dietary absorption, bone formation and resorption, equilibration with intracellular stores, and renal excretion.[2] When kidney function is impaired, phosphate excretion declines. Without specific treatment, hyperphosphataemia occurs almost universally, despite dietary phosphate restriction and conventional dialysis treatment.[2][3] In patients on dialysis, hyperphosphataemia is an independent risk factor for fractures, cardiovascular disease and mortality.[4][5] Abnormalities in phosphate metabolism such as hyperphosphatemia are included in the definition of the new chronic kidney disease–mineral and bone disorder (CKD-MBD).[5]
Structure and mechanism of action
Sucroferric oxyhydroxide comprises a polynuclear iron(III)-oxyhydroxide core that is stabilised with a carbohydrate shell composed of sucrose and starch.[6][7] The carbohydrate shell stabilises the iron(III)-oxyhydroxide core to preserve the phosphate adsorption capacity.
Dietary phosphate binds strongly to sucroferric oxyhydroxide in the gastrointestinal (GI) tract. The bound phosphate is eliminated in the faeces and thereby prevented from absorption into the blood. As a consequence of the decreased dietary phosphate absorption, serum phosphorus concentrations are reduced.
Medical uses
Sucroferric oxyhydroxide is approved by the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the control of serum phosphorus levels in patients with chronic kidney disease (CKD) on dialysis.[1][8]
Adverse effects
The most frequently reported adverse drug reactions reported from trials were diarrhoea and discoloured faeces.[1][8] The vast majority of gastrointestinal adverse events occurred early during treatment and abated with time under continued dosing.[1]
Interactions
Drug-interaction studies and post hoc analyses of Phase 3 studies showed no clinically relevant interaction of sucroferric oxyhydroxide with the systemic exposures to losartan, furosemide, omeprazole, digoxin, and warfarin,[9] the lipid-lowering effects of statins,[10] and oral vitamin D receptor agonists.[11] According to the European label (Summary of Product Characteristics), medicinal products that are known to interact with iron (e.g. doxycycline) or have the potential to interact with Velphoro should be administered at least one hour before or two hours after Velphoro.[1] This allows sucroferric oxyhydroxide to bind phosphate as intended and be excreted without coming into contact with medications in the gut that it might interact with. According to the US prescribing information, Velphoro should not be prescribed with oral levothyroxine.[8] The combination of sucroferric oxyhydroxide and levothyroxine is contraindicated because sucroferric oxyhydroxide contains iron, which may cause levothyroxine to become insoluble in the gut, thereby preventing the intestinal absorption of levothyroxine.[12]
Chewability
The chewability of sucroferric oxyhydroxide compares well with that of Calcimagon, a calcium containing tablet used as a standard for very good chewability.[13] Tablets of sucroferric oxyhydroxide easily disintegrated in artificial saliva.
Effectiveness and phosphate binding
Clinical Phase 3 studies showed that sucroferric oxyhydroxide achieves and maintains phosphate levels in compliance with the KDOQI guidelines.[14][15] The reduction in serum phosphate levels of sucroferric oxyhydroxide-treated patients was non-inferior to that in sevelamer-treated patients. The required daily pill burden was lower with sucroferric oxyhydroxide.[14]
Sucroferric oxyhydroxide binds phosphate under empty and full stomach conditions and across the physiologically relevant pH range of the GI tract.[7]
In a retrospective, real-world study, hyperphosphatemic peritoneal dialysis patients who were prescribed to switch to sucroferric oxyhydroxide from sevelamer, lanthanum carbonate, or calcium acetate had significant reductions in serum phosphorus levels, along with a 53% decrease in the prescribed daily pill burden.[16]
Sucroferric oxyhydroxide nonproprietary drug name
1. February 27, 2013. N13/36. STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN (ZZ-19). SUCROFERRIC …
The US Food and Drug Administration has given the green light to Vifor Fresenius Medical Care Renal Pharma’s hyperphosphatemia drug Velphoro.
The approval for Velphoro (sucroferric oxyhydroxide), formerly known as PA21, is based on Phase III data demonstrated that the drug successfully controls the accumulation of phosphorus in the blood with the advantage of a much lower pill burden than the current standard of care in patients with chronic kidney disease on dialysis, namely Sanofi’s Renvela (sevelamer carbonate). read this at
http://www.pharmatimes.com/Article/13-11-28/FDA_okays_Vifor_Fresenius_phosphate_binder_Velphoro.aspx
Velphoro (PA21) receives US FDA approval for the treatment of hyperphosphatemia in Chronic Kidney Disease Patients on dialysis
Velphoro (sucroferric oxyhydroxide) has received US Food and Drug Administration (FDA) approval for the control of serum phosphorus levels in patients with Chronic Kidney Disease (CKD) on dialysis. Velphoro will be launched in the US by Fresenius Medical Care North America in 2014.
Velphoro (previously known as PA21) is an iron-based, calcium-free, chewable phosphate binder. US approval was based on a pivotal Phase III study, which met its primary and secondary endpoints. The study demonstrated that Velphoro® successfully controls hyperphosphatemia with fewer pills than sevelamer carbonate, the current standard of care in patients with CKD on dialysis. The average daily dose to control hyperphosphatemia was 3.3 pills per day after 52 weeks.
Velphoro was developed by Vifor Pharma. In 2011, all rights were transferred to Vifor Fresenius Medical Care Renal Pharma, a common company of Galenica and Fresenius Medical Care. In the US, Velphorowill be marketed by Fresenius Medical Care North America, a company with a strong marketing and sales organization, and expertise in dialysis care. The active ingredient of Velphoro is produced by Vifor Pharma in Switzerland.
