NEW DRUG APPROVALS FAST APPROACHING 20 LAKH VIEWS MARK…….https://newdrugapprovals.org/
Filed under: BLOGS Tagged: blogs








NEW DRUG APPROVALS FAST APPROACHING 20 LAKH VIEWS MARK…….https://newdrugapprovals.org/
![]()

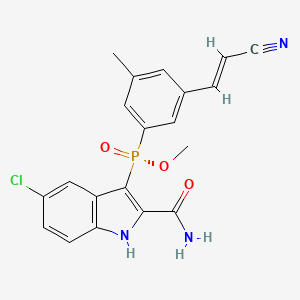
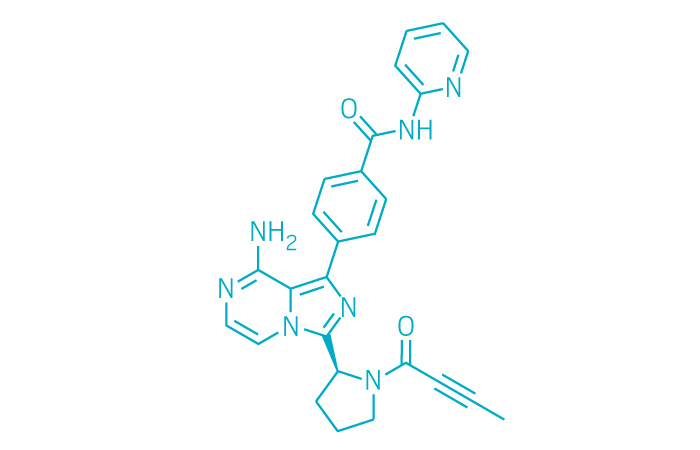
WO-2018001353, APREMILAST, NEW PATENT, ZHEJIANG HUAHAI PHARMACEUTICAL CO., LTD
(WO2018001353) METHOD FOR PREPARING APREMILAST
ZHEJIANG HUAHAI PHARMACEUTICAL CO., LTD
DU, Xiaoqiu; (CN).
ZHOU, Lianchao; (CN).
LIU, Jiegen; (CN)
EN)Method one: (S) -1- (3-ethoxy-4-methoxyphenyl) -2- (methylsulfonyl) ethylamine N-acetyl-L-leucine salt of formula II is reacted with 3-acetylaminophthalic anhydride of formula III in an aprotic solvent to produce the compound of formula I; method two: (S) -1- (3-ethoxy-4-methoxyphenyl) -2- (methylsulfonyl) ethylamine N-acetyl-L- leucine salt of formula II is reacted with 3-acetylaminophthalic anhydride of formula III in an organic solvent in the presence of an organic alkaline or an alkali metal hydride to produce the compound of formula I. The method for preparing apremilast requires inexpensive raw materials and reagents , is suitable for industrialized production, and has great economic effects.
////////////WO 2018001353, APREMILAST, NEW PATENT, ZHEJIANG HUAHAI PHARMACEUTICAL CO., LTD
Trilaciclib dihydrochloride
1977495-97-8
In phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin
![]()
PATENT, WO 2014144326, Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
| Norman Sharpless | |
|---|---|
 |
|
| Born | Norman Edward Sharpless September 20, 1966 Greensboro, North Carolina |
| Nationality | American |
| Other names | Ned Sharpless |
| Occupation | Director, Lineberger Comprehensive Cancer Center Founder, G1 Therapeutics ($GTHX) |
| Notable work | Wellcome Distinguished Professor, American Society of Clinical Investigation Member, Association of American Cancer Institute board of directors, |
NCI Director Dr. Norman E. Sharpless, Credit: National Institutes of Health
Norman E. “Ned” Sharpless, M.D., was officially sworn in as the 15th director of the National Cancer Institute (NCI) on October 17, 2017. Prior to his appointment, Dr. Sharpless served as the director of the University of North Carolina (UNC) Lineberger Comprehensive Cancer Center, a position he held since January 2014.
Dr. Sharpless was a Morehead Scholar at UNC–Chapel Hill and received his undergraduate degree in mathematics. He went on to pursue his medical degree from the UNC School of Medicine, graduating with honors and distinction in 1993. He then completed his internal medicine residency at the Massachusetts General Hospital and a hematology/oncology fellowship at Dana-Farber/Partners Cancer Care, both of Harvard Medical School in Boston.
After 2 years on the faculty at Harvard Medical School, he joined the faculty of the UNC School of Medicine in the Departments of Medicine and Genetics in 2002. He became the Wellcome Professor of Cancer Research at UNC in 2012.
Dr. Sharpless is a member of the Association of American Physicians as well as the American Society for Clinical Investigation (ASCI), the nation’s oldest honor society for physician–scientists, and served on the ASCI council from 2011 to 2014. Dr. Sharpless was an associate editor of Aging Cell and deputy editor of the Journal of Clinical Investigation. He has authored more than 150 original scientific papers, reviews, and book chapters, and is an inventor on 10 patents. He cofounded two clinical-stage biotechnology companies: G1 Therapeutics and HealthSpan Diagnostics.
In addition to serving as director of NCI, Dr. Sharpless continues his research in understanding the biology of the aging process that promotes the conversion of normal self-renewing cells into dysfunctional cancer cells. Dr. Sharpless has made seminal contributions to the understanding of the relationship between aging and cancer, and in the preclinical development of novel therapeutics for melanoma, lung cancer, and breast cancer.
| Record ID | Title | Status | Phase |
|---|---|---|---|
| NCT03041311 | Carboplatin, Etoposide, and Atezolizumab With or Without Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Extensive Stage Small Cell Lung Cancer (SCLC) | Recruiting | 2 |
| NCT02978716 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Gemcitabineand Carboplatin in Metastatic Triple Negative Breast Cancer (mTNBC) | Recruiting | 2 |
| NCT02514447 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Patients With Previously Treated Extensive Stage SCLC Receiving Topotecan Chemotherapy | Recruiting | 2 |
| NCT02499770 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Etoposide and Carboplatin in Extensive Stage Small Cell Lung Cancer (SCLC) | Active, not recruiting | 2 |
WO 2016040858


Trilaciclib (G1T28)
Trilaciclib is a potential first-in-class short-acting CDK4/6 inhibitor in development to preserve hematopoietic stem cells and enhance immune system function during chemotherapy. Trilaciclib is administered intravenously prior to chemotherapy and has the potential to significantly improve treatment outcomes.
G1 is currently evaluating trilaciclib in four Phase 2 clinical trials: three studies in patients with small-cell lung cancer (SCLC), and one study in patients with triple-negative breast cancer (TNBC). Preliminary data from the SCLC trials were presented at the American Society of Clinical Oncology 2017 Annual Meeting and at the 2016 World Conference on Lung Cancer.
Data from a Phase 1 trial in healthy volunteers were presented at the American Society of Clinical Oncology 2015 Annual Meeting and published in Science Translational Medicine. Trilacicilib has been extensively studied in animals; these preclinical data have been presented at several scientific meetings and published in Molecular Cancer Therapeutics, Science Translational Medicine, and Cancer Discovery.
Trilaciclib is a small molecule, competitive inhibitor of cyclin dependent kinases 4 and 6 (CDK4/6), with potential antineoplastic and chemoprotective activities. Upon intravenous administration, trilaciclib binds to and inhibits the activity of CDK4/6, thereby blocking the phosphorylation of the retinoblastoma protein (Rb) in early G1. This prevents G1/S phase transition, causes cell cycle arrest in the G1 phase, induces apoptosis, and inhibits the proliferation of CDK4/6-overexpressing tumor cells. In patients with CDK4/6-independent tumor cells, G1T28 may protect against multi-lineage chemotherapy-induced myelosuppression (CIM) by transiently and reversibly inducing G1 cell cycle arrest in hematopoietic stem and progenitor cells (HSPCs) and preventing transition to the S phase. This protects all hematopoietic lineages, including red blood cells, platelets, neutrophils and lymphocytes, from the DNA-damaging effects of certain chemotherapeutics and preserves the function of the bone marrow and the immune system. CDKs are serine/threonine kinases involved in the regulation of the cell cycle and may be overexpressed in certain cancer cell types. HSPCs are dependent upon CDK4/6 for proliferation.
Trilaciclib (G1T28) is a CDK4/6 inhibitor in phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin. Also, phase II trials are ongoing in newly diagnosed, treatment-naive small-cell lung cancer patients, in combination with carboplatin, etoposide, and atezolizumab and phase I trials in previously treated small-cell lung cancer patients, in combination with topotecan.
U.S. Patent Nos. 8,822,683; 8,598,197; 8,598,186, 8,691,830, 8,829,102, 8,822,683, 9, 102,682, 9,499,564, 9,481,591, and 9,260,442, filed by Tavares and Strum and assigned to Gl Therapeutics describe a class of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amine cyclin dependent kinase inhibitors including those of the formula with variables as defined therein):

U.S. Patent Nos. 9,464,092, 9,487,530, and 9,527,857 which are also assigned to Gl Therapeutics describe the use of the above pyrimidine-based agents in the treatment of cancer.
These patents provide a general synthesis of the compounds that is based on a coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. Such coupling reactions are sometimes referred to as Buchwald coupling (see WO Ί56 paragraph 127; reference WO 2010/020675). The lactam of the fused chloropyrimidine, for example, a 2-chloro-spirocyclo-pyrrolo[2,3-d]pyrimidine-one such as Intermediate K as shown below can be prepared by dehydration of the corresponding carboxylic acid. The reported process to prepare intermediate IK requires seven steps.

