
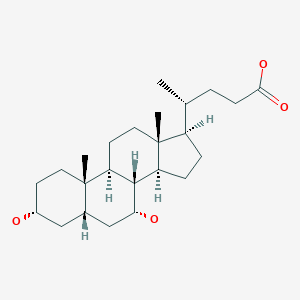
Chenodeoxycholic acid
Chenodiol
- Molecular FormulaC24H40O4
- Average mass392.572
|
Chenodeoxycholate;
Chenodeoxycholic acid; 3alpha,7alpha-Dihydroxy-5beta-cholanic acid; Chenodiol |
Synthesis ReferenceHenry Francis Frost, Fritz Fabian, Christopher James Sharpe, William Arthur Jones, “Process for preparing chenodeoxycholic acid.” U.S. Patent US4022806, issued October, 1974. US4022806
First ref
- By Windaus, A.; Bohne, A.; Schwarzkopf, E.
- From Z. physiol. Chem. (1924), 140, 177-85
- By Wieland, Heinrich; Reverey, Gustav
- From Z. physiol. Chem. (1924), 140, 186-202.

Chenodeoxycholic acid (also known as chenodesoxycholic acid, chenocholic acid and 3α,7α-dihydroxy-5β-cholan-24-oic acid) is a bile acid. It occurs as a white crystalline substance insoluble in water but soluble in alcohol and acetic acid, with melting point at 165–167 °C. Salts of this carboxylic acid are called chenodeoxycholates. Chenodeoxycholic acid is one of the main bile acids produced by the liver.[1]
It was first isolated from the bile of the domestic goose, which gives it the “cheno” portion of its name (Greek: χήν = goose).[2]
Chenodeoxycholic acid and cholic acid are the two primary bile acids in humans. Some other mammals have muricholic acid or deoxycholic acid rather than chenodeoxycholic acid.[1]
Chenodeoxycholic acid is synthesized in the liver from cholesterol by a process which involves several enzymatic steps.[1] Like other bile acids, it can be conjugated in the liver with taurine or glycine, forming taurochenodeoxycholate or glycochenodeoxycholate. Conjugation results in a lower pKa. This means the conjugated bile acids are ionized at the usual pH in the intestine and will stay in the gastrointestinal tract until reaching the ileum where most will be reabsorbed. Bile acids form micelles which facilitate lipid digestion. After absorption, they are taken up by the liver and resecreted, so undergoing an enterohepatic circulation. Unabsorbed chenodeoxycholic acid can be metabolised by bacteria in the colon to form the secondary bile acid known as lithocholic acid.
Chenodeoxycholic acid is the most potent natural bile acid at stimulating the nuclear bile acid receptor, farnesoid X receptor (FXR).[3]The transcription of many genes is activated by FXR.
Indication
Chenodiol is indicated for patients with radiolucent stones in well-opacifying gallbladders, in whom selective surgery would be undertaken except for the presence of increased surgical risk due to systemic disease or age. Chenodiol will not dissolve calcified (radiopaque) or radiolucent bile pigment stones.
Associated Conditions
Pharmacodynamics
It acts by reducing levels of cholesterol in the bile, helping gallstones that are made predominantly of cholesterol to dissolve. Chenodeoxycholic acid is ineffective with stones of a high calcium or bile acid content.
Mechanism of action
Chenodiol suppresses hepatic synthesis of both cholesterol and cholic acid, gradually replacing the latter and its metabolite, deoxycholic acid in an expanded bile acid pool. These actions contribute to biliary cholesterol desaturation and gradual dissolution of radiolucent cholesterol gallstones in the presence of a gall-bladder visualized by oral cholecystography. Bile acids may also bind the the bile acid receptor (FXR) which regulates the synthesis and transport of bile acids.
EMA
On 16 December 2014, orphan designation (EU/3/14/1406) was granted by the European Commission to Sigma-Tau Pharma Ltd, United Kingdom, for chenodeoxycholic acid for the treatment of inborn errors in primary bile acid synthesis.
The sponsorship was transferred to sigma-tau Arzneimittel GmbH, Germany, in May 2015.
Chenodeoxycholic acid has been authorised in the EU as Chenodeoxycholic acid sigma-tau since 10 April 2017.
The name of the product changed to Chenodeoxycholic acid Leadiant in May 2017.
The sponsorship was transferred to Leadiant GmbH, Germany, in June 2017.
On 16 February 2017, the Committee for Orphan Medicinal Products (COMP) concluded its review of the designation EU/3/14/1406 for Chenodeoxycholic acid sigma-tau (chenodeoxycholic acid) as an orphan medicinal product for the treatment of inborn errors in primary bile acid synthesis. The COMP assessed whether, at the time of marketing authorisation, the medicinal product still met the criteria for orphan designation. The Committee looked at the seriousness and prevalence of the condition, and the existence of other methods of treatment. As other methods of treatment are authorised in the European Union (EU), the COMP also considered whether the medicine is of significant benefit to patients with inborn errors in primary bile acid synthesis. The COMP recommended that the orphan designation of the medicine be maintained1.