Hyperphosphatemia, an abnormal elevation of phosphorus levels in the blood, is a common and serious condition in CKD patients on dialysis. Most dialysis patients are treated with phosphate binders. However, despite the availability of a number of different phosphate binders, up to 50% of patients depending on the region are still unable to achieve and maintain their target serum phosphorus levels. In some patients, noncompliance due to the high pill burden and poor tolerability appear to be key factors in the lack of control of serum phosphorus levels. On average, dialysis patients take approximately 19 pills per day with phosphate binders comprising approximately 50% of the total daily pill burden. The recommended starting dose of Velphoro is 3 tablets per day (1 tablet per meal).
Full results from the pivotal Phase III study involving more than 1,000 patients were presented at both the 50th ERA-EDTA (European Renal Association European Dialysis and Transplant Association) Congress in Istanbul, Turkey, in May 2013, and the American Society of Nephrology (ASN) Kidney Week in Atlanta, Georgia, in November 2013. Velphorowas shown to be a potent phosphate binder, with lower pill burden and a good safety profile.
Based on these data, Vifor Fresenius Medical Care Renal Pharma believes that Velphoro offers a new and effective therapeutic option for the control of serum phosphorus levels in patients with chronic kidney disease on dialysis.
The regulatory processes in Europe, Switzerland and Singapore are ongoing and decisions are expected in the first half 2014. Further submissions for approval are being prepared.
PATENT
https://patents.google.com/patent/WO2016038541A1/en
Hyperphosphatemia is associated with significant increase in morbidity and mortality, and may induce severe complications, such as hypocalcemia, decreasing of vitamin-D production and metastatic calcification. Hyperphosphatemia is also contributing to the increased incidence of cardiovascular disease among dialysis-dependent patients. The phosphate binding capacity of iron oxide hydroxides is known in the art. The possible medical application of iron hydroxides and iron oxide hydroxides as phosphate adsorbents is also described.
US 4,970,079 patent discloses a method of controlling serum phosphate level in patients by iron oxy-hydroxides which bind to ingested phosphate. US 5,514,281 patent also discloses a process for the selective elimination of inorganic phosphate from body fluids by using a polynuclear metal oxyhydroxide preferably iron (III) oxyhydroxide.
US 6,174,442 patent describes an adsorbent for phosphate and a process for the preparation thereof, which contains polynuclear β-iron hydroxide stabilized by carbohydrates and/or humic acid.
In order to obtain an iron-based compound which can be used as a pharmaceutical, it is necessary to have an iron-based compound which is stable. It is known that iron oxide- hydroxide is not a stable compound with time ageing occurs. Ageing usually not only involves crystallization but also particle enlargement. Such ageing may alter the phosphate binding of an iron oxide -hydroxide based phosphate adsorbent. Accordingly, there exists a need for a process for manufacturing of an iron containing phosphate adsorbent. The process needs to be scalable, robust and consistently producing an iron containing phosphate adsorbent of the required pharmaceutical grade.
Examples
In examples which are intended to illustrate embodiments of the invention but which are not intended to limit the scope of the invention: ) Method of Making an Iron Containing Phosphate Adsorbent
To a solution of 1.96 kg sodium carbonate dissolved in 12.5 liter water, solution of 2.5 kg iron (III) chloride hexahydrate dissolved in 17.5 liter water was added at a temperature of 5 – 10°C. The resulting mixture was stirred for 90 to 120 minutes at 5 – 10°C. (25.0×3) liter water was added to the reaction mass and raised the temperature at 15 – 20°C with stirring. Stopped the stirring, settled precipitate and the supernatant water was removed. The precipitate was filtered and washed with 1.25 liter water. A suspension of the precipitate was prepared in water. To this, 875.0 gm sucrose and 695.0 gm potato starch were added and stirred for 120 minutes at 25 – 35°C. Cooled the reaction mass at 10 – 15°C and stirred for 90 to 120 minutes. 25.0 liters cold acetone was added to the reaction mass at 10 – 15°C and stirred for 90 to 120 minutes. The final product was filtered and washed with 1.25 liter cold acetone and further dried under vacuum at 30-35°C.
Yield: 2.08 kg ) Large-scale Method of Making an Iron Containing Phosphate Adsorbent
An aqueous solution of sodium carbonate and an aqueous solution of iron (III) chloride hexahydrate were mixed at a temperature of 5 – 10°C, optionally in the presence of solvent- 1. A volume of aqueous solution of sodium carbonate necessary to maintain the pH at about 7.0 to form a colloidal suspension of ferric hydroxide. The resulting mixture was stirred for 90 to 120 minutes at 5 – 10°C. Water was added to the reaction mass with stirring. Stopped the stirring, settled precipitated product and the water was decanted or siphoned. The precipitated product was further filtered and washed with using water. Suspension of the precipitated product was prepared in the water. Subsequently, sucrose and starch were added in to the suspension and stirred for 120 minutes at 25 – 35°C. Cooled the reaction mixture at 10 – 15°C and stirred for 90 to 120 minutes. Solvent-2 was added to the reaction mixture at 10 – 15°C and stirred for 90 to 120 minutes. The product was filtered and washed with the solvent-2 and further dried under vacuum at 30-35°C. Few illustrative examples provided in Table- 1, wherein the iron containing phosphate adsorbents were prepared according to the process of example-2 using the respective combination of Solvent- 1 and Solvent-2 as given in the table:
Table-1