(Intermediate IK; page 60, paragraph 215 of WO Ί56)
WO 2013/148748 (U.S. S.N. 61/617,657) entitled “Lactam Kinase Inhibitors” filed by Tavares, and also assigned to Gl Therapeutics likewise describes the synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines via the coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine.
WO 2013/163239 (U.S. S.N. 61/638,491) “Synthesis of Lactams” describes a method for the synthesis of this class of compounds with the variation that in the lactam preparation step, a carboxylic acid can be cyclized with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. The purported improvement is that cyclization can occur without losing the protecting group on the amine before cyclization. The typical leaving group is “tBOC” (t-butoxycarbonyl). The application teaches (page 2 of WO 2013/163239) that the strong acid is, for example, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride or mixed anhydrides. An additional step may be necessary to take off the N-protecting group. The dehydrating agent can be a carbodiimide-based compound such as DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-dimethylaminopropyl)carbodiimide, or DIC (Ν,Ν-diisopropylcarbodiimide). DCC and DIC are in the same class of reagents-carbodiimides. DIC is sometimes considered better because it is a liquid at room temperature, which facilitates reactions.
WO 2015/061407 filed by Tavares and licensed to Gl Therapeutics also describes the synthesis of these compounds via the coupling of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. WO ‘407 focuses on the lactam production step and in particular describes that the fused lactams of these compounds can be prepared by treating the carboxylic acid with an acid and a dehydrating agent in a manner that a leaving group on the amine is not removed during the amide-forming ring closing step.
Other publications that describe compounds of this general class include the following. WO 2014/144326 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of normal cells during chemotherapy using pyrimidine based CDK4/6 inhibitors. WO 2014/144596 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of hematopoietic stem and progenitor cells against ionizing radiation using pyrimidine based CDK4/6 inhibitors. WO 2014/144847 filed by Strum et al. and assigned to Gl Therapeutics describes HSPC-sparing treatments of abnormal cellular proliferation using pyrimidine based CDK4/6 inhibitors. WO2014/144740 filed by Strum et al. and assigned to Gl Therapeutics describes highly active anti -neoplastic and anti-proliferative pyrimidine based CDK 4/6 inhibitors. WO 2015/161285 filed by Strum et al. and assigned to Gl Therapeutics describes tricyclic pyrimidine based CDK inhibitors for use in radioprotection. WO 2015/161287 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for the protection of cells during chemotherapy. WO 2015/161283 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use in HSPC-sparing treatments of RB-positive abnormal cellular proliferation. WO 2015/161288 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use as anti -neoplastic and anti-proliferative agents. WO 2016/040858 filed by Strum et al. and assigned to Gl Therapeutics describes the use of combinations of pyrimidine based CDK4/6 inhibitors with other anti-neoplastic agents. WO 2016/040848 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for treating certain Rb-negative cancers with CDK4/6 inhibitors and topoisomerase inhibitors.
Other biologically active fused spirolactams and their syntheses are described, for example, in the following publications. Griffith, D. A., et al. (2013). “Spirolactam-Based Acetyl-CoA Carboxylase Inhibitors: Toward Improved Metabolic Stability of a Chromanone Lead Structure.” Journal of Medicinal Chemistry 56(17): 7110-7119, describes metabolically stable spirolactams wherein the lactam resides on the fused ring for the inhibition of acetyl-CoA carboxylase. WO 2013/169574 filed by Bell et al. describes aliphatic spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2007/061677 filed by Bell et al. describes aryl spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2008/073251 filed by Bell et al. describes constrained spirolactam compounds wherein the lactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031606 filed by Bell et al. describes carboxamide spirolactam compounds wherein the spirolactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031610, WO 2006/031491, and WO 2006/029153 filed by Bell et al. describe anilide spirolactam compounds wherein the spirolactam resides on the spiro ring; WO 2008/109464 filed by Bhunai et al. describes spirolactam compounds wherein the lactam resides on the spiro ring which is optionally further fused.
Given the therapeutic activity of selected N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines, it would be useful to have additional methods for their preparation. It would also be useful to have new intermediates that can be used to prepare this class of compounds.
PATENT
WO 2014144596
PATENT
Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
EXAMPLES
Intermediates B, E, K, L, 1A, IF and 1CA were synthesized according to US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C..
The patents WO 2013/148748 entitled Lactam Kinase Inhibitors to Tavares, F.X., WO 2013/163239 entitled Synthesis of Lactams to Tavares, F.X., and US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C. are incorporated by reference herein in their entirety. Example 1
Synthesis of tert-butyl N- [2- [(5-bromo-2-chloro-pyrimidin-4yl)amino] ethyl] carbamate, Compound 1
To a solution of 5-bromo-2,4-dichloropyrimidine (3.2 g, 0.0135 mol) in ethanol (80 mL) was added Hunig’s base (3.0 mL) followed by the addition of a solution of N-(tert- butoxycarbonyl)-l,2-diaminoethane (2.5 g, 0.0156 mole) in ethanol (20 mL). The contents were stirred overnight for 20 hrs. The solvent was evaporated under vacuum. Ethyl acetate (200 mL) and water (100 mL) were added and the layers separated. The organic layer was dried with magnesium sulfate and then concentrated under vacuum. Column chromatography on silica gel using hexane/ethyl acetate (0- 60%) afforded tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4- yl)amino]ethyl]carbamate. 1HNMR (d6-DMSO) δ ppm 8.21 (s, 1H), 7.62 (brs, 1H), 7.27 (brs, 1H), 3.39 (m, 2H), 3.12 (m, 2H), 1.34 (s, 9H). LCMS (ESI) 351 (M + H).
Example 2
Synthesis of tert-butyl N-[2-[[2-chloro-5-(3,3-diethoxyprop-l-ynyl)pyrimidin-4- yl] amino] ethyl] carbamate, Compound 2
To tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4-yl)amino]ethyl]carbamate (1.265 g, 6 mmol) in THF (10 mL) was added the acetal (0.778 mL, 5.43 mmol), Pd(dppf)CH2Cl2 (148 g), and triethylamine (0.757 mL, 5.43 mmol). The contents were degassed and then purged with nitrogen. To this was then added Cul (29 mg). The reaction mixture was heated at reflux for 48 hrs. After cooling, the contents were filtered over CELITE and concentrated. Column chromatography of the resulting residue using hexane/ethyl acetate (0- 30%) afforded tert-butyl N- [2- [ [2-chloro-5 -(3 ,3 -diethoxyprop- 1 -ynyl)pyrimidin-4-yl]amino] ethyl] carbamate. 1HNMR (d6-DMSO) δ ppm 8.18 (s, 1H), 7.63 (brs, 1H), 7.40 (brs, 1H), 5.55 (s, 1H), 3.70 (m, 2H), 3.60 (m, 2H), 3.42 (m, 2H), 3.15 (m, 2H), 1.19 – 1.16 (m, 15H). LCMS (ESI) 399 (M + H).
and concentrated. Column chromatography of the resulting residue using hexane/ethyl acetate (0- 30%) afforded tert-butyl N- [2- [ [2-chloro-5 -(3 ,3 -diethoxyprop- 1 -ynyl)pyrimidin-4-yl]amino] ethyl] carbamate. 1HNMR (d6-DMSO) δ ppm 8.18 (s, 1H), 7.63 (brs, 1H), 7.40 (brs, 1H), 5.55 (s, 1H), 3.70 (m, 2H), 3.60 (m, 2H), 3.42 (m, 2H), 3.15 (m, 2H), 1.19 – 1.16 (m, 15H). LCMS (ESI) 399 (M + H).
Example 3
Synthesis of tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7- yl] ethyl] carbamate, Compound 3
To a solution of the coupled product (2.1 g, 0.00526 mole) in THF (30 mL) was added TBAF solid (7.0 g). The contents were heated to and maintained at 65 degrees for 2 hrs. Concentration followed by column chromatography using ethyl acetate/hexane (0-50%) afforded tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7-yl]ethyl]carbamate as a pale brown liquid (1.1 g). 1FiNMR (d6-DMSO) δ ppm 8.88 (s, 1H), 6.95 (brs, 1H), 6.69 (s, 1H), 5.79 (s, 1H), 4.29 (m, 2H), 3.59 (m, 4H), 3.34 (m, 1H), 3.18 (m, 1H), 1.19 (m, 9H), 1.17 (m, 6H). LCMS (ESI) 399 (M + H).
Example 4
Synthesis of tert-buty\ N-[2-(2-chloro-6-formyl-pyrrolo [2,3-d] pyrimidin-7- yl)ethyl] carbamate, Compound 4
To the acetal (900 mg) from the preceeding step was added AcOH (8.0 mL) and water
(1.0 mL). The reaction was stirred at room temperature for 16 hrs. Cone, and column chromatography over silica gel using ethyl acetate/hexanes (0- 60%) afforded tert-butyl N-[2-(2- chloro-6-formyl-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl]carbamate as a foam (0.510 g). 1HNMR (d6-DMSO) δ ppm 9.98 (s, 1H), 9.18 (s, 1H), 7.66 (s, 1H), 6.80 (brs, 1H), 4.52 (m, 2H), 4.36 (m, 2H), 1.14 (s, 9H). LCMS (ESI) 325 (M + H).
Example 5
Synthesis of 7- [2-(teri-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3-d] pyrimidine-6- carboxylic acid, Compound 5
To the aldehyde (0.940 g) from the preceeding step in DMF (4 mL) was added oxone (1.95 g, 1.1 eq). The contents were stirred at room temp for 7 hrs. Silica gel column chromatography using hexane/ethyl acetate (0- 100%) afforded l-\2-(tert- butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g). 1HNMR (d6-DMSO) δ ppm 9.11 (s, 1H), 7.39 (s, 1H), 4.38 (m, 2H), 4.15 (m, 2H), 1.48 (m, 9H). LCMS (ESI) 341(M + H).
Example 6
Synthesis of methyl 7-[2-(teri-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate, Compound 6
To a solution of 2-chloro-7-propyl-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g, 0.00156 mole) from the preceeding step in toluene (3.5 mL) and MeOH (1 mL) was added TMS- diazomethane (1.2 mL). After stirring overnight at room temperature, the excess of TMS- diazomethane was quenched with acetic acid (3 mL) and the reaction was concentrated under vacuum. The residue was purified by silica gel column chromatography with hexane/ethyl acetate (0- 70%) to afford methyl 7-[2-(tert-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate as an off white solid (0.52 g). 1HNMR (d6-DMSO) δ ppm 9.10 (s, 1H), 7.45 (s, 1H), 6.81 (brs, 1H) 4.60 (m, 2H), 3.91 (s, 3H), 3.29 (m, 2H), 1.18 (m, 9H) LCMS (ESI) 355 (M + H).
Example 7
Synthesis of Chloro tricyclic amide, Compound 7
To methyl 7- [2-(tert-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3 -d]pyrimidine-6- carboxylate (0.50 g, 0.0014 mole) from the preceeding step in dichloromethane (2.0 mL) was added TFA (0.830 mL). The contents were stirred at room temperature for 1 hr. Concentration under vacuum afforded the crude amino ester which was suspended in toluene (5 mL) and Hunig’s base (0.5 mL). The contents were heated at reflux for 2 hrs. Concentration followed by silica gel column chromatography using hexane/ethyl acetate (0- 50%) afforded the desired chloro tricyclic amide (0.260 g). 1HNMR (d6-DMSO) δ ppm 9.08 (s, 1H), 8.48 (brs, 1H), 7.21 (s, 1H) 4.33 (m, 2H), 3.64 (m, 2H). LCMS (ESI) 223 (M + H).
Example 8
Synthesis of chloro-N-methyltricyclic amide, Compound 8
To a solution of the chloro tricycliclactam, Compound 7, (185 mg, 0.00083 mole) in DMF (2.0 mL) was added sodium hydride (55% dispersion in oil, 52 mg). After stirring for 15 mins, methyl iodide (62 μί, 1.2 eq). The contents were stirred at room temperature for 30 mins. After the addition of methanol (5 mL), sat NaHCOs was added followed by the addition of ethyl acetate. Separation of the organic layer followed by drying with magnesium sulfate and concentration under vacuum afforded the N-methylated amide in quantitative yield. 1FiNMR (d6-DMSO) δ ppm 9.05 (s, 1H), 7.17 (s, 1H) 4.38 (m, 2H), 3.80 (m, 2H), 3.05 (s, 3H). LCMS (ESI) 237 (M + H). Example 9
Synthesis of l-methyl-4-(6-nitro-3-pyridyl)piperazine, Compound 9
To 5-bromo-2-nitropyridine (4.93 g, 24.3 mmole) in DMF (20 mL) was added N- methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 degrees for 24 hrs. After addition of ethyl acetate (200 mL), water (100 mL) was added and the layers separated. Drying followed by concentration afforded the crude product which was purified by silica gel column chromatography using (0-10%) DCM/Methanol. 1HNMR (d6-DMSO) δ ppm 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
Example 10
Synthesis of 5-(4-methylpiperazin-l-yl)pyridin-2-amine, Compound 10
To l-methyl-4-(6-nitro-3-pyridyl)piperazine (3.4 g) in ethyl acetate (100 mL) and ethanol (100 mL) was added 10%> Pd/C (400 mg) and then the reaction was stirred under hydrogen (10 psi) overnight. After filtration through CELITE, the solvents were evaporated and the crude product was purified by silica gel column chromatography using DCM/ 7N ammonia in MeOH (0- 5%) to afford 5-(4-methylpiperazin-l-yl)pyridin-2-amine (2.2 g). 1HNMR (d6-DMSO) δ ppm 7.56 (1H, d, J = 3 Hz), 7.13 (1H, m), 6.36 (1H, d, J = 8.8 Hz), 5.33 (brs, 2H), 2.88 (m, 4H), 2.47 (m, 4H), 2.16 (s, 3H).
Example 11
Synthesis of tert-butyl 4-(6-amino-3-pyridyl)piperazine-l-carboxylate, Compound 11
This compound was prepared as described in WO 2010/020675 Al .
Synthesis of Compound 89 (also referred to as Compound T)
Compound 89 was synthesized in a similar manner to that described for compound 78 and was converted to an HCl salt. 1HNMR (600 MHz, DMSO-d6) δ ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS (ESI) 447 (M + H)
PATENT
WO 2014144740
PATENT
Preparation of Active Compounds
Syntheses
The disclosed compounds can be made by the following general schemes:

Scheme 1
In Scheme 1, Ref-1 is WO 2010/020675 Al; Ref-2 is White, J. D.; et al. J. Org. Chem. 1995, 60, 3600; and Ref-3 Presser, A. and Hufher, A. Monatshefte fir Chemie 2004, 135, 1015.

Scheme 2
In Scheme 2, Ref-1 is WO 2010/020675 Al; Ref-4 is WO 2005/040166 Al; and Ref-5 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.