1 The maintenance of the orphan designation at time of marketing authorisation would, except in specific situations, give an orphan medicinal product 10 years of market exclusivity in the EU. This means that in the 10 years after its authorisation similar products with the same therapeutic indication cannot be placed on the market.
http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2015/02/WC500183233.pdf
Therapeutic applications
Chenodeoxycholic acid has been used as medical therapy to dissolve gallstones.[4]
Chenodeoxycholic acid can be used in the treatment of cerebrotendineous xanthomatosis.[5]
The Australian biotechnology company Giaconda has tested a treatment for Hepatitis C infection that combines chenodeoxycholic acid with bezafibrate.[6]
As diarrhea is a complication of chenodeoxycholic acid therapy, it has also been used to treat constipation.[7][8]
In supramolecular chemistry, molecular tweezers based on a chenodeoxycholic acid scaffold is a urea receptor that can contain anionsin its binding pocket in order of affinity: H2PO4− (dihydrogen phosphate) > Cl− > Br− > I− reflecting their basicities (tetrabutylammonium counter ion).[9]
![Molecular tweezer based on chenodeoxycholic acid]()
- PAPER
- 1H and 13C NMR characterization and stereochemical assignments of bile acids in aqueous media
Lipids (2005), 40, (10), 1031-1041. - https://onlinelibrary.wiley.com/doi/abs/10.1007/s11745-005-1466-1

PAPER
Improved Chemical Synthesis, X-Ray Crystallographic Analysis, and NMR Characterization of (22R)-/(22S)-Hydroxy Epimers of Bile Acids
Lipids (2014), 49, (11), 1169-1180.
Improved Chemical Synthesis, X‐Ray Crystallographic Analysis, and NMR Characterization of (22R)‐/(22S)‐Hydroxy Epimers of Bile Acids
A Practical and Eco-friendly Synthesis of Oxo-bile Acids
By Han, Young Taek and Yun, HwayoungFrom Organic Preparations and Procedures International, 48(1), 55-61; 2016
DOI:10.1080/00304948.2016.1127101
General Procedure
An aqueous solution of 0.2 M NaBrO3 (1.5 equiv. per hydroxy group) was added dropwise to a slurry of bile acid (1 equiv.) and ceric ammonium nitrate (0.05 equiv.) in 20% aqueous acetonitrile (0.2 M) at 80°C over 20 min. The bile acid slowly dissolved in a few minutes, and then the color of the reaction mixture changed to orange. The reaction mixture was stirred at the same temperature and the progress of the reaction was monitored by TLC on silica gel (1:20 MeOH-CH2Cl2) until disappearance of the starting material and partially oxidized intermediates. It was then cooled in an ice bath and quenched with aqueous Na2S2O3 solution. Water was added slowly to the resulting white suspension until no more oxo-bile acid precipitated. The white solid was collected, washed with water until the filtrate was colorless, and then dried in vacuo at 50°C. Methyl 3,7α-Diacetoxy-12-oxo-5β-cholanoate(3),21 was obtained in 92% yield (275 mg) as a white solid from 300 mg (0.590 mmol) of 2 via the general procedure. mp. 176-178°C, lit.22 mp. 178-179°C, IR (thin film, neat): 2947 (m), 2873 (s), 1736 (w), 1706 (w), 1436 (s), 1365 (m) cm-1; 1H-NMR (400 MHz, CDCl3): δ 4.96 (m, 1H, 7-CH), 4.55 (m, 1H, 3-CH), 3.64 (s, 3H), 2.49 (t, 1H, J = 12.6 Hz), 2.41-0.80 (m, 23H), 2.01 (s, 3H), 2.00 (s, 3H), 1.01 (s, 3H, 18-CH3), 1.00 (s, 3H, 19-CH3), 0.83 (d, 3H, J = 6.6 Hz, 21-CH3); 13C-NMR (CDCl3, 100 MHz): δ 214.0 (12-C), 174.6 (24-C), 170.7 (C = O), 170.2 (C = O), 73.5 (3-C), 70.5 (7-C), 57.1 (13-C), 53.1 (14-C), 51.5 (CH3O), 46.3 (17-C), 40.5 (5-C), 37.9 (11-C), 37.8 (4-C), 37.6 (8-C), 35.54 (9-C), 35.52 (20-C), 34.9 (1-C), 34.5 (10-C), 31.3 (6-C), 31.2 (22-C), 30.4 (23-C), 27.4 (16-C), 26.5 (2-C), 23.8 (15-C), 22.1 (19-C), 21.51 (CH3CO2), 21.46 (CH3CO2), 18.6 (21-C), 11.5 (18-C); LR-MS (FABC) m/z 505 (M+H +). HR-MS (FABC): Calcd for C29H45O7 (M+H +): 505.3165. Found 505.3161.