3) Physical Properties of an Iron Containing Phosphate Adsorbents prepared as per above example-2.
> BET active Surface Area:
· Instrument : Surface area analyzer
• Condition : Surface area (m2/gm) at N2.P/P0 = 10%
Table-2

> Phosphate Binding Capacity at pH 3.0:
· Method : Ion Chromatography Instrument : Metrohm IC equipped with pump, Injector, conductivity detector and recorder.
Column Dionex Ion Pac AS-11 (4.0 x 250mm), 13μπι
Guard column Dionex Ion Pac AG-11 (4.0 x 50mm), 13μπι
Buffer preparation Weigh accurately about 2.118g of Sodium carbonate and 180mg of Sodium hydroxide in 1700mL water.
Mobile phase preparation : Buffer and acetonitrile (1700:300).
Results: Phosphate binding of an iron containing phosphate adsorbents obtained by following the process of the present invention found in the range of 30 mg/gm to 60 mg/gm. Particle Size Distribution:
Instrument Model : Malvern Mastersizer 2000 Particle size analyzer
Sampling Unit : Hydro 2000S
Analysis Model : General Purpose
Dispersant : 0.1% Span 85 in n-Hexane
Dispersant RI : 1.380
Stirrer Speed : 2200 RPM
Absorption : 1
Particle RI : 1.5
Obscuration : 10% to 20%
Sample Measurement time : 12 seconds
Background Measurement time : 12 seconds Table-3
Particle size distribution
Example no.
d(0.9) (μηι)
3d 43.67
3e 65.37
3f 37.75