92


93 
3) Pd/C/H2 
Scheme 6

Scheme 7
NHfOH

Scheme 8
In Scheme 8, Ref-1 is WO 2010/020675 Al; Ref-2 is WO 2005/040166 Al; and Ref-3 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.
Alternatively, the lactam can be generated by reacting the carboxylic acid with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. Examples of strong acid anhydrides include, but are not limited to, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride, or mixed anhydrides. The dehydrating agent can be a carbodiimide based compound such as but not limited to DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-
dimethylaminopropyl)carbodiimide or DIC (Ν,Ν-diisopropylcarbodiimide). An additional step may be necessary to take off the N-protecting group and the methodologies are known to those skilled in the art.
Alternatively, the halogen moiety bonded to the pyrimidine ring can be substituted with any leaving group that can be displaced by a primary amine, for example to create an intermediate for a final product such as Br, I, F, SMe, SO2Me, SOalkyl, SO2alkyl. See, for Exmaple PCT /US2013/037878 to Tavares.
Other amine intermediates and final amine compounds can be synthesized by those skilled in the art. It will be appreciated that the chemistry can employ reagents that comprise reactive functionalities that can be protected and de-protected and will be known to those skilled in the art at the time of the invention. See for example, Greene, T.W. and Wuts, P.G.M., Greene’s Protective Groups in Organic Synthesis, 4th edition, John Wiley and Sons.

Scheme 9
CDK4/6 Inhibitors of the present invention can be synthesized according to the generalized Scheme 9. Specific synthesis and characterization of the Substituted 2-aminopyrmidines can be found in, for instance, WO2012/061156.
Compounds T, Q, GG, and U were prepared as above and were characterized by mass spectrometry and NMR as shown below:
Compound T
1H NMR (600 MHz, DMSO- d6) ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS ESI (M + H) 447.
PATENT
Synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines. The application appears to be particularly focused on methods for the preparation of trilaciclib and an analog of it. Trilaciclib is the company’s lead CDK4/6 inhibitor presently in phase II trials against small-cell lung cancer and triple negative breast cancer. Interestingly, the company is working on a second CDK4/6 inhibitor, G1T38 , which is in a phase II trial against breast cancer.
GENERAL METHODS
The structure of starting materials, intermediates, and final products was confirmed by standard analytical techniques, including NMR spectroscopy and mass spectrometry. Unless otherwise noted, reagents and solvents were used as received from commercial suppliers. Proton nuclear magnetic resonance spectra were obtained on a Bruker AVANCE 500 at 500 MHz in DMSO-dis. HPLC analyses were performed on a Waters HPLC using the below HPLC method.
HPLC Method
Column: Atlantis T3 (150 χ 4.6, 3 μιη)
Column Temperature: 40°C
Flow Rate: 1 mL/min
Detection: UV @ 275 nm
Analysis Time: 36 min
Mobile Phase A: Water (with 0.1% Trifluoroacetic Acid)
Mobile Phase B : Acetonitrile (with 0.1% Trifluoroacetic Acid)
Sample preparation: dissolve PC sample, wet or dry solid (~1 mg of active compound) in acetonitrile/water (1/1) to achieve complete dissolution.
HPLC Method Gradient

Example 1. General Routes of Synthesis

Scheme 1-1 : Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3, Step 4, Step 5, or Step 6. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the hydroxyl group of the fused spirolactam is converted to a leaving group.
In Step 5 the leaving group is dehydrated to afford a compound of Formula IV. In Step 6 the compound of Formula IV is optionally deprotected.

Scheme 1-2: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3 or Step 4. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the compound of Formula IV is optionally deprotected.

Scheme 1-3 : Starting from an appropriately substituted alkyl glycinate, compounds of the present invention can be prepared. In Step 1 the appropriately substituted alkyl glycinate is subjected to cyclohexanone and TMSCN in the presence of base to afford a cyano species. In Step 2 the appropriately substituted cyanospecies is reduced and subsequently cyclized to afford a compound of Formula I.
Scheme 1-4

Scheme 1-4: Starting from an appropriately substituted l-(aminomethyl)cyclohexan-l-amine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted l-(aminomethyl)cyclohexan-l -amine is reductively aminated with an aldehyde. In Step 2 the appropriately substituted cyclohexane amine is optionally deprotected (i.e.: the group selected from R2 if not H is optionally replaced by H). In Step 3 the cyclohexane amine is cyclized to afford a compound of Formula I. In Step 4 the compound of Formula I is optionally protected.
1-5

Conversion

Scheme 1-5: Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a
substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2, Step 3, Step 4, or Step 5. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the hydroxyl group of the fused spirolactam is converted to a leaving group. In Step 4 the leaving group is dehydrated to afford a compound of Formula IV. In Step 5 the compound of Formula IV is optionally deprotected.
S

Scheme 1-6: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2 or Step 3. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the compound of Formula IV is optionally deprotected.

Scheme 1-7: Starting from compound of Formula IV a CDK4/6 inhibitor can be prepared. In Step 1 a heteroaryl amine is subjected to a base and a compound of Formula IV is added slowly under chilled conditions to afford a nucleophilic substitution reaction. The compound of Formula IV can previously be prepared as described in the schemes herein.
Example 2. Representative Routes of Synthesis
Scheme 2-1

quant, yield 2 steps
isolated

70% yield 2 steps 75% yield 95% yield
isolated isolated isolated
Scheme 2-1 : An ester route is one embodiment, of the present invention. Ideally, the best synthesis scheme would afford crystalline intermediates to provide material of consistent purity without column chromatography, and high yielding steps while using safe and cost effective reagents when possible.
The first step in the ester route is a SNAr nucleophilic substitution of CI group in commercially available ester 3 using spirolactam 4. Due to low reactivity of 4, a reaction temperature of 85-95 °C was required. Because of the temperature requirements, DIPEA and dimethylacetamide were selected as the base and solvent, respectively. The reaction follows second-order kinetics and usually stalls after -85% conversion. Therefore, the reaction was typically stopped after 60 hours by first cooling it to room temperature at which point solid formation was observed. The mixture was then partitioned between MTBE and water and product was filtered with excellent purity with -53% yield of the desired product 5. The obtained
compound 5 was protected with a Boc group using Boc anhydride and DMAP as the catalyst and dichloromethane as the solvent. The intermediate 6 was obtained in a quantitative yield. Due to the semi-solid nature of compound 6, the material was taken to the next step without further purification. The Dieckmann condensation was initially performed with strong bases such as LiHMDS and tBuOK. A similar result to the aldehyde route (Scheme 2-2) was obtained: a partial deprotection of Boc group was observed that required column chromatography. However, the best results were obtained when DBU was used as base and THF as solvent. The reaction outcome was complete, clean conversion of 6 to 7. Moreover, the product crystallized from the reaction mixture upon seeding, and a quantitative yield was obtained for the two steps.
The hydroxyl group of 7 was removed via a two-step procedure. First, compound 7 was converted completely into triflate 8 using triflic anhydride and triethylamine in dichloromethane. The reaction was found to proceed well at 0°C. Due to the potential instability of the triflate intermediate, it was not isolated. It was immediately taken to the next step and reduced with triethylsilane and palladium tetrakis to afford the product 9 after ethyl acetate crystallization in -70% yield. The Boc group of 9 was removed using trifluoroacetic acid in dichloromethane to afford 10. Intermediate 10 was converted into the final sulfone 11 using Oxone in acetonitrile/water solvent system.
The obtained sulfone 11 was use-tested in the coupling step and was found to perform well. In conclusion, the route to sulfone 11 was developed which eliminated the use of column chromatography with good to excellent yields on all steps.
Scheme 2-2

Molecular Weight: 421 
Scheme 2-2: The first step of Scheme 2-2 consistently afforded product 13 contaminated with one major impurity found in substantial amount. Thorough evaluation of the reaction impurity profile by LC-MS and 2D MR was performed, which showed the impurity was structurally the condensation of two aldehyde 12 molecules and one molecule of lactam 4. Therefore, column chromatography was required to purify compound 13, which consistently resulted in a modest 30% yield. A solvent screen revealed that sec-butanol, amyl alcohol, dioxane, and tert-butanol can all be used in the reaction but a similar conversion was observed in each case. However, tert-butanol provided the cleanest reaction profile, so it was selected as a solvent for the reaction. Assessing the impact of varying the stoichiometric ratio of 4 and 12 on the reaction outcome was also investigated. The reaction was performed with 4 equivalents of amine 4 in an attempt to disrupt the 2: 1 aldehyde/amine composition of the impurity. The result was only a marginal increase in product 13 formation. The temperature impact on the reaction outcome was evaluated next. The coupling of aldehyde 12 and 4 was investigated at two different temperatures: 50 °C and 40 °C with 1 : 1 ratio of aldehyde/amine. Reactions were checked at 2 and 4 hours and then every 12 hours. The reaction progress was slow at 50°C and was accompanied by growth of other impurities. The reaction at 40°C was much cleaner; however the conversion was lower in the same time period. The mode of addition of the reagents was investigated as well at 80°C with a slow addition (over 6 hours) of either aldehyde 12 or amine 4 to the reaction mixture. The product distribution did not change and an about 1 to 1 ratio was observed between product and impurity when amine 4 was added slowly to the reaction mixture containing aldehyde 12 and
DIPEA at reflux. The product distribution did change when aldehyde 12 was added slowly to the mixture of amine 4 and DIPEA. However, the major product of the reaction was the undesired impurity. Other organic bases were tried as well as different ratios of DIPEA. No product was observed when potassium carbonate was used as a base. The results of the experiments are presented in Table 1 below.
Table 1

Compound 13 was successfully formed in three cases: triethylamine, 2,6-lutidine and DIPEA, with the DIPEA result being the best. The use of Boc protected spirolactam 4 had no effect on the impurity formation as well. Its utilization was speculated to be beneficial in performing the coupling step together with the following step, preparation of compound 14.
The major impurity formed during Step 1 of Scheme 2-2 is:

Chemical Formula:€2)Η;Μ(¾ 6( 2ί>2
Molecular Weight: 527.4903
The second step (Boc protection of the free lactam) proceeded well using DMAP as a catalyst in dichloromethane at room temperature. The product 14 is a thick oil, and, therefore, cannot be purified by crystallization. The Boc protected intermediate 14 was cyclized successfully into the desired pentacyclic structure 10 upon treatment with a strong base such as LiHMDS or tBuOK. Surprisingly, the Boc group was partially removed during the reaction. The level of deprotection was independent from the internal reaction temperature and was positively correlated with excess of base used. Therefore the mixture of the desired product 10 and 10-Boc compound was treated with acid to completely deprotect Boc group. The conversion of methyl sulfide into the final sulfone 11 was carried out with Oxone. Initially a mixture of methanol and water was used for the reaction. As the result, a partial displacement of sulfone by methoxy group was detected. The methanol was replaced with acetonitrile and the sulfone displacement was eliminated.
In summary, the ester route (Scheme 2-1) is preferred because:
1. Formation of the impurity during the first step of Scheme 2-2 was unavoidable and resulted in yields of < 35%.
2. Column purification was required to isolate intermediate 14.
3. The aldehyde starting material was not commercially available and required two synthetic steps from the corresponding ester.

Scheme 2-3 : Starting with cyclohexanone, compounds of the present invention can be prepared. In Step 1 the methyl glycinate is subjected to cyclohexanone and TMSCN in the presence of tri ethyl amine in DCM to afford 15. In Step 2 15 hydrogenated with hydrogen gas in the presence of catalytic platinum oxide and subsequently undergoes an intramolecular cyclization to afford compound 16 which is used in the schemes above.

Scheme 2-4: Starting with compound 17, compounds of the present invention can be prepared. In Step 1 compound 17 is subjected to ethyl 2-oxoacetate in the presence platinum on carbon and hydrogen gas to afford compound 18. In Step 2 compound 18 is Boc-deprotected with hydrochloric acid. In Step 3 compound 18 is cyclized to afford compound 16 which is used in the schemes above.
Scheme 2-5

11 19
Scheme 2-5: Starting from compound 11 the CDK 4/6 inhibitor 19 can be prepared. In Step 1 5-(4-methylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 19. Compound 11 can be prepared as described in the schemes herein.