next step
R:KOH, R:N2H4

NOTE STARTING IS BILE ACID AS BELOW
Cholan-24-oic acid, 3,7-bis(acetyloxy)-12-oxo-, methyl ester, (3α,5β,7α)-
- 5β-Cholan-24-oic acid, 3α,7α-dihydroxy-12-oxo-, methyl ester, diacetate (8CI)
- 5β-Cholanic acid, 3α,7α-dihydroxy-12-oxo-, methyl ester, diacetate (6CI,7CI)
- 3α,7α-Diacetoxy-12-oxo-5β-cholan-24-oic acid methyl ester
- Methyl 3α,7α-diacetoxy-12-oxo-5β-cholan-24-oate
- Methyl 3α,7α-diacetoxy-12-oxo-5β-cholanate
![]()
-
- The structures of the principal human bile acids









PAPER
https://pubs.acs.org/doi/pdf/10.1021/jo01091a623
Journal of Organic Chemistry
Volume24
Pages1367-8
Journal
1959
DOI:10.1021/jo01091a623
Chenodeoxycholic acid (V). Five hundred mg. of the above ester IV was hydrolyzed with 80 ml. of ethanolic 5% potassium hydroxide for 4 hr. After partial concentration of the volume and addition of water, the reaction product was acidified with hydrochloric acid. The resulting precipitate was collected, dried, and crystallized from ethyl acetate. A quantitative crop (400 mg.) of prisms melting at 143- 145° were obtained. Recrystallization from the same solvent yielded a product of m.p. 145-146°, [ ]2 +10.7° (dioxane). Anal. Caled, for C24H40O4: C, 73.43; H, 10.27. Found: C, 73.49; H, 10.31.

NOTE I IS BILE ACID
Cholan-24-oic acid, 3,7-bis(acetyloxy)-12-oxo-, methyl ester, (3α,5β,7α)-
- 5β-Cholan-24-oic acid, 3α,7α-dihydroxy-12-oxo-, methyl ester, diacetate (8CI)
- 5β-Cholanic acid, 3α,7α-dihydroxy-12-oxo-, methyl ester, diacetate (6CI,7CI)
- 3α,7α-Diacetoxy-12-oxo-5β-cholan-24-oic acid methyl ester
- Methyl 3α,7α-diacetoxy-12-oxo-5β-cholan-24-oate
- Methyl 3α,7α-diacetoxy-12-oxo-5β-cholanate
![]()
-
- The structures of the principal human bile acids
PATENT
https://patents.google.com/patent/CN102060902A/en
chenodeoxycholic acid (3 α, 7 α – dihydroxy _5 β – cholestane-24-oic acid) Chenodeoxycholic Ac id (referred to as CDCA), clinically used to correct dissolving cholesterol calculi and bile saturation drugs, the main function is to reduce the cholesterol in the bile saturation, large doses can inhibit the synthesis of cholesterol CDCA and increasing bile gallstone patients cholesterol level in a non-saturated, thereby preventing the formation of cholesterol gallstones of cholesterol and promote stone dissolve and fall off. It also has significant anti-asthmatic, anti-inflammatory, antitussive and expectorant effects.
[0003] Synthesis of chenodeoxycholic acid or ursodeoxycholic acid (3 α, 7β_ -5β_ dihydroxy-cholestane-24-oic acid, ursodeoxycholic Acid, referred UDCA), a key intermediate. Ursodeoxycholic acid is the main active ingredient of precious Chinese medicine bear bile, used in a variety of clinical hepatobiliary disease and dyspepsia. Currently we bear bile resources are scarce, mainly used synthetic chemical ursodeoxycholic acid as a clinical treatment. Therefore, the preparation of chenodeoxycholic acid is also important for the preparation of ursodeoxycholic acid.
[0004] CDCA mainly come from poultry or livestock bile extraction. Traditional extraction process complicated operation, low yield, (pharmaceutical industry, 1987,18 (9), 416; Chinese Journal of Biochemical Pharmaceutics, 1996,17 (1), 17; Applied Technology, 1998, (4), 9; CN1850846A ) can not meet the needs of modern industry. Chemical synthesis of chenodeoxycholic acid have also been reported (Japanese Journal of Chemistry 1955,76 (3), 297 -J Org Chem 1982,47 (2): 2331; Journal of Biochemical Pharmaceutics 1987,1,6 -, Tap Chi Duoc ^ oc2004 , 44 (1), 11; CN1869043A), but lower yield widespread pollution major problem, especially in the oxidation reaction is often used to expensive, and polluting agents.Therefore, to reduce pollution, reduce environmental hazards, streamline operations, improve yield, reduce costs, important for the synthesis of chenodeoxycholic acid.
n particular by the following steps:
(1) Preparation of cholate: bile acid in alcohol, concentrated hydrochloric acid as catalyst, at reflux, cooling and crystallization, filtration, and washed with methanol.
[0008] (2) Preparation of 3α, 7α- diacetyl hydroxy -12α- cholate: bile acid ester was dissolved in dichloromethane and triethylamine was added with stirring acetic anhydride and the catalyst N, N- dimethyl pyridine, methylene chloride was distilled off, poured into water, filtered to give 3α, 7α- diacetyl -12 α – hydroxy cholate.
[0009] (3) 3α, 7α- diacetyl -12– Preparation oxo chenodeoxycholic acid ester: Take 3 α, 7 α – diacetyl -12 α – hydroxy cholate dissolved in ethyl acetate and methanol, bromide and tetrabutylammonium bromide as catalyst, and acetic acid was added dropwise under stirring hypochlorite, the organic solvent was distilled off and filtered, to give 12-oxo-3,7-diacetyl Chenodeoxy cholate.