References
- ^ Jump up to:a b c d e “Velphoro (sucroferric oxyhydroxide). Summary of Product Characteristics”(PDF). EMA. Archived from the original on October 21, 2014. Retrieved 24 October 2014.
- ^ Jump up to:a b Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, Saran R, Wang AY, Yang CW (July 2013). “Chronic kidney disease: global dimension and perspectives”. Lancet. 382(9888): 260–72. doi:10.1016/S0140-6736(13)60687-X. PMID 23727169.
- Jump up^ Hutchison AJ, Smith CP, Brenchley PE (September 2011). “Pharmacology, efficacy and safety of oral phosphate binders”. Nature Reviews. Nephrology. 7 (10): 578–89. doi:10.1038/nrneph.2011.112. PMID 21894188.
- Jump up^ Isakova T, Gutiérrez OM, Chang Y, Shah A, Tamez H, Smith K, Thadhani R, Wolf M (February 2009). “Phosphorus binders and survival on hemodialysis”. Journal of the American Society of Nephrology. 20 (2): 388–96. doi:10.1681/ASN.2008060609. PMC 2637053
 . PMID 19092121.
. PMID 19092121. - ^ Jump up to:a b “KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD)”. Kidney International Supplement. 76 (113): S1–130. August 2009. doi:10.1038/ki.2009.188. PMID 19644521.
- Jump up^ Vifor Fresenius Medical Care Renal Pharma. Product Monograph 2015.
- ^ Jump up to:a b Wilhelm M, Gaillard S, Rakov V, Funk F (April 2014). “The iron-based phosphate binder PA21 has potent phosphate binding capacity and minimal iron release across a physiological pH range in vitro”. Clinical Nephrology. 81 (4): 251–8. doi:10.5414/cn108119. PMID 24656315.
- ^ Jump up to:a b c “Highlights of Prescribing information for Velphoro”. Fresenius. September 2014.
- Jump up^ Chong E, Kalia V, Willsie S, Winkle P (December 2014). “Drug-drug interactions between sucroferric oxyhydroxide and losartan, furosemide, omeprazole, digoxin and warfarin in healthy subjects”. Journal of Nephrology. 27 (6): 659–66. doi:10.1007/s40620-014-0080-1. PMC 4242982
 . PMID 24699894.
. PMID 24699894. - Jump up^ Levesque V, Chong EMF, Moneuse P (2013). “Post-hoc analysis of pharmacodynamic interaction of PA21 with statins in a Phase 3 study of PA21 in dialysis patients with hyperphosphatemia”. J Am Soc Nephrol. 24: 758A.
- Jump up^ Floege J, Botha J, Chong E et al. (31 May 2014). PA21 does not interact with oral vitamin D receptor agonists: a post hoc analysis of a Phase 3 study. ERA-EDTA congress. Amsterdam, The Netherlands. Abstract no. SP257.
- Jump up^ Prescribing Information. Synthroid (levothyroxine). Chicago, IL: Abbott Laboratories. March 1, 2008.
- Jump up^ Lanz M, Baldischweiler J, Kriwet B, Schill J, Stafford J, Imanidis G (December 2014). “Chewability testing in the development of a chewable tablet for hyperphosphatemia”. Drug Development and Industrial Pharmacy. 40 (12): 1623–31. doi:10.3109/03639045.2013.838583. PMID 24010939.
- ^ Jump up to:a b Floege J, Covic AC, Ketteler M, Rastogi A, Chong EM, Gaillard S, Lisk LJ, Sprague SM (September 2014). “A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients”. Kidney International. 86 (3): 638–47. doi:10.1038/ki.2014.58. PMC 4150998
 . PMID 24646861.
. PMID 24646861. - Jump up^ Floege J, Covic AC, Ketteler M, Mann JF, Rastogi A, Spinowitz B, Chong EM, Gaillard S, Lisk LJ, Sprague SM (June 2015). “Long-term effects of the iron-based phosphate binder, sucroferric oxyhydroxide, in dialysis patients”. Nephrology, Dialysis, Transplantation. 30(6): 1037–46. doi:10.1093/ndt/gfv006. PMC 4438742
 . PMID 25691681.
. PMID 25691681. - Jump up^ Kalantar-Zadeh K, Parameswaran V, Ficociello LH, Anderson L, Ofsthun NJ, Kwoh C, Mullon C, Kossmann RJ, Coyne DW (2018). “Real-World Scenario Improvements in Serum Phosphorus Levels and Pill Burden in Peritoneal Dialysis Patients Treated with Sucroferric Oxyhydroxide”. American Journal of Nephrology. 47 (3): 153–161. doi:10.1159/000487856. PMC 5906196
 . PMID 29514139.
. PMID 29514139.