Scheme 2-6: Starting from compound 11 the CDK 4/6 inhibitor 20 can be prepared. In Step 1 5-(4-isopropylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 20. Compound 11 can be prepared as described in the schemes herein.
Preparation of Compound 5:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate 3 (49.2 g, 0.21 mol, 1.00 equiv.), spirolactam 4 (39.2 g, 0.23 mol, 1.10 equiv.), DIPEA (54.7 g, 0.42 mol, 2.00 equiv.), and DMAc (147.6 mL, 3 vol). The batch was heated to 90-95 °C, and after 60 h, IPC confirmed -14% (AUC) of ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate remained. The batch was cooled to RT, and precipitate formation was observed. The suspension was diluted with MTBE (100 mL, 2 vol) and water (442 mL, 9 vol) and stirred for 2 h at RT. The product was isolated by vacuum filtration and washed with MTBE (49 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford compound 5 [41.0 g, 53% yield] as an off-white solid with a purity of >99% AUC. ¾ MR (CDCh): δ 8.76 (d, J = 2.0 Hz, 1H), 6.51-6.29 (br, 1H), 4.33 (q, J = 7.0 Hz, 2H), 3.78 (s, 2H), 3.58 (s, 2H), 2.92 (s, 2H), 2.53 (s, 3H), 1.63-1.37 (m, 12H). LCMS (ESI, m/z = 365.3 [M+H]).
Preparation of Compound 6:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 5 [41.0 g, 0.11 mol, 1.00 equiv.], Boc-anhydride (36.8 g, 0.17 mol, 1.50 equiv.), DMAP (1.37 g, 0.01 mol, 0.10 equiv.), and dichloromethane (287 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained (AUC). The batch was concentrated into a residue under reduced pressure and taken to the next step (a quantitative yield is assumed for this step). An aliquot (200 mg) was purified by column chromatography (heptanes/ethyl acetate 0 to 100%) to afford compound 6. 1H MR (CDCh): δ 8.64 (s, 1H), 4.31 (q, J = 7.0 Hz, 2H), 4.07 (s, 2H), 3.83 (S, 2H), 3.15 (m, 2H), 2.56 (s, 3H), 172 (m, 3H), 1.59 (m, 15H), 1.42 (t, J= 7.0 Hz, 3H). LCMS (ESI, m/z = 465.2 [M+H]).
Preparation of Compound 7:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 6 [residue from a previous step, quantitative yield assumed, 52.2 g, 0.11 mol, 1.00 equiv.], and THF (261 mL, 5 vol). The batch was cooled to 0°C and 1,8-diazabicyclo[5.4.0]un-dec-7-ene (17.1 g, 0.11 mmol, 1.00 equiv.) was added keeping the internal temperature in 0-10°C range. After the addition was complete, the cooling bath was removed and the reaction mixture was allowed to warm up to RT and after 2 h, IPC confirmed no starting material remained. The batch was seeded with the product (1.0 g) and was cooled to 0°C. The slurry was stirred at 0°C for 2 h. The product was isolated by vacuum filtration and washed with cold (0°C) THF (50 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 7 [47 g, quantitative yield] as a light orange solid with a purity of >99% AUC. The color of the product changed into yellow once the batch was exposed to air for an extended period of time (~ 1 day). Material was isolated with substantial amount DBU, according to proton NMR. However, it did not interfere with the next step. 1H MR (CDCh): δ 8.71 (s, 1H), 4.03 (s, 2H), 2.57 (s, 3H), 1.85 (m, 10H), 1.51 (s, 9H). LCMS (ESI, m/z = 419.2 [M+H]).
Preparation of Compound 8:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 7 [40.8 g, 0.10 mol, 1.00 equiv.], triethylamine (31.5 g, 0.31 mol, 3.20 equiv.), and dichloromethane (408 mL, 10 vol). The batch was purged with N2 for 15 min and was cooled to 0°C. Triflic anhydride (44.0 g, 0.16 mol, 1.60 equiv.) was added keeping the
internal temperature in 0-10°C range. The batch was stirred at 0°C and after 3 h, IPC confirmed -7.0% (AUC) of 7 remained. [It was speculated that the product was hydrolyzing back into starting material during the analysis.] Once the reaction was deemed complete, the batch was transferred to a 1 L, separatory funnel and was washed with 50% saturated sodium bicarbonate (200 mL, 5 vol). [It was prepared by mixing saturated sodium bicarbonate (100 mL) with water (100 mL)).] The aqueous layer was separated and was extracted with DCM (2×40 mL, 1 vol). The organic layers were combined and concentrated into a residue under reduced pressure and taken to the next step. LCMS (ESI, m/z = 551.6 [M+H]).
Preparation of Compound 9:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 8 [residue from a previous step, quantitative yield assumed, 53.7 g, 0.10 mol, 1.00 equiv.], and THF (110 mL, 2 vol). The solvent was removed under vacuum distillation and the procedure was repeated two times. The flask was charged with triethylsilane (22.7 g, 0.20 mol, 2.00 equiv.), and DMF (268 mL, 5 vol). The batch was degassed by five cycles of evacuation, followed by backfilling with nitrogen. The flask was charged with tetrakis(triphenylphosphine)palladium(0) (11.3 g, 0.01 mol, 0.1 equiv.). The batch was heated to 45-50°C, and after 14 h, IPC confirmed no starting material remained. The batch was transferred to a 500 mL, separatory funnel while still warm. The reaction was partitioned between water (5 vol) and ethyl acetate (5 vol). The aqueous layer was extracted with ethyl acetate (3 x3 vol). The organic layers were combined and concentrated down to 2 volumes. The precipitate was filtered and washed with ethyl acetate (2x 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 9 [27.5 g, 70% yield] as a yellow solid with a purity of -98% AUC. Proton NMR showed some triphenylphosphine oxide present. ¾ NMR (DMSO-i¾):5 9.01 (s, 1H), 7.40 (s, 1H), 4.30 (s, 2H), 2.58 (m, 2H), 2.58 (s, 3H), 1.81 (m, 5H), 1.51 (s, 9H). LCMS (ESI, m/z = 403.4 [M+H]).
Preparation of Compound 10 from the Scheme 2-1 route:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged 9 (12.8 g, 31.8 mmol, 1.00 equiv.) and dichloromethane (64 mL, 5 vol). Trifluoroacetic acid (18.2 g, 159 mmol, 5.00 equiv.) was added over 20 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (200 mL). The aqueous layer was extracted with dichlorom ethane (3 x3 vol). The organic layers were combined and concentrated down to 1 volume. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [6.72 g, 70% yield] as an off-white solid with a purity of 99.1% AUC. ¾ NMR (DMSO-dis): δ 8.95 (s, 1H), 8.32 (s, 1H), 7.15 (s, 1H), 3.68 (d, J = 1.0 Hz, 2H), 2.86 (m, 2H), 2.57 (s, 3H), 1.92 (m, 2H), 1.73 (m, 3H), 1.39 (m, 3H). LCMS, ESI, m/z = 303.2 [M+H]).
Preparation of Compound 10 from Scheme 2-2 route:
A 50 mL, three-neck flask equipped with a magnetic stirring bar, thermocouple, N2 inlet was charged 14 (680 mg, 1.62 mmol, 1.00 equiv.) and THF (6.8 mL, 10 vol). A I M solution of potassium tert-butoxide (3.2 mL, 3.24 mmol, 2.00 equiv.) in THF was added over 10 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was acidified with 4 N hydrogen chloride solution in dioxane (2.4 mL, 9.72 mmol, 6.00 equiv.) and stirred for additional 1 h. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (100 mL). The aqueous layer was extracted with ethyl acetate (3 x20 vol). The organic layers were combined and concentrated down to 3volumes and product precipitated. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [489 mg, quantitative yield] as an off-white solid.
Preparation of Compound 11 :
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 10 (9.00 g, 29.8 mmol, 1.00 equiv.), and acetonitrile (180 mL, 20 vol). A solution of Oxone (45.9 g, 0.15 mol, 5.00 equiv.) in water (180 mL, 20 vol) was added to the batch over 20 min. The batch was stirred for 2 h and IPC confirmed the reaction was complete. The batch was concentrated down to ½ of the original volume and was extracted with dichloromethane DCM (4x 10 vol). The organic layers were combined; polish filtered and concentrated down to -10 vol of DCM. The product was slowly crystallized out by addition of heptanes (-30 vol). The mixture was cooled to 0°C and the product was filtered and dried under vacuum at 40 °C for 16 h to afford 11 [9.45 g, 95% yield] as an off-white solid with a purity of >99% AUC. ¾ NMR (CDCb): 5 9.24 (s, 1H), 7.78 (s, 1H), 7.46 (s, 1H), 3.89 (d, J= 2.0 Hz, 2H), 3.43 (s, 3H), 2.98 (m, 2H), 2.10 (m, 2H), 1.86 (m, 3H), 1.50 (m, 3H). LCMS (ESI, m/z = 335.2 [M+H]).
Preparation of Compound 13:
A 250 mL, single-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with 4-chloro-2-(methylthio)pyrimidine-5-carbaldehyde (2.00 g, 10.6 mmol, 1.00 equiv.), spirolactam 4 (1.96 g, 11.7 mmol, 1.10 equiv.), DIPEA (2.74 g, 21.2 mmol, 2.00 equiv.), and fert-butanol (20 mL, 10 vol). The batch was heated to 80-85 °C, and after 24 h, IPC confirmed no aldehyde 12 remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 13 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 14:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 13 [0.98 g, 3.00 mmol, 1.00 equiv.], Boc-anhydride (4.90 g, 21.5 mmol, 7.00 equiv.), DMAP (36 mg, 0.30 mmol, 0.10 equiv.), and dichloromethane (7 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 14 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 15:
To a suspension of methyl glycinate (500 g, 3.98 mol, 1 eq) in DCM (10 L) was added
TEA dropwise at rt under nitrogen atmosphere, followed by the addition of cyclohexanone (781 g, 7.96 mol, 2 eq) dropwise over 15 min. To the resulting mixture was added TMSCN (591 g, 5.97 mol, 1.5 eq) dropwise over 1 hour while maintaining the internal reaction temperature below 35
°C. After stirred at rt for 2 hrs, the suspension became a clear solution. The progress of the reaction was monitored by H- MR.
When the methyl glycinate was consumed completely as indicated by H-NMR analysis, the reaction was quenched by water (5 L). The layers were separated. The aqueous layer was extracted with DCM (1 L). The combined organic phase was washed with water (5 L X 2) and
dried over Na2S04 (1.5 Kg). After filtration and concentration, 1.24 Kg of crude 15 was obtained as oil.
The crude 15 was dissolved in IPA (4 L). The solution was treated with HC1/IPA solution (4.4 mol/L, 1.1L) at RT. A large amount of solid was precipitated during the addition. The resulting suspension was stirred for 2 hrs. The solid product was collected by vacuum filtration and rinsed with MTBE (800 mL). 819 g of pure 15 was obtained as a white solid. The yield was 88.4%. ¾- MR (300 MHz, CD3OD) 4.20 (s, 2H), 3.88 (s, 3H), 2.30-2.40 (d, J = 12 Hz, 2H), 1.95-2.02 (d, J = 12 Hz, 2H), 1.55-1.85 (m, 5H), 1.20-1.40 (m, 1H).
Preparation of Compound 16:
To a solution of 15 (10 g, 43 mmol) in MeOH (100 mL) was added methanolic hydrochloride solution (2 .44 mol/L, 35.3 mL, 2 eq) and Pt02 (0.5 g, 5 wt %). The reaction suspension was stirred with hydrogen bubble at 40 °C for 6 hours. H- MR analysis showed consumption of 15. To the reaction mixture was added K2CO3 (15 g, 108 mmol, 2.5 eq) and the mixture was stirred for 3 hrs. The suspension was filtered and the filtrate was concentrated to dryness. The residual oil was diluted with DCM (100 mL) and resulting suspension was stirred for 3 hrs. After filtration, the filtrate was concentrated to provide crude 16 (6.6 g) as an oil. The crude 16 was diluted with EtOAc/hexane (1 : 1, 18 mL) at rt for 2 hrs. After filtration, 16 (4 g) was isolated. The obtained 16 was dissolved in DCM (16.7 mL) and hexane (100 mL) was added dropwise to precipitate the product. After further stirred for 1 h, 2.8 g of the pure 16 was isolated as a white solid. The yield was 39%. HPLC purity was 98.3%; MS (ESI): 169.2 (MH+); 1 H-NMR (300 MHz, D2O) 3.23 (s, 3H), 3.07 (s, 3H), 1.37-1.49 (m, 10H).
Preparation of compound 19:
5-(4-methylpiperazin-l-yl)pyridin-2-amine (803.1 g; 3.0 equivalents based on sulfone 11) was charged to a 22 L flask. The flask was blanketed with N2 and anhydrous THF added (12.4 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (4.7 L; 1.2 equivalents based on sulfone 11) was added via an addition funnel in three equal additions to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C. The sulfone 11 (455.1 g; 1.00 equivalents) was added in five additions. Reaction proceeded until HPLC analysis of an IPC sample indicated less than 3% of sulfone 11 remained.
To quench the reaction, the contents of the 22L flask were transferred to a 100 L flask containing water. After stirring for 30 minutes at <30°C, the crude product was collected by filtration, washed with water and dried to afford 19 (387 g, 99.1% purity, 63.7% yield).
Preparation of compound 20:
5-(4-isopropylpiperazin-l-yl)pyridin-2-amine (1976.2 g; 3.0 equivalents based on sulfone 11) was charged to a 50 L flask. The flask was blanketed with N2 and anhydrous THF added (10.7 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (9.6 kg; 3.6 equivalents based on sulfone) was added via an addition funnel at a rate to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C over 120 minutes by removing the ice bath. The sulfone (1000 g; 1.00 mol) was added in five additions. The reaction proceeded until HPLC analysis of an IPC sample indicated less than 1% of sulfone 11 remained. After completion of the reaction, ammonium chloride was added to the reaction mixture. The mixture stirred at < 32°C for at least 30 minutes and the solids collected by filtration to afford 20 (900 g, 99.1% purity, 64.2% yield).
Alternate Route to Spirolactam via cyclohexanone:
Scheme 2-7