[0010] (4) i2 – Preparation oxo chenodeoxycholic acid: 3,7-diacetyl-12-oxo-chenodeoxycholic acid ester added ethanol – sodium hydroxide solution, at reflux.PH adjusted with hydrochloric acid value of the reaction system acidic, ethanol was distilled off, and filtered to give 12- oxo crude chenodeoxycholic acid, fine recrystallization.
[0011] Preparation of chenodeoxycholic acid (5): 12- oxo take chenodeoxycholic acid, ethylene glycol and solid sodium hydroxide, hydrated corpus, refluxed for 2 hours, gradually warming evaporated partially hydrated corpus, continue to heat up to 150 ° C, continued to reflux, cooled to room temperature, poured into water, adjusting the PH with hydrochloric acid, the white precipitate was filtered, washed with water to give crude chenodeoxycholic acid, recrystallization
Azusa mouth
M ο not mesh
[0012] Step (1): cholic acid to alcohol weight to volume ratio of 1: 2 ~ 5, the volume ratio of concentrated hydrochloric acid to alcohol is 10 wide: 100, 5-5 hours reflux time was 0.5.
[0013] Step (2): cholate: acetic anhydride molar ratio = 1: 2 ~ 5, the reaction temperature, time; Tl2O hours; cholate was added per mole of N, N- dimethylpyridine wide 5g.
[0014] Step (; 3): The hypochlorite is sodium hypochlorite or calcium hypochlorite; bromide is sodium bromide, potassium bromide and the like.
[0015] Step (4): recrystallization from a solvent with an alcohol such: as methanol or ethanol.
[0016] Step (5): recrystallization solvent is a water-miscible organic solvents, such as: methanol, ethanol, acetonitrile, acetone and the like.
[0017] Step (cholate was used ¾ of methyl cholate, ethyl cholate, cholic acid or cholic acid propyl ester; Step (3) used as 3 [alpha], 7 α – diacetyl -12 α – hydroxy cholate as 3 α, 7 α – diacetyl -12 α – hydroxy methyl cholate, 3 α, 7α- diacetyl -12 α – hydroxy bile acid ethyl ester, 3 α, 7α- diacetyl yl -12 α – hydroxy acid or ester 3α, 7α- diacetyl -12 α – hydroxy acid ester.
[0018] The invention has the advantages: in cholic acid as raw materials, and the choice of bromide tetrabutylammonium bromide as catalyst, in a non-polluting oxidizing agent is hypochlorite, Intermediate 3 α, 7 α – Diacetyl _12_ oxo chenodeoxycholic acid ester yield of 90% or more, thereby improving the yield of the final product of chenodeoxycholic acid, 99% yield, low cost and no pollution, very convenient for industrial production. detailed description
[0019] The present invention will be better described, for example is as follows:
(1) Preparation of methyl cholate: bile acid 5. lg, 15ml of anhydrous methanol, heating the whole solution. Refluxed for 3 hours, was added 0. 4ml concentrated hydrochloric acid, the reaction was stopped after 30min, after slow cooling, and filtered to give methyl cholate 5. 05g, 95% yield. 1HNMR (CDCl3):. Δ 0. 70 (s, 3H, 18- CH3), 0.90 (s, 3H, 19- CH3), 0.98 (d, 3H, 21-CH3), 3 50 (m, 1H, 3 β -H), 3. 67 (s, 3H, OCH3), 3. 87 (s, 1H, 7 β -H), 3. 99 (s, 1H, 12 β -H).
[0020] (2) Preparation of 3α, 7α- methyl cholate diacetyl-hydroxy -12α-: bile acid methyl ester 4. 71g (Ilmmol) IOOml was placed in a flask, was added methylene chloride 30ml, triethylamine 3 . Chiu 1, stirred at room temperature, was added dropwise acetic anhydride 2. 7ml (28. 6mmo 1), followed by addition of 20mg N, N- dimethylpyridine catalyst, the reaction time of 7 hours, methylene chloride was distilled off, into the water, filtered to give a white solid. The crude product was recrystallized from methanol to give white crystals 4. 05g, yield 67.2%. 1H NMR (CDCl3) δ: 4.90 (m, 1H, 7 β -H), 4. 59 (s, 1H, 3 β -H), 4 01 (s, 1H, 12 β -H), 3 67.. (s, 3Η, OCH3), 2. 08 (s, 3Η, CH3CO), 2. 02 (s, 3Η, CH3CO), 0. 98 (s, 3Η, 21-CH3), 0. 93 (s, 3Η , 19-CH3), 0.69 (s, 3Η, 18_CH3).