| Clinical data | |
|---|---|
| Trade names | Velphoro |
| AHFS/Drugs.com | Consumer Drug Information |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral (chewable tablets) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Chemical and physical data | |
| Formula | Varies |
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6174442 |
| Expiration | Dec 19, 2018 |
| Applicant | VIFOR FRESENIUS |
| Drug Application | N205109 (Prescription Drug: VELPHORO. Ingredients: SUCROFERRIC OXYHYDROXIDE) |
/////////////Sucroferric oxyhydroxide, EU 2014, Iron sugar, Saccharated iron, Sucroferric oxyhydroxide, Saccharated iron oxide, Saccharated ferric oxide, Ferrivenin, 含糖酸化鉄, スクロオキシ水酸化鉄 , NDC 49230-645-51
C(C1C(C(C(C(O1)OC2(C(C(C(O2)CO)O)O)CO)O)O)O)O.O.O.O.[OH-].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[Na+].[Na+].[Fe+3].[Fe+3].[Fe+3].[Fe+3].[Fe+3]
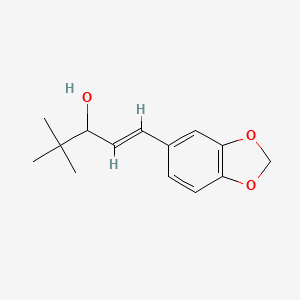
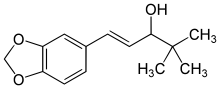



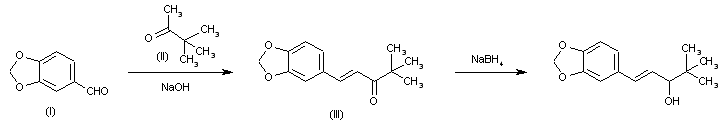
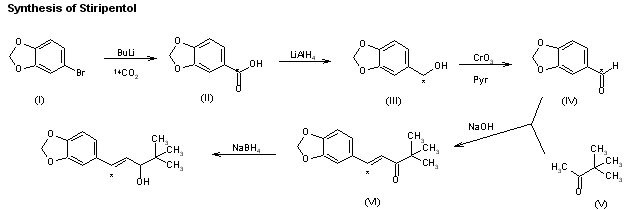



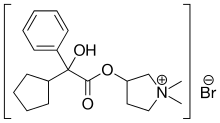
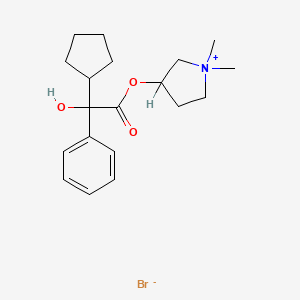