26
In one embodiment the spirolactam is made via the synthetic scheme above. By reducing the nitrile group before addition of the glycinate group the reaction sequence proceeds in higher yield. The chemistry used in Step 1 is described in the literature (J. Org. Chem. 2005, 70,8027-8034), and was performed on a kilogram scale. The chemistry to convert Compound 24 into the
spirolactam was also demonstrated on kilogram scale. The Boc protection of Compound 23, is carried out at -70°C in order to limit formation of the di-Boc protected product. Experimental details of a 200 g pilot run are described below.
Step 1

200 g of cyclohexanone 21 was converted to 22 using Ti(Oi-Pr)4 /TMSCN/NH3. After work-up, 213 g of 22 was obtained. The H- MR was clean. The yield was 84%. The titanium salts were removed by vacuum filtration. In one embodiment, the titanium salts are removed by centrifugation or Celite filtration.
Step 2

190 g of 22 was mixed with LAH (2 eq) in MTBE for 30 minutes at 45°C. After work-up, 148 g of crude 23 was obtained.
Step 3

136 g of the crude 23 from step 2 was converted to 24 with 0.9 eq of B0C2O at -70°C. The reaction was completed and worked up. After concentration, 188 g of 24 was obtained. The yield was 86%. The H-NMR and C-NMR spectra confirmed that the compound was pure.
Step 4

188 g of 24 was subjected to methyl 2-bromoacetate and K2CO3 in acetonitrile to afford 25. 247 g of crude 25 was obtained.
Step 5

247 g of 25 was subjected to TFA in DCE heated to reflux to afford 26. After work-up, 112 g of 6 as TFA salt was obtained. H- MR was clean.
Step 6

26 27
Compound 26 was stirred in EtOH in the presence at room temperature overnight to afford 27. In one embodiment DCM is used as the solvent instead of EtOH.
Example 3. Purge of residual palladium from Step 5 Scheme 2-1:
Since palladium was used in Step 5 of Scheme 2-1, the levels of residual Pd present in the subsequent synthetic steps was determined. Table 2 below and Figure 3 show the palladium levels in the isolated solids.
Table 2

The material after Step 5 was isolated containing 1.47% (14700 ppm) of residual palladium. This data represents the highest level of palladium in the worst case scenario. The workup conditions of the latter steps purged nearly all of the palladium and the final product, 19 bis HC1 salt, contained 14 ppm of Pd, which is below the standard 20 ppm guidline. The Pd levels will likely be even lower once the catal st loading is optimized in Step 5.

19
The process developed in this route was a significant improvement over the one used for the first generation synthesis. Overall, the scheme consists of seven steps with five isolations, all by crystallization. No silica column chromatography is employed in the synthesis, which makes the process highly scalable. The process workup conditions can successfully purge the 1.47% of residual palladium after step 5 of Scheme 2-1.
///////////////TRILACICLIB, G1T28, G1T 28, SHR 6390, PHASE 2, G1 Therapeutics, Inc.
CN1CCN(CC1)C2=CN=C(C=C2)NC3=NC=C4C=C5C(=O)NCC6(N5C4=N3)CCCCC6
![]()

BICTEGRAVIR, NEW PATENT, WO 2018005328, CONCERT PHARMA
WO2018005328) DEUTERATED BICTEGRAVIR
CONCERT PHARMACEUTICALS, INC.
TUNG, Roger, D.; (US)

Concert CEO Roger Tung
Novel deuterated forms of bictegravir is claimed. Gilead Sciences is developing the integrase inhibitor bictegravir as an oral tablet for the treatment of HIV-1 infection.
This invention relates to deuterated forms of bictegravir, and pharmaceutically acceptable salts thereof. In one aspect, the invention provides a compound of Formula (I) or a pharmaceutically acceptable salt thereof, wherein each of Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11b is independently hydrogen or deuterium; provided that if each Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, and Y11 is hydrogen, then Y11b is deuterium.

Many current medicines suffer from poor absorption, distribution, metabolism and/or excretion (ADME) properties that prevent their wider use or limit their use in certain indications. Poor ADME properties are also a major reason for the failure of drug candidates in clinical trials. While formulation technologies and prodrug strategies can be employed in some cases to improve certain ADME properties, these approaches often fail to address the underlying ADME problems that exist for many drugs and drug candidates. One such problem is rapid metabolism that causes a number of drugs, which otherwise would be highly effective in treating a disease, to be cleared too rapidly from the body. A possible solution to rapid drug clearance is frequent or high dosing to attain a sufficiently high plasma level of drug. This, however, introduces a number of potential treatment problems such as poor patient compliance with the dosing regimen, side effects that become more acute with higher doses, and increased cost of treatment. A rapidly metabolized drug may also expose patients to undesirable toxic or reactive metabolites.
[3] Another ADME limitation that affects many medicines is the formation of toxic or biologically reactive metabolites. As a result, some patients receiving the drug may experience toxicities, or the safe dosing of such drugs may be limited such that patients receive a suboptimal amount of the active agent. In certain cases, modifying dosing intervals or formulation approaches can help to reduce clinical adverse effects, but often the formation of such undesirable metabolites is intrinsic to the metabolism of the compound.
[4] In some select cases, a metabolic inhibitor will be co-administered with a drug that is cleared too rapidly. Such is the case with the protease inhibitor class of drugs that are used to treat HIV infection. The FDA recommends that these drugs be co-dosed with ritonavir, an inhibitor of cytochrome P450 enzyme 3A4 (CYP3A4), the enzyme typically responsible for their metabolism (see Kempf, D.J. et al., Antimicrobial agents and chemotherapy, 1997, 41(3): 654-60). Ritonavir, however, causes adverse effects and adds to the pill burden for HIV patients who must already take a combination of different drugs. Similarly, the
CYP2D6 inhibitor quinidine has been added to dextromethorphan for the purpose of reducing rapid CYP2D6 metabolism of dextromethorphan in a treatment of pseudobulbar affect. Quinidine, however, has unwanted side effects that greatly limit its use in potential combination therapy (see Wang, L et al., Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt 1): 659-67; and FDA label for quinidine at http://www.accessdata.fda.gov).
[5] In general, combining drugs with cytochrome P450 inhibitors is not a satisfactory strategy for decreasing drug clearance. The inhibition of a CYP enzyme’s activity can affect the metabolism and clearance of other drugs metabolized by that same enzyme. CYP inhibition can cause other drugs to accumulate in the body to toxic levels.
[6] A potentially attractive strategy for improving a drug’s metabolic properties is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the formation of undesirable metabolites by replacing one or more hydrogen atoms with deuterium atoms. Deuterium is a safe, stable, non-radioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the ADME properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
[7] Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, MI et al, J Pharm Sci, 1975, 64:367-91; Foster, AB, Adv Drug Res 1985, 14:1-40 (“Foster”); Kushner, DJ et al, Can J Physiol Pharmacol 1999, 79-88; Fisher, MB et al, Curr Opin Drug Discov Devel, 2006, 9:101-09 (“Fisher”)). The results have been variable and unpredictable. For some compounds deuteration caused decreased metabolic clearance in vivo. For others, there was no change in metabolism. Still others demonstrated increased metabolic clearance. The variability in deuterium effects has also led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting adverse metabolism (see Foster at p.35 and Fisher at p.101).
[8] The effects of deuterium modification on a drug’s metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem.1991, 34, 2871-76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
Exemplary Synthesis
[72] Deuterium-modified analogs of bictegravir can be synthesized by means known in the art of organic chemistry. For instance, using methods described in US Patent No.9,216,996 (Haolun J. et al., assigned to Gilead Sciences, Inc. and incorporated herein by reference), using deuterium-containing reagents provides the desired deuterated analogs.
[73] Such methods can be carried out utilizing corresponding deuterated and optionally, other isotope-containing reagents and/or intermediates to synthesize the compounds delineated herein, or invoking standard synthetic protocols known in the art for introducing isotopic atoms to a chemical structure.
[74] A convenient method for synthesizing compounds of Formula I is depicted in the Schemes below.
 [75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[76] In a manner analogous to the procedure described in US 9,216,996, acetal deprotection of carboxylic acid 7 followed by cyclization with appropriately deuterated aminocyclopentanol 9 provides carboxylic acid intermediate 10. Amide coupling with appropriately deuterated benzylamine 11 followed by deprotection of the methyl ether ultimately affords a compound of Formula I in eight overall steps from compound 1.
[77] Use of appropriately deuterated reagents allows deuterium incorporation at the Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11bpositions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and/or Y11b.
[78] Appropriately deuterated intermediates 2a and 2b, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 2 below.
S h 2 S th i f C d 2 d 2b

[79] Synthesis of compound 2a (wherein Y3=H) by acetal formation of N,N-dimethylformamide (DMF) with dimethylsulfate has been described in Mesnard, D. et. al. J. Organomet. Chem.1989, 373, 1-10. Replacing DMF with N,N-dimethylformamide-d1 (98-99 atom % D; commercially available from Cambridge Isotope Laboratories) in this reaction would thereby provide compound 2b (wherein Y3=D).
[80] Use of appropriately deuterated reagents allows deuterium incorporation at the Y3 position of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at Y3.
[81] Appropriately deuterated intermediates 4a-4d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 3 below.

[82] As described in Malik, M. S. et. al. Org. Prep. Proc. Int.1991, 26, 764-766, acetaldehyde is converted to alkylhalide 14a via reaction with chlorine gas and subsequent acetal protection with CaCl2 in methanol. As described in CN 103739506, reaction of 14a with aqueous ammonia and then sodium hydroxide provides primary amine 4a (wherein Y9=Y10a=Y10b=H). Replacing acetaldehyde with acetaldehyde-d1, acetaldehyde-2,2,2-d3, or acetaldehyde-d4 (all commercially available from CDN Isotopes with 98-99 atom % D) in the sequence then provides access to compounds 4b (Y9=D, Y10a=Y10b=H), 4c (Y9=H,
Y10a=Y10b=D) and 4d (Y9=Y10a=Y10b=D) respectively (Schemes 3b-d).
[83] Use of appropriately deuterated reagents allows deuterium incorporation at the Y9, Y10a, and Y10b positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y9, Y10a, and/or Y10b.
[84] Appropriately deuterated intermediates 9a-9d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 4 below.
 [85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[86] As depicted in Scheme 4b, aminocyclopentanol 9b (Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b= Y8=D) is obtained through an analogous synthetic sequence using cyclopentadiene-d6 and performing the penultimate hydrogenation with D2 in place of H2. Cyclopentadiene-d6 is prepared according to the procedure described in Cangoenuel, A. et. al. Inorg. Chem.2013, 52, 11859-11866.
[87] Alternatively, as shown in Scheme 4c, the meso-diol obtained in Scheme 4a is oxidized to the diketone following the procedure reported by Rasmusson, G.H. et. al. Org. Syn.1962, 42, 36-38. Subsequent mono-reduction with sodium borodeuteride and CeCl3 then affords the D1-alcohol in analogy to the method described in WO 2001044254 for the all-protio analog using sodium borohydride. Reduction of the remaining ketone using similar conditions provides the meso-D2-diol in analogy to the method reported in Specklin, S. et. al. Tet. Lett.2014, 55, 6987-6991 for the all protio analog using sodium borohydride. The meso-D2-diol is then converted to 9c (Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=H, Y6=Y8=D) following the same procedures outlined in Scheme 4a.
[88] Likewise, the meso-diol obtained in Scheme 4b may be converted to 9d
(Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=D, Y6=Y8=H) in an analogous manner as depicted in Scheme 4d. The use of deuterated solvents such as D2O or MeOD may be considered to reduce the risk of D to H exchange for ketone containing intermediates.
[89] Use of appropriately deuterated reagents allows deuterium incorporation at the Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and Y8 positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and/or Y8.
[90] Appropriately deuterated intermediates 11a-11d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 5 below.
Scheme 5. Synthesis of Benzylamines 11a-11d

//////////////////


UV Spectroscopy The UV absorption spectrum of carbamazepine in methanol shown in Fig. 1 was recorded using Shimadzu UV–vis Spectrometer 1601 PC. The compound exhibited maxima at 288 and 259 nm. Clarke reported the following: methanol—237 and 285 nm (A 1%, 1 cm¼490) [1].
1 A.C. Moffat (Ed.), Clarke’s Isolation and Identification of Drugs, second ed.,
The Pharmaceutical Press, London, 1986, p. 428.