[0021] (3) 3α, 7α – 12-oxo-diacetyl chenodeoxycholic acid methyl ester prepared: Take 3 α, 7 α – diacetyl -12 α- hydroxy methyl cholate 1.917 g ( 3. 79mmol) was placed in a 50ml round bottom flask, 12ml of ethyl acetate was added, 5ml methanol, stirring at room temperature, was added 0. 25g 0. Ig of potassium bromide and tetrabutylammonium bromide. Was added dropwise a solution of acetic acid and 6g of sodium hypochlorite (7%) (5.62mmol), for 10 hours. Methanol was distilled off under reduced pressure and ethyl acetate, filtered, washed with water, and dried to give crude 1.915g, 1.75g as a white solid after recrystallization from methanol, yield 91.2%. 1H bandit R (CDCl3) δ:.. 4. 99 (d, 1H, 7 β-H), 4 60 (m, 1H, 3 β-H), 3 67 (s, 3H, OCH3), 2. 07 (s, 6H, CH3CO), 1. 03 (s, 6H, I8-CH3 and 19-CH3), 0. 82 (d, 3H, 21-CH3) ο
[0022] (4) 12- oxo chenodeoxycholic acid Preparation: Take 3 α, 7 α – diacetyl _12_ oxo chenodeoxycholic acid methyl ester 1. 56g, was dissolved in 30ml 95% ethanol was added 3. 2g of sodium hydroxide, heated at reflux for 5 hours. PH adjusted with hydrochloric acid value of the reaction system, most of the ethanol was distilled off, filtered, washed with water, and dried to give a white solid 12- oxo-1 crude chenodeoxycholic acid, recrystallized from methanol ^ g 1. 25g, yield rate of 96%. Tun bandit R (CDCl3) δ:. 3.96 (d, 1H, 7 β-H), 3 47 (m, 1H, 3 β-H), 1.03 (s, 3H, 19_CH3), 0.89 (s, 3H, 18_CH3 ), 0 · 70 (d, 3 H, 21_CH3).
[0023] Preparation of chenodeoxycholic acid (5): 12- oxo take chenodeoxycholic acid 0. 9g, 15ml ethylene glycol was added solid sodium hydroxide and 1. 5g, 15ml hydrated corpus (80%) , 120 ° C reflux for 2 hours, change return device is a distillation apparatus, was gradually warmed evaporated amount hydrated corpus, continue to heat up to 150 ° C, continuing reflux for 4h, cooled to room temperature, poured into water, adjusted with HCl of PH3, white precipitated, was filtered cake was washed with water, and dried to give crude chenodeoxycholic acid 0. 92g, recrystallized from methanol to give 0. 86g, 99 (s, 1H, C00H).
Paper
https://pubs.acs.org/doi/abs/10.1021/ja01168a045
Reactions of 2-Arylcyclohexanones. IV. Michael Addition of Malonic Ester to 2-Phenyl-Δ2-cyclohexenone
The Preparation of Chenodeoxycholic Acid and Its Glycine and Taurine Conjugates.Hofmann, Alan F.


Preparation of Chenodeoxycholic Acid
Further studies on the synthesis of thienamycin: a facile and stereoselective synthesis of a bicyclic .beta.-keto ester by 1,3-dipolar cycloaddition




Chenodeoxycholic acid (3α, 7α- -5β- dihydroxy-cholestane acid) Chenodeoxycholic Acid (referred to as CDCA), a medicine for treating gallstones. 1848 first discovered in goose bile, 1924, known as the CDCA. By reducing cholesterol absorption, synthesis, the bile cholesterol decreased, thereby suppressing cholesterol gallstone formation and promote dissolution, and can reduce cholesterol saturation.
Chenodeoxycholic acid addition pharmaceutically itself, but also as the preparation of ursodeoxycholic acid (3α, 7β- -5β- dihydroxy bile acid, abbreviated UDCA) starting material. Ursodeoxycholic acid is the main active ingredient contained bile valuable medicine, in clinical treatment of various gastrointestinal diseases and bladder diseases. But the limited sources of bear bile medicine, and contrary to the principles of animal protection. So, dwindling source of natural bear bile, can not meet the medical requirements. Therefore, the preparation of chenodeoxycholic acid is also of great significance for further preparation of ursodeoxycholic acid.
CDCA bile extracted mainly from poultry or animal bile extraction methods in the past as it involves toxic chemicals (animal biological pharmacy, 1981, People’s Medical Publishing House, P259; pharmaceutical industry, 1987,18 (2): 75-76; ) or unsafe to use a large amount of organic solvent (Chinese Journal of biochemical Pharmaceutics, 1996,17 (1): 17; application technology, 1998,4: 9-10; US Patent, 3,965,131; US Patent, 4,331,607; USPatent, 4,163,017), can not be meet the requirements of modern industry, CDCA and low purity prepared costly.
PATENT
https://patents.google.com/patent/WO2007069814A1/en
Chenodeoxycholic acid is generally contained in bile of cow, swine, bear, or poultry such as chicken or goose, as well as in bile of human. Chenodeoxycholic acid is used as starting material for the preparation of ursodeoxycholic acid which is effective to alleviate biliary system diseases, hyperlipidemia, cholelithiasis, and chronic liver diseases, and a typical process for preparing ursodeoxycholic acid known in the art is as follows.