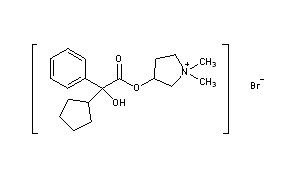









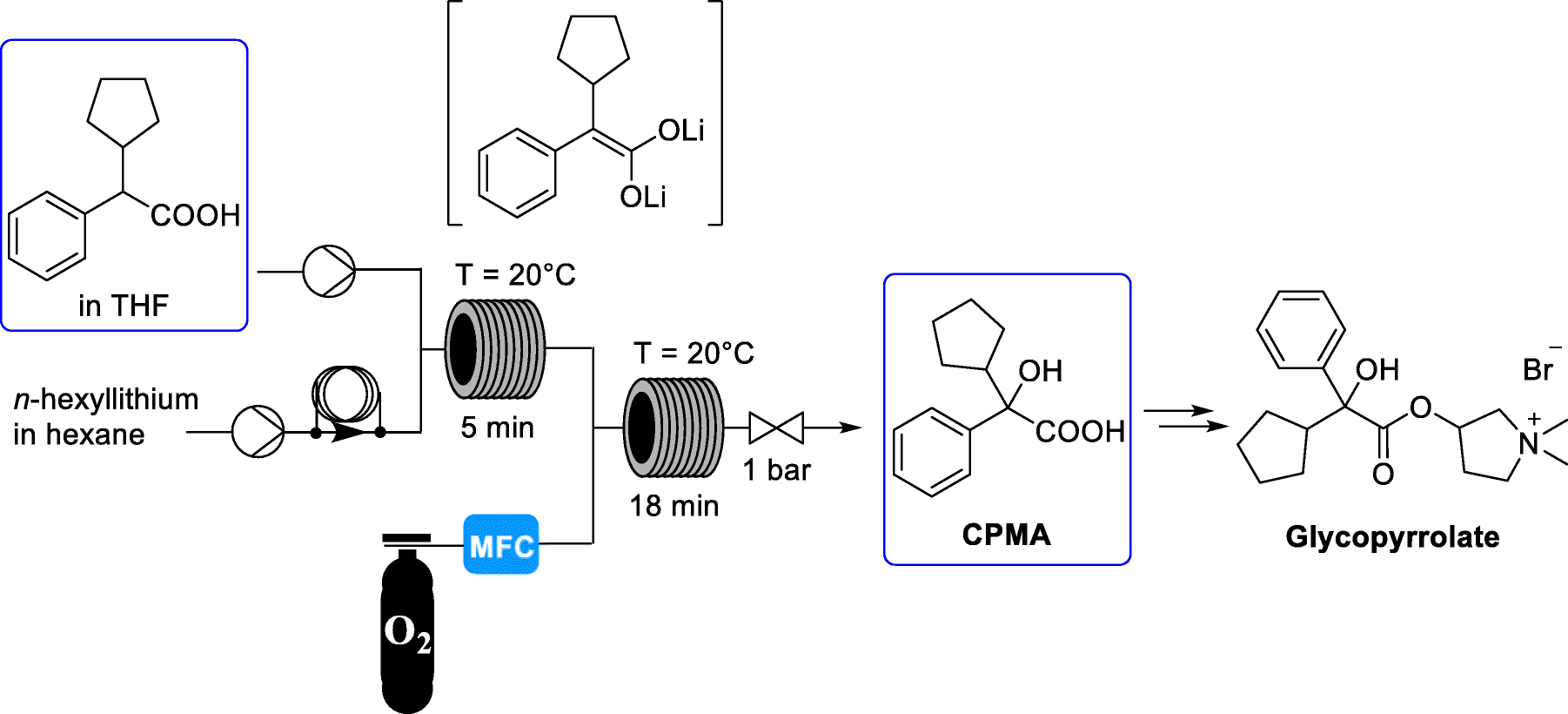





















 C6H6O3S : 547.07
C6H6O3S : 547.07


























 , a new antimalarial combination of OZ277 and piperaquine, against different developmental stages of Schistosoma mansoni. Acta Trop. 2015 Mar;143:36-46. doi: 10.1016/j.actatropica.2014.12.005. PubMed PMID: 25530543.
, a new antimalarial combination of OZ277 and piperaquine, against different developmental stages of Schistosoma mansoni. Acta Trop. 2015 Mar;143:36-46. doi: 10.1016/j.actatropica.2014.12.005. PubMed PMID: 25530543.
















 Diazoxide
Diazoxide