Vibrational Spectroscopy The FT-infrared absorption spectrum of carbamazepine was obtained in a KBr pellet using a Perkin-Elmer FT-infrared spectrophotometer. FTinfrared spectrum is shown in Fig. 2, where the principal peaks are observed at 3465, 3157, 1675, 1604, 1594, 1488, 1381, 1307, 870, 800, 762, and 724 cm1 .

1 H NMR Spectra The proton nuclear resonance (1 H NMR) spectra of carbamazepine were obtained using a Bruker instrument operating at 500 MHz. Standard Bruker software was used to execute the recording of the 1D and 2D spectra. The sample was dissolved in DMSO-d6 and all resonance bands were referencedto tetramethylsilane (TMS) as internal standard. The entire proton spectra are shown in Figs. 3 and 4. A singlet resonates at δ 5.54 representing the two protons of the amino group. An additional singlet which resonates at δ 6.99 ppm is assigned to the olefinic protons at positions 10 and 11. The two multiplets which resonate at δ 7.30–7.34 and δ 7.41–7.43 ppm are assigned to the aromatic protons of the two phenyl rings.

13C NMR Spectra A noise-modulated, broadband decoupling 13C NMR spectrum (Fig. 5) showed 11 carbon absorptions in accordance with what is anticipated for the structure of carbamazepine. Carbon resonance bands at δ 127.1, 129.0, 129.2, 129.3, 129.8, 130.3, 131.0, and 134.8 ppm account for the CH functions. A carbon band at δ 140.6 ppm represents the ethylene carbons. The carbonyl carbon resonates at δ 156.3 ppm. A DEPT experiment (Fig. 6) permitted the identification and confirmation of the methyl and methine carbons. Another confirmation was obtained through the HSQC experiment (Fig. 7).