A typical process for preparing chenodeoxycholic acid comprises the steps of: esterifying cholic acid (3α,7α,12θ!-trihydroxy cholic acid) with methyl; protecting the hydroxyl group of 3α and Ia position by acetylating them with anhydrous acetic acid; oxidizing the hydroxyl group of 12α position to carbonyl group by using chromic acid, and then removing the carbonyl group by Wolff-kichner reduction reaction; hydrolyzing and deprotecting the obtained product to yield chenodeoxycholic acid. The above process requires the reaction to be maintained at a high temperature of more than 200 °C , and the supply of raw material may be interrupted by bovine spongiform encephalopathy, etc. Bile ,of poultry contains chenodeoxycholic acid, lithocholic acid, and a small amount of cholic acid. Thus, the process for separating chenodeoxycholic acid from poultry is well known in the art, but is not economically reasonable due to the supply decrease of raw material and low yield [see, Windhaus et al, I Physiol. Chem., 140, 177-185 (1924)].
US Patent No. 4,186,143 disclosed a process for purely separating and purifying chenodeoxycholic acid from chenodeoxycholic acid mixture derived from natural swine bile. This process comprises the major steps of: pre-treatment to remove 3ohydroxy-6- oxo-5/3-cholic acid by saponification of bile; esterification of bile acid; acetylation of bile acid ester; removal of intermediate product by using non-polar organic solvent; crystallization of acetylated ester of formula I; deprotection; and production of the compound of formula I by using crystallization in organic solvent. However, this patent does not describe HPLC content for acetylated ester of formula I, and the purity of the final product is very low since the specific rotatory power is [ofo25 +13.8° (c=l, CHCl3), and the melting point is 119-121 °C [STD: [α]D 25 +15.2°(c=l, CHCl3), melting point 127- 129 “C]. Also, the crystallization for purifying the final product requires a very long time (i.e., 16-48 hours), and the entire process is complex as eight (8) steps. Thus, when purifying the compound of formula I by using the above process, the yield of the final product becomes low, and the reaction time is as long as 12 days. Therefore, the process is not economically reasonable.
Step 6: Deprotection and crystallization of chenodeoxycholic acid
To 220ml of water were added 24.5g of chenodeoxycholic acid-diacetate-ester and 29.5g of sodium hydroxide, and then the solution was stirred with reflux for 4 hours. To the solution was added 370ml of water. The solution’s pH is adjusted to 2.0-3.0 by using 59ml of hydrochloric acid. Then, the solution was stirred at 35-45 °C for 1 hour, and then filtered. The filtered material was washed with 24.5ml of water and dried in vacuum at 70 °C to obtain 19.5g of pure chenodeoxycholic acid, m.p.: 160-161 °C, [α]o25 +13.0°(c=l, CHCl3).
Step 8: Production of the compound of formula I
The reaction solution was extracted by using ethyl acetate, and aqueous layer was discarded therefrom. Ethyl acetate layer in the solution was washed with 6% saline, and the solution was distilled to about 90ml. This solution was cooled, kept cool for one day after adding 90ml of hexane, and filtered. Thus filtered material was washed with 20ml of hexane, and dried in vacuum at 60 °C to produce 12.7g of chenodeoxycholic acid. m.p. 142-1450C; [α]D 25 +13.0°(c=l, CHCl3). INDUSTRIAL APPLICABILITY The present invention can purify chenodeoxycholic acid of formula I from swine bile solid in high yield and purity. Also, the present invention is suitable for industrial purification by reducing the purification time.
PATENT
https://patents.google.com/patent/CN102060902A/en
chenodeoxycholic acid (3 α, 7 α – dihydroxy _5 β – cholestane-24-oic acid) Chenodeoxycholic Ac id (referred to as CDCA), clinically used to correct dissolving cholesterol calculi and bile saturation drugs, the main function is to reduce the cholesterol in the bile saturation, large doses can inhibit the synthesis of cholesterol CDCA and increasing bile gallstone patients cholesterol level in a non-saturated, thereby preventing the formation of cholesterol gallstones of cholesterol and promote stone dissolve and fall off. It also has significant anti-asthmatic, anti-inflammatory, antitussive and expectorant effects.
[0003] Synthesis of chenodeoxycholic acid or ursodeoxycholic acid (3 α, 7β_ -5β_ dihydroxy-cholestane-24-oic acid, ursodeoxycholic Acid, referred UDCA), a key intermediate. Ursodeoxycholic acid is the main active ingredient of precious Chinese medicine bear bile, used in a variety of clinical hepatobiliary disease and dyspepsia. Currently we bear bile resources are scarce, mainly used synthetic chemical ursodeoxycholic acid as a clinical treatment. Therefore, the preparation of chenodeoxycholic acid is also important for the preparation of ursodeoxycholic acid.
[0004] CDCA mainly come from poultry or livestock bile extraction. Traditional extraction process complicated operation, low yield, (pharmaceutical industry, 1987,18 (9), 416; Chinese Journal of Biochemical Pharmaceutics, 1996,17 (1), 17; Applied Technology, 1998, (4), 9; CN1850846A ) can not meet the needs of modern industry. Chemical synthesis of chenodeoxycholic acid have also been reported (Japanese Journal of Chemistry 1955,76 (3), 297 -J Org Chem 1982,47 (2): 2331; Journal of Biochemical Pharmaceutics 1987,1,6 -, Tap Chi Duoc ^ oc2004 , 44 (1), 11; CN1869043A), but lower yield widespread pollution major problem, especially in the oxidation reaction is often used to expensive, and polluting agents.Therefore, to reduce pollution, reduce environmental hazards, streamline operations, improve yield, reduce costs, important for the synthesis of chenodeoxycholic acid.