PATENT

https://www.google.com/patents/US6670472
Oxcarbazepine is an anticonvulsant drug (as described in U.S. Pat. No. 3,642,775), and has been proposed for use as an anti-epileptical agent in the treatment of AIDS-related neural disorders (as described in PCT patent specification no. WO 94/20110); and for the treatment of Parkinson’s disease and/or Parkinsonian syndromes (as described in U.S. Pat. No. 5,658,900 and European patent specification no. 678 026).
Various processes for preparing oxcarbazepine have been described in the prior art. For example, U.S. Pat. No. 3,642,775 describes the preparation of oxcarbazepine from 10-methoxyiminostilbene (Scheme-1), which is first phosgenated in toluene, followed by amidation (ethanol and ammonia) and hydrolysis in an acidic medium to furnish the desired product. The main drawback of this process is the use of phosgene (COCl2), a toxic and hazardous substance.
Canadian patent specification no. 1 112 241 describes an alternative preparation of oxcarbazepine from the catalysed re-arrangement of 10,11-epoxycarbamazepine, which itself may be prepared from carbamazepine by reaction with m-chloroperbenzoic acid (CPBA) (Scheme-2). However, the drawbacks of this process are: use of carbamazepine, an expensive raw material; and converting this into its corresponding epoxide in poor yields and quality.
Another process, disclosed in European patent specification no. 028 028, starts from 5-cyanoiminostilbene through nitration, reduction and hydrolysis stages (Scheme-3). However, the drawback of the process is in the preparation of the 5-cyanoiminostilbene itself, which can be made from iminostilbene and cyanogen chloride. The latter is also toxic, hazardous and difficult to handle.
Another alternative is described in Swiss patent specification no. 642 950 and comprises hydrolysis, using concentrated sulphuric acid, of the corresponding chloride (10-chloro-5H-dibenz[b,f]azepin-5carboxamide) to form the oxcarbazepine.
More recently, a process has been described in PCT patent specification no. WO 96/21649 (Scheme-4), which starts with 10-methoxyiminostilbene and treats it with an alkali or alkaline earth metal cyanate and acid to produce 10-methoxycarbamazepine which, on acid hydrolysis, furnishes oxcarbazepine. Alternatively, 10-methoxyiminostilbene is first hydrolysed to produce 10-oxo-iminodibenzyl (10-keto-iminodibenzyl) which, upon condensation with chlorosulphonyl isocyanate followed by hydrolysis, furnishes oxcarbazepine. Chlorosulphonyl isocyanate is a very costly, highly moisture-sensitive and toxic reagents which is the main drawback of this latter process.
The biggest problem with the former process is that 10-methoxyiminostilbene undergoes two kinds of competitive reactions when an alkali metal cyanate and an acid are added. The enol-ether moiety of the compound undergoes hydrolysis to give the corresponding ketone (“oxo” compound), which does not undergo a carboxamidation reaction with HOCN, whereas the imino function of the intact 10-methoxyiminostilbene does undergo a carboxamidation reaction. Therefore, the end result is that a mixture of oxcarbazepine, oxo-iminodibenzyl and impurities are obtained, after hydrolysis, making the subsequent crystallization process highly tedious and uneconomical.
The acids that are used in this reaction (Scheme-4), according to the Examples of WO 96/21649, include acetic acid, mono-, di- and tri-chloroacetic acids, dry HCl and concentrated sulphuric acid etc. The general description teaches that concentrated mineral acids are to be used, optionally in solution in the organic acids. Nevertheless, all these acids produce substantial quantities of side products, ie oxo-iminodibenzyl and impurities formed therefrom. Due to this, although the conversion is high, the selectivity leading to the carboxamidation reaction is poor.
Furthermore, international patent specification no. WO 01/56992 describes the use of acetic acid in the absence of an additional solvent in this process, which is stated to result in an improved yield. Nothing about the purity of the end-product (oxcarbazepine) is mentioned, however, and the specific example given shows that the yield thereof is less than or equal to 78% after hydrolysis with water and sulphuric acid in the absence of a solvent such as toluene.
All the known methods therefore suffer from disadvantages, in particular, the requirement to use “environmentally unfriendly” reactants, and/or result in poor yields due to side reactions as mentioned above. In particular, the method described in WO 01/56992 precludes the use of a solvent, which imposes unfavourable limitations on the subsequent processing of the intermediate in the preparation of the end-product.
We have surprisingly found that reaction of 10-methoxyiminostilbene with cyanic acid (HOCN) in the presence of a mild acidic reagent, especially an aromatic acid, enables the disadvantages of the prior art preparation of 10-methoxycarbamazepine to be overcome. In particular, it allows for the use of a solvent in the subsequent reaction steps, which has advantages as will be further described hereinbelow
Accordingly, the present invention provides a process for the preparation of 10-methoxycarbamazepine, which process comprises reacting 10-methoxyiminostilbene with HOCN in a solvent therefor in the presence of a mild acidic reagent. It is important that the mild acidic reagent be chosen so that the enol-ether function is not rapidly hydrolysed. Accordingly, this reagent is preferably a weak acid, such as an aromatic acid. Preferred aromatic acids include weak, non-aliphatic organic acids, such as benzoic acid and substituted benzoic acids; suitable substituents being halo, especially chloro eg para-chlorobenzoic acid. Suitably, the acid has a pKa value in the range of from about 10−4 to 10−5.
Furthermore, the mild acidic reagent is preferably relatively insoluble in the solvent, especially at room temperature but also preferably at the temperature of the reaction, compared to other acids, such as acetic acid. Suitably, the mild acidic reagent has a solubility in the solvent of less than 75%, preferably less than 50% and more preferably less than 25% in the solvent. Especially preferred is when the mild acidic reagent has a solubility of less than about 10-12%, even at elevated temperatures, such as at the temperature of the reaction, and particularly preferred is when the mild acidic reagent has a solubility of less than about 1% at room/ambient Temperature. In this context, it is to be understood that ‘room temperature’ is less than 35° C. and more usually about 20-25° C., such as 21-22° C. Of all the aromatic acids, benzoic acid is the most suitable acid in terms of selectivity (by ‘selectivity’ in this context is meant preference for the carboxamidation reaction over the enol-ether hydrolysis).
Excess molar quantity of the weak acid is preferably used in comparison to the 10-methoxyiminostilbene starting material; for example, in the range of from 2 to 10 molar excess, more preferably about 5 to 8 times, eg 6-7 times, benzoic acid is most preferably employed in the reaction. Most of the acidic reagent can be easily recovered and re-used, such as up to 90-95% can be re-cycled. Such acids less readily hydrolyse the enol-ether moiety present in the 10-methoxyiminostilbene, while nevertheless being able readily to catalyse the reaction between the 10-methoxyiminostilbene and the HOCN.
In another aspect, the present invention provides a process for the preparation of 10-methoxycarbamazepine, which process comprises reacting 10-methoxyiminostilbene with HOCN in the absence of a strong acid. In particular, the present invention provides a process for the preparation of 10-methoxycarbamazepine, which process comprises reacting 10-methoxyiminostilbene with HOCN in the absence of an acid having a high solubility in the solvent. In this context, a strong acid is one that would rapidly hydrolyse the enol-ether function of the starting material, such as aliphatic organic acids (including acetic acid, which also has a high solubility in solvents such as toluene) and mineral acids. For example, when aliphatic acids, such as acetic acid, monochloro-acetic acid, ethylhexanoic acid and phenylacetic acid etc, were used in the reaction, the percentage formation of 10-methoxycarbamazepine was very poor, varying from 26% to 51%. Worse still, when mineral acids, such as hydrochloric acid and sulphuric acid, were tried, the percentage formation of 10-methoxycarbamazepine was even more poor (˜1%). In all the above reactions (ie when aliphatic acids or mineral acids were used), a significant percentage of 10-oxo-iminostilbene and impurities were formed. Table 1 below shows the results, using sodium cyanate in all reactions and 10 volumes of toluene per part of 10-methoxyiminostilbene.
| TABLE 1 | ||||||
| HPLC Analysis | ||||||
| % of 10- | % of | |||||
| Reflux | Conversion | methoxycarba- | Oxo- | Total | % of Unreacted | |
| Acid used | (hours) | (%) | mazepine | IDB | Impurity | 10-methoxy ISB |
| Hydrochloric acid | 4 | 89.63 | 0.24 | 70.19 | 19.19 | 10.37 |
| Sulphuric acid | 4 | 99.48 | 1.12 | 93.67 | 4.69 | 0.52 |
| Acetic acid | 12 | 59.05 | 26.22 | 12.97 | 19.86 | 40.95 |
| Monochloro-acetic acid | 12 | 96.32 | 51.5 | 24.00 | 20.82 | 3.68 |
| Ethylhexanoic acid | 22 | 44.14 | 22.86 | 12.93 | 8.35 | 55.86 |
| Benzoic acid | 12 | 98.00 | 75.50 | 9.10 | 13.40 | 2.00 |
| p-Chlorobenzoic acid | 12 | 99.66 | 56.44 | 20.00 | 23.22 | 0.34 |
| o-Chlorobenzoic acid | 12 | 98.13 | 31.25 | 54.77 | 12.11 | 1.87 |
| 2,4-Dichlorobenzoic acid | 6 | 98.48 | 55.45 | 30.04 | 12.99 | 1.52 |
| Phenylacetic acid | 6 | 72.88 | 34.38 | 18.36 | 20.14 | 27.12 |
On the contrary, when the aromatic acids such as mentioned above are used, the selectivity of the main reaction (ie the carboxamidation reaction as compared to hydrolysis of the enol-ether moiety) can increase to more than 75%. This results in improved efficiency and eventually in simpler methods of purification of the end product oxcarbazepine, resulting in easier commercialization of the process.
The carboxamidation of the 10-methoxyiminostilbene according to the present invention is preferably carried out in an organic medium, most preferably under reflux conditions. The organic medium is suitably an aromatic hydrocarbon solvent or an aliphatic chlorinated solvent, such as benzene, toluene, xylene, dichloromethane, chloroform and dichloroethane etc, including others described in relation to the Scheme-4 synthesis mentioned above and in WO 96/21649. The solvent(s) used in the carboxamidation reaction also play an important role in the selectivity and completion of reaction. We have found that toluene is the best solvent both in terms of selectivity and completion of reaction. It is important that the solvent is chosen such that the starting material and the HOCN are both soluble therein. Furthermore, as indicated above, it is important that the weak acid is relatively insoluble therein.
The HOCN reacts with the imino function to produce desired intermediate, 10-methoxycarbamazepine, which can afford the pharmacologically active end-product, ie oxcarbazepine, after hydrolysis.
The HOCN may be generated in situ by reaction of an alkali metal cyanate with the mild acidic reagent. Suitable cyanates include sodium and potassium, preferably sodium, cyanates. However, other methods of generating the HOCN, such as from cyanuric acid (as described in the Merck Index or by Linhard in Anorg Allgem Chem 236 200 (1938)) or other means may be used. Nevertheless, we have found that the method using sodium cyanate and an aromatic organic acid, especially benzoic acid, is commercially the most viable. In the preferred method of this invention, therefore, the mild acidic reagent is also capable of reacting with an alkali metal cyanate to produce cyanic acid (HOCN).
Accordingly, the present invention in a preferred aspect provides a process for the preparation of 10-methoxycarbamazepine, which process comprises reacting 10-methoxyiminostilbene with an alkali metal cyanate and a mild acidic reagent, as defined above.
Accordingly, the present invention further provides an improved method for preparing oxcarbazepine from 10-methoxystilbene, wherein the improvement comprises preparing the intermediate 10-methoxycarbamazepine according to the method described above.
The intermediate 10-methoxycarbamazepine is then preferably hydrolysed with an acid, more preferably a dilute mineral acid, such as hydrochloric and sulphuric acids, especially hydrochloric acid (HCI) to furnish oxcarbazepine. Finally, the oxcarbazepine thus obtained may be purified in a mixture of solvent systems selected from both a protic solvent with either an aromatic hydrocarbon solvent or a halogenated aliphatic solvent and an aromatic hydrocarbon solvent with a halogenated aliphatic solvent. Preferably, the mixed solvent system is one wherein the oxcarbazepine is soluble at elevated temperatures, suitably in the range of from 45 to 75° C., but crystallizes therefrom upon cooling. The oxcarbazepine may not be appreciably soluble in any of these solvents individually, but may be soluble in the mixture at elevated temperature. Examples of suitable mixtures include those such as methanol:toluene; dichloromethane:toluene; dichloroethane:toluene; dichloromethane:methanol; and dichloroethane:methanol.
Hydrolysis of the methoxycarbamazepine is preferably carried out in a biphasic system chosen such that the oxcarbazepine is substantially insoluble in both phases, whereas the by-products or impurities are soluble in at least one of the phases. The biphasic system comprises an organic phase and an aqueous phase in which the organic phase preferably comprises the solvent used in the carboxylation reaction eg toluene. Preferably, an excess of this solvent, compared with the amount of impurity or by-product to be produced, is used in the process of this invention. The preferred aqueous phase comprises an aqueous solution of the acid for the hydrolysis step and is therefore most preferably dilute hydrochloric acid. The advantage of this biphasic system is that oxcarbazepine formed in the reaction is thrown out from both the solvents, whereas the impurities remain soluble in the toluene.
Accordingly, the present invention further provides an improved method of hydrolyzing 10-methoxycarbazepine, which improvement comprises carrying out the hydrolysis in a biphasic system as described above.
Especially preferred is when both improved processes of the invention are used, consecutively. The improved processes of the invention enable the oxcarbazepine thereby produced to be purified in a single step.
An especially preferred method according to this invention comprises reaction of 10-methoxy-5H-dibenz[b,f]azepine with benzoic acid and sodium cyanate in toluene at reflux temperature to give 10-methoxy-5H-dibenz[b,f]azepine carboxamide as a major product (such as about 75%), along with 10-oxo-iminodibenzyl and other impurities. The reaction mixture is thereafter filtered and washed with water, and the toluene layer taken as such for hydrolysis in a biphasic system (aqueous hydrochloric acid/toluene) to furnish oxcarbazepine, which is purified just once (whereas twice at least is needed when the prior art process is carried out) in a mixture of methanol and dichloromethane (Scheme-5).
10-methoxyiminostilbene, the key starting material in the following Examples, maybe prepared according to the process disclosed in Belgian patent specification no. 597 793 and Swiss patent specification no. 392 515.
EXAMPLE A Using Monochloro-Acetic Acid and Sodium CyanateA mixture of 100 gms of 10-methoxyiminostilbene in 1000 mL of toluene containing 106 gms of monochloro-acetic acid and 73 gms of sodium cyanate were heated to 40° C. under stirring and maintained for 4 hours. After completion of the reaction (monitored by HPLC and/or TLC), the mixture was cooled to room temperature, filtered and washed with 5% sodium carbonate solution followed by water. The toluene layer was then added to 1000 mL of 2N hydrochloric acid, and the mixture was heated to 75-80° C. and maintained for 2 hours under good agitation. It was then cooled to 0-5° C. and maintained for 2 hours, and the product oxcarbazepine was separated by filtration. This was then purified twice in toluene:methanol followed by methanol:dichloromethane solvent mixture to furnish 28 gms of pure oxcarbazepine.
EXAMPLE 1 Using benzoic acid and sodium cyanateA mixture of 100 gms of 10-methoxyiminostilbene in 2000 mL of toluene containing 274 gms of benzoic acid and 370 gms of sodium cyanate were heated to reflux temperature under stirring and maintained for 12 hours. The reaction mixture was then cooled to room temperature and filtered. The clear toluene filtrate was washed with 5% sodium carbonate solution followed by water. The toluene layer was then added to 1000 mL of 2N hydrochloric acid and the mixture was heated at 75-90° C. for a period of 2 hours under good agitation. It was then cooled to 0-5° C., maintained for 2 hours and the product oxcarbazepine was separated by filtration. This was then purified once in a dichloromethane:methanol mixture to furnish 46 gms of pure oxcarbazepine. Purity was determined by HPLC to be 99.45%.
EXAMPLE 2 Using para-chlorobenzoic acid and sodium cyanateA mixture of 100 gms of 10-methoxyiminostilbene in 1000 mL of toluene containing 351 gms of para-chlorobenzoic acid and 370 gms of sodium cyanate were heated to reflux and refluxed for 12 hours. The reaction mixture was then cooled to room temperature and filtered. The clear toluene filtrate was then washed with 5% sodium carbonate solution followed by water. The toluene layer was then added to 1000 mL of 2N hydrochloric acid and the mixture was heated at 75-80° C. for a period of 2 hours under good agitation. It was then cooled to 0-5° C., maintained for 2 hours and the product oxcarbazepine was separated by filtration. This was then purified once in a dichloromethane methanol mixture to furnish 44 gms of pure oxcarbazepine.
EXAMPLE 3 Alternative Use of benzoic acid and sodium cyanateThe method of Example 1 was repeated, but using 1000 ml toluene; 164 g benzoic acid and 44 g of sodium cyanate, which were heated to 85-90° C. for 14 hours with the 10-methoxyiminostilbene to result in 55 gms of pure oxcarbazepine, found to be 99.45% pure by HPLC.
EXAMPLE 4 Using 2,4-dichloro benzoic acid and sodium cyanateA mixture of 100 gms of 10-methoxyiminostilbene in 1000 mL of toluene containing 430 gms of 2,4-dichlorobenzoic acid and 370 gms of sodium cyanate were heated to reflux and refluxed for 6 hours. The reaction mixture was then cooled to room temperature and filtered. The clear toluene filtrate was then washed with 5% sodium carbonate solution followed by water. The toluene layer was then added to 1000 mL of 2N hydrochloric acid and the mixture was heated at 75-80° C. for a period of 2 hours under good agitation. It was then cooled to 0-5° C., maintained for 2 hours and the product oxcarbazepine was separated by filtration. This was then purified once in a dichloromethane:methanol mixture to furnish 40 gms of pure oxcarbazepine.
EXAMPLE 5 Using benzoic acid and potassium cyanateThe method was carried out according to that described in Example 1, but replacing sodium cyanate with potassium cyanate (461.5 gm) and reflux maintained for 24 hrs to complete consumption of starting material. Following the similar process for hydrolysis and purification produced 32.00 gm of pure oxcarbazepine. Purity was determined according to Example 1 and found 98.80%.
EXAMPLE 6The method was carried out according to that described in Example 1, but replacing 2N hydrochloric with 2N sulphuric acid (1000 mL). Following a similar process of carboxamidation and purification produced 25.00 gm of pure oxcarbazepine. Purity was determined according to Example 1 and found 98.50%.
EXAMPLE 7 Hydrolysis Step Using 2N monochloro-acetic acidThe method was carried out according to that described in Example 1, but replacing 2N hydrochloric acid with 2N monochloro-acetic acid (1000 mL). The reaction mixture was heated to 75° C. to 80° C. and maintained for 24 hrs (after which 20% of unreacted methoxy ISB was found to be present). Under similar conditions for the carboxamidation reaction and purification step, this comparative Example produced 20.00 gm of pure oxcarbazepine. Purity was determined according to Example 1 and found to be 98.00%.
EXAMPLE 8 Purification Using toluene:methanol solvent systemThe method was carried out according to that described in Example 1, but replacing dichloromethane with toluene. Following a similar process of carboxamidation and hydrolysis produced 47.0 gm of pure oxcarbazepine. Purity was determined according to Example 1 and found 98.50%.
EXAMPLE 9 Purification Using toluene:dichloromethane solvent systemThe method was carried out according to that described in Example 1, but replacing methanol with toluene. Following a similar process of carboxamidation and hydrolysis produced 45.00 gm of pure oxcarbazepine. Purity was determined according to Example 1 and found to be 98.00%.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1996021649A1 | Jan 3, 1996 | Jul 18, 1996 | Trifarma, S.R.L. | A PROCESS FOR THE PREPARATION OF 10-OXO-10,11-DIHYDRO-5H-DIBENZ(b,f)AZEPIN-5-CARBOXAMIDE |
| WO2001056992A2 | Feb 7, 2001 | Aug 9, 2001 | Novartis Ag | Dibenzo (b,f) azepine intermediates |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7091339 | Jun 13, 2003 | Aug 15, 2006 | Taro Pharmaceuticals Usa, Inc. | Method of preparing a 5H-dibenz(b,f)azepine-5-carboxamide |
| US7125987 | Jun 16, 2005 | Oct 24, 2006 | Apotex Pharmachem Inc. | Process for the preparation of oxcarbazepine and related intermediates |
| US7183272 | Feb 12, 2002 | Feb 27, 2007 | Teva Pharmaceutical Industries Ltd. | Crystal forms of oxcarbazepine and processes for their preparation |
| US7459553 | Mar 11, 2005 | Dec 2, 2008 | Glenmark Generics Ltd. | Process for the preparation of carboxamide compounds |
| US7722898 | Apr 13, 2007 | May 25, 2010 | Supernus Pharmaceuticals, Inc. | Modified-release preparations containing oxcarbazepine and derivatives thereof |
| US7723514 | Jun 29, 2006 | May 25, 2010 | Taro Pharmaceuticals U.S.A., Inc. | Method of preparing a 5H-dibenz(b,f)azepine-5-carboxamide |
| US8530647 | May 6, 2009 | Sep 10, 2013 | Mylan Laboratories Limited | Process for the preparation of oxcarbazepine |
| US20030004154 * | Feb 12, 2002 | Jan 2, 2003 | Judith Aronhime | New crystal forms of oxcarbazepine and processes for their preparation |
| US20040044200 * | Jun 13, 2003 | Mar 4, 2004 | Daniella Gutman | Method of preparing a 5H-dibenz(b,f)azepine-5-carboxamide |
| US20050203297 * | Mar 11, 2005 | Sep 15, 2005 | Sivakumar Bobba V. | Process for the preparation of carboxamide compounds |
| US20050282797 * | Jun 16, 2005 | Dec 22, 2005 | Apotex Pharmachem Inc. | Process for the preparation of oxcarbazepine and related intermediates |
| US20060241292 * | Jun 29, 2006 | Oct 26, 2006 | Taro Pharmaceuticals Usa, Inc | Method of preparing a 5H-dibenz(b,f)azepine-5-carboxamide |
| US20070254033 * | Apr 13, 2007 | Nov 1, 2007 | Supernus Pharmaceuticals, Inc. | Modified-release preparations containing oxcarbazepine and derivatives thereof |
| US20110065917 * | May 6, 2009 | Mar 17, 2011 | Matrix Laboratories Ltd | process for the preparation of oxcarbazepine |
| WO2009139001A2 * | May 6, 2009 | Nov 19, 2009 | Matrix Laboratories Ltd | An improved process for the preparation of oxcarbazepine |
| WO2009139001A3 * | May 6, 2009 | Jan 27, 2011 | Matrix Laboratories Ltd | An improved process for the preparation of oxcarbazepine |
| WO2014049550A1 | Sep 26, 2013 | Apr 3, 2014 | Ranbaxy Laboratories Limited | Process for the preparation of oxcarbazepine and its use as intermediate in the preparation of eslicarbazepine acetate |
////////////
OLINCIGUAT
cas 1628732-62-6
Chemical Formula: C21H16F5N7O3
UNII-PD5F4ZXD21
Molecular Weight: 509.4
Olinciguat is a guanylate cyclase activator drug candidate.
(2R)-3,3,3-trifluoro-2-{[(5-fluoro-2-{1-[(2-fluorophenyl)methyl]- 5-(1,2-oxazol-3-yl)-1H-pyrazol- 3-yl}pyrimidin-4-yl)amino]methyl}-2-hydroxypropanamide
![]()
Currently in Phase II Clinical Development
Achalasia and Sickle Cell Disease
Dysregulation of the nitric oxide-soluble guanylate cyclase-cyclical guanosine monophosphate (NO-sGC-cGMP) signaling pathway is believed to be linked to multiple vascular and fibrotic diseases, such as achalasia and sickle cell disease.
IW-1701 is an investigational soluble guanylate cyclase (sGC) stimulator from Ironwood’s diverse library of sGC stimulators, which are being investigated for their potential effects on vascular and fibrotic diseases. The compound has been shown in nonclinical studies to modulate the NO-sGC-cGMP signaling pathway and is currently being evaluated in a Phase II study in achalasia. IW-1701 is wholly-owned by Ironwood Pharmaceuticals.
 IW-1701 is being evaluated to determine its potential effects on vascular and fibrotic diseases.
IW-1701 is being evaluated to determine its potential effects on vascular and fibrotic diseases.
sGC is the primary receptor for NO in vivo. sGC can be activated via both NO-dependent and NO-independent mechanisms. In response to this activation, sGC converts Guanosine-5′-triphosphate (GTP) into the secondary messenger cGMP. The increased level of cGMP, in turn, modulates the activity of downstream effectors including protein kinases, phosphodiesterases (PDEs) and ion channels.
In the body, NO is synthesized from arginine and oxygen by various nitric oxide synthase (NOS) enzymes and by sequential reduction of inorganic nitrate. Three distinct isoforms of NOS have been identified: inducible NOS (iNOS or NOS II) found in activated macrophage cells; constitutive neuronal NOS (nNOS or NOS I), involved in neurotransmission and long term potentiation; and constitutive endothelial NOS (eNOS or NOS III) which regulates smooth muscle relaxation and blood pressure. Experimental and clinical evidence indicates that reduced concentrations orbioavailability of NO and/or diminished responsiveness to endogenously produced NO contributes to the development of disease.
NO-independent, heme -dependent sGC stimulators, have shown several important differentiating characteristics, when compared to sGC activators, including crucial dependency on the presence of the reduced prosthetic heme moiety for their activity, strong synergistic enzyme activation when combined with NO and stimulation of the synthesis of cGMP by direct stimulation of sGC, independent of NO. The benzylindazole compound YC-1 was the first sGC stimulator to be identified. Additional sGC stimulators with improved potency and specificity for sGC have since been developed.
Compounds that stimulate sGC in an NO-independent manner offer considerable advantages over other current alternative therapies that target the aberrant NO pathway. There is a need to develop novel, well-characterized stimulators of sGC. Compound I is an sGC stimulator that has demonstrated efficacy for the treatment of a number of NO related disorders in preclinical models. Compound I was previously described in WO2014144100, Example 1, as a light orange solid. Compound I may be present in various crystalline forms and may also form several pharmaceutically acceptable salts.
Compounds which enhance eNOS transcription: for example those described in WO
02/064146, WO 02/064545, WO 02/064546 and WO 02/064565, and corresponding patent documents such as US2003/0008915, US2003/0022935, US2003/0022939 and US2003/0055093. Other eNOS transcriptional enhancers including those described in US20050101599 (e.g. 2,2-difluorobenzo[l,3]dioxol-5-carboxylic acid indan-2-ylamide, and 4-fluoro-N-(indan-2-yl)-benzamide), and Sanofi-Aventis compounds AVE3085 and AVE9488 (CA Registry NO. 916514-70-0; Schafer et al., Journal of Thrombosis and Homeostasis 2005; Volume 3, Supplement 1 : abstract number P 1487);
NO independent heme-independent sGC activators, including, but not limited to: -2667 (see patent publication DE19943635)

HMR-1766 (ataciguat sodi

S 3448 (2-(4-chloro-phenylsulfonylamino)-4,5-dimethoxy-N-(4-(thiomoφholine-4-sulfonyl)-phenyl)-benzamide (see patent publi

HMR-1069 (Sanofi-Aventis).
(7) Heme-dependent sGC stimulators including, but not limited to:
YC-1 (see patent publications EP667345 and DE19744026)

Riociguat (BAY 63-2521, Adempas, commercial product, described in DE19834044)

Neliciguat (BAY 60-4552, described in WO 2003095451)

Vericiguat (BAY 1021189, clinical backup to Riociguat),
BAY 41-2272 (described in DE19834047 and DE19942809)

BAY 41-8543 (described in DE I 9834044)

Etriciguat (described in WO 2003086407)

CFM-1571 (see patent publicatio
A-344905, its acrylamide analo analogue A-778935.

A-344905;

Compounds disclosed in one of publications: US20090209556, US8455638, US20110118282 (WO2009032249), US20100292192, US20110201621, US7947664, US8053455 (WO2009094242), US20100216764, US8507512, (WO2010099054) US20110218202 (WO2010065275),
US20130012511 (WO2011119518), US20130072492 (WO2011149921), US20130210798
(WO2012058132) and other compounds disclosed in Tetrahedron Letters (2003), 44(48): 8661-8663.
Pictorial synthesis
FROM PATENTS
CONSTRUCT YOUR OWN




SIDE CHAIN SHOWN ABOVE

FINAL STEP SHOWN ABOVE OLINCIGUAT










PATENT
WO2014144100, Example 1
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016044447
Compound 195

lntermediate-36 Compound 195
[00463] lntermediate-36 (35 mg, 0.09 mmol),
(R)-2-(aminomethyl)-3,3,3-trifluoro-2-hydroxypropanamide (60 mg, 0.35 mmol) and
N-ethyl-N-isopropylpropan-2-amine (0.10 mL, 0.56 mmol) were mixed in dimethylsulfoxide (1.5 mL) and heated at 95°C for 8 hr. The solution was cooled to room temperature, diluted with water (2 mL) and the pH taken to 2-3 with 1 N (aq) HC1. The solution was mixed with ethyl acetate (50 mL) and the organic phase was washed with water (2 x 5 mL), brine, then dried over Na2S04, filtered and concentrated by rotary evaporation. The residue was subjected to preparative reverse phase HPLC
. . . t . + + . using a giauiciu ui water acetonitri e . tni uoroacetic aci as e uant to give me iouu i s a wnite solid (11 mg, 23% yield). ¾-NMR (400 MHz, CD3OD) δ 8.83 (br s, 1H), 8.27 (br s, 1H), 7.49 (br s,
1H), 6.9-7.0 (m, 2H), 6.5-6.6 (m, 2H), 5.86 (s, 2H), 4.35 (d, 1H), 4.16 (d, 1H) ppm. Note: exchangable protons all appeared under the residual HOD peak at 4.91 ppm.
PATENT
WO-2018009609
Novel crystalline solid forms of olinciguat (presumed to be IW-1701), an SGC stimulator and their salts, such as hydrochloride acid (designated as Forms A, B, D, E, F, H and G), processes for their preparation and compositions comprising them are claimed. Also claimed are processes for preparing the crystalline forms. Further claimed are their use for treating cancer, sickle cell disease, osteoporosis, dyspepsia, Duchenne muscular dystrophy, amyotrophic lateral sclerosis and spinal muscle atrophy
In one aspect, the invention relates to crystalline solid forms of Compound I, depicted below:

Compound I
[0009] For purposes of this disclosure, “Compound I,” unless otherwise specifically indicated, refers to the free base or to the hydrochloric acid salt of the structure denoted above. Compound I, as its crystalline free base, is highly polymorphic and known to have seven crystalline forms (Forms A, B, D, E, F, G and H) as well as multiple solvates. Compound I was previously described in
WO2014144100, Example 1, as a light orange solid.
[0010] In one embodiment, the crystalline solid forms of Compound I here disclosed are polymorphs of the free base. In another embodiment, a crystalline solid form of Compound I is the hydrochloric acid salt. In one embodiment, the polymorphs of Compound I are crystalline free base forms. In another embodiment, they are solvates.
[001 1] In another aspect, also provided herein are methods for the preparation of the above described crystalline free forms and salts of Compound I.
[0012J In another aspect, the invention relates to pharmaceutical compositions comprising one or more of the polymorphs of Compound I herein disclosed, or the hydrochloric acid salt of Compound I, and at least one pharmaceutically acceptable excipient or carrier. In another embodiment, the invention relates to pharmaceutical dosage forms comprising said pharmaceutical compositions.
[0013] In another embodiment, the invention relates to a method of treating a disease, health condition or disorder in a subject in need thereof, comprising administering, alone or in combination therapy, a therapeutically effective amount of a polymorph of Compound I herein disclosed, or a mixture of polymorphs thereof, or its hydrochloric acid salt , to the subject; wherein the disease or disorder is one that may benefit from sGC stimulation or from an increase in the concentration of NO and/or cGMP.
EXAMPLES
Example 1: Preparation of crude Compound I
i): Coupling of Compound (1′) and 7V,0-Dimethylhydroxylamine to provide N-methoxy-N-methylisoxazole-3-carboxamide (2′)
[00238] Isooxazole-3-carboxylic acid ((l’)> 241.6 g, 2137 mmoles, 1.0 equiv.), toluene (1450 mL) and DMF (7.8 g, 107 mmoles, 0.05 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The resulting slurry was heated to 45-50 °C. Oxalyl chloride (325 g, 2559 mmoles, 1.2 equiv.) was then charged via an addition funnel over the course of 2 h while maintaining the reaction temperature between 45 to 50 °C and a vigorous gas evolution was observed. A brown mixture was obtained after addition. The brown mixture was heated to 87 to 92 °C over 1 h and stirred at 87 to 92 °C for 1 h. The reaction was completed as shown by HPLC. During heating, the brown mixture turned into a dark solution. The reaction was monitored by quenching a portion of the reaction mixture into piperidine and monitoring the piperidine amide by HPLC. The dark mixture was cooled to 20-25 °C and then filtered through a sintered glass funnel to remove any insolubles. The dark filtrate was concentrated under reduced pressure to a volume of 400 mL dark oil.
[00239] Potassium carbonate (413 g, 2988 mmoles, 1.4 equiv.) and water (1000 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The reaction solution was cooled to -10 to -5 °C. N,0-dimethylhydroxyamine hydrochloride (229 g, 2348 mmoles, 1.1 equiv.) was charged to a suitable reaction vessel and dissolved in water (1000 mL). The N,0-dimethylhydroxyamine solution and dichloromethane (2500 mL) were then charged to the potassium carbonate solution.
[00240] The above dark oil (400 mL) was then charged slowly via an addition funnel while maintaining the reaction temperature -10 to 0 °C. The addition was slightly exothermic and a brown mixture was obtained after addition. The mixture was stirred at 0 to 5 °C over 20 min. and then warmed to 20 to 25 °C. The bottom organic layer was collected and the top aq. layer was extracted with dichloromethane (400 mL). The combined organic layers were washed with 15% sodium chloride solution (1200 mL). The organic layer was dried over magnesium sulfate and then filtered. The filtrate was concentrated under reduced pressure to give intermediate (2′) as a dark oil (261.9 g, 97 wt%, 76% yield, 3 wt