Preparation of chenodeoxycholic acid.
[0007]

Cholic acid esters prepared by (1) Weigh 50 g of cholic acid, dissolved in 150 ml of anhydrous methanol was added 5 ml of concentrated hydrochloric acid was refluxed for 30 minutes, cooled slowly into the freezer, the available capacity methyl cholate It was 95%.
(2) hydroxy -12α- diacetyl – Preparation of methyl cholate methyl cholate weighed 50 g, was dissolved in 100 ml of pyridine was purified, dissolved completely, 100 ml of acetic anhydride was stirred at room temperature for 3 to 4 hours, poured into 500 ml of water, a white precipitate in the refrigerator, filtered the next day, diacetyl -12α- available hydroxy – methyl cholate, yield 40%.
(3) 3α, 7α–diacetoxy-12-oxo – Preparation of methyl cholanic acid prepared above was weighed 25 g of crude product, dissolved in 250 ml of acetone, filtered to remove insolubles, the stirring conditions , the Jones reagent was slowly added, at room temperature for 30 minutes, filtered, water was added to the filtrate precipitated white precipitate was filtered available 3α, 7α–diacetoxy-12-oxo – methyl-cholanic acid. The yield was 100%.
(4) 12- oxo – Preparation of chenodeoxycholic acid in ethanol 10% – sodium hydroxide solution and saponified for 1 hour at room temperature, the solution was acidified, poured into water to give 12- oxo – chenodeoxycholic acid , 100% yield.Recrystallized in absolute ethanol.
Preparation of chenodeoxycholic acid (5) was weighed 12- oxo – chenodeoxycholic acid, 20 grams, was added 300 ml of ethylene glycol and 30 g of solid sodium hydroxide and 300 ml of hydrazine hydrate (85%), 100 ℃ refluxed for 2 hours, warming gradually raised to 130. ℃, generated by hydrazine hydrate was distilled off, continue to heat up to 185 ~ 190 ℃, continued reflux for 4 hours, cooled to a lower temperature, poured into water and heat, PH adjusted with hydrochloric acid (20%) 3, a white precipitate was filtered cake was washed with water to give chenodeoxycholic acid.
(6) Purification of chenodeoxycholic acid obtained weighed amount of chenodeoxycholic acid, dissolved with a small amount of ethanol, was impregnated on a silica gel column petroleum ether, liquid flow linear velocity by column chromatography 1 ~ 5cm / control points, with petroleum ether: acetone = 2, begins to elute, detected by TLC chromatography therebetween, Junichi appearance of spots to be chenodeoxycholic acid appears to start collecting the eluate until no Chenodeoxy acid spots, distillation under reduced pressure and dried to give pure higher chenodeoxycholic acid.
PATENTS
References
- ^ Jump up to:a b c Russell DW (2003). “The enzymes, regulation, and genetics of bile acid synthesis”. Annu. Rev. Biochem. 72: 137–74. doi:10.1146/annurev.biochem.72.121801.161712. PMID 12543708.
- Jump up^ Carey MC (December 1975). “Editorial: Cheno and urso: what the goose and the bear have in common”. N. Engl. J. Med. 293 (24): 1255–7. doi:10.1056/NEJM197512112932412. PMID 1186807.
- Jump up^ Parks DJ, Blanchard SG, Bledsoe RK, et al. (May 1999). “Bile acids: natural ligands for an orphan nuclear receptor”. Science. 284 (5418): 1365–8. doi:10.1126/science.284.5418.1365. PMID 10334993.
- Jump up^ Thistle JL, Hofmann AF (September 1973). “Efficacy and specificity of chenodeoxycholic acid therapy for dissolving gallstones”. N. Engl. J. Med. 289 (13): 655–9. doi:10.1056/NEJM197309272891303. PMID 4580472.
- Jump up^ Berginer VM, Salen G, Shefer S (December 1984). “Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid”. N. Engl. J. Med. 311 (26): 1649–52. doi:10.1056/NEJM198412273112601. PMID 6504105.
- Jump up^ Giaconda. “Press release”. Retrieved 5 April 2014.
- Jump up^ Bazzoli F, Malavolti M, Petronelli A, Barbara L, Roda E (1983). “Treatment of constipation with chenodeoxycholic acid”. J. Int. Med. Res. 11 (2): 120–3. PMID 6852359.
- Jump up^ Rao AS, Wong BS, Camilleri M, et al. (November 2010). “Chenodeoxycholate in females with irritable bowel syndrome-constipation: a pharmacodynamic and pharmacogenetic analysis”. Gastroenterology. 139 (5): 1549–58, 1558.e1. doi:10.1053/j.gastro.2010.07.052. PMC 3189402
 . PMID 20691689.
. PMID 20691689. - Jump up^ Ki Soo Kim, Hong-Seok Kim Molecular Tweezer Based on Chenodeoxycholic Acid:Synthesis, Anion Binding Properties. Bulletin of the Korean Society 1411-1413 2004 Article ArchivedSeptember 27, 2007, at the Wayback Machine.

|
|
 |
|
| Names | |
|---|---|
| IUPAC names
chenodiol
OR 3α,7α-dihydroxy-5β-cholanic acid OR 5β-cholanic acid-3α,7α-diol OR (R)-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoic acid |
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| DrugBank | |
| ECHA InfoCard | 100.006.803 |
| EC Number | 207-481-8 |
| KEGG | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C24H40O4 | |
| Molar mass | 392.57 g/mol |
| Melting point | 165 to 167 °C (329 to 333 °F; 438 to 440 K) |
| Pharmacology | |
| A05AA01 (WHO) | |
| License data | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
 verify (what is verify (what is   ?) ?) |
|
| Infobox references | |
////////////////////Chenodeoxycholic acid, ケノデオキシコール酸 , orphan designation
[H][C@@]1(CC[C@@]2([H])[C@]3([H])[C@H](O)C[C@]4([H])C[C@H](O)CC[C@]4(C)[C@@]3([H])CC[C@]12C)[C@H](C)CCC(O)=O


































 30, Pd EnCat
30, Pd EnCat

















































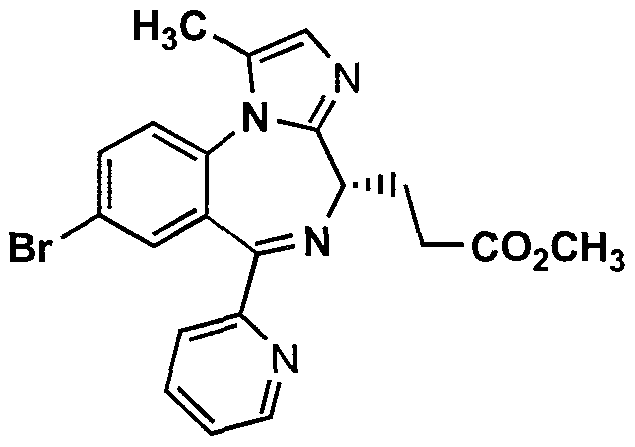


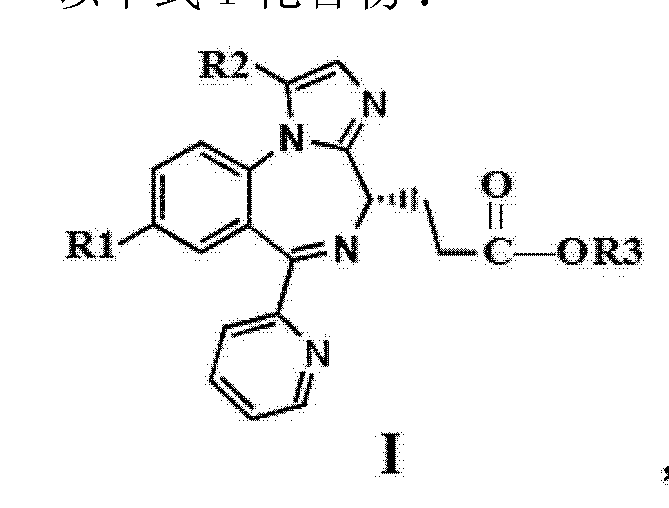




























































































 11
11
 0.10
0.10 to +0.10
to +0.10







 3) The methylation of 3-(2-oxo-1,2-dihydroquinolin-4-yl)-(R,S)-alanine (IX) with SOCl2 and methanol yields the corresponding methyl ester (X), which is submitted to optical resolution with D-(-)-mandelic acid, affording adducts (XII) and (XIII). The hydrolytic treatment of (XII) and (XIII) with HCl and propylene oxide finally yields isomers (VIIr) and (VIIs), already obtained. Racemic OPC-12759 can also be resolved into its optical isomers by treatment with brucine and fractionated crystallization.
3) The methylation of 3-(2-oxo-1,2-dihydroquinolin-4-yl)-(R,S)-alanine (IX) with SOCl2 and methanol yields the corresponding methyl ester (X), which is submitted to optical resolution with D-(-)-mandelic acid, affording adducts (XII) and (XIII). The hydrolytic treatment of (XII) and (XIII) with HCl and propylene oxide finally yields isomers (VIIr) and (VIIs), already obtained. Racemic OPC-12759 can also be resolved into its optical isomers by treatment with brucine and fractionated crystallization.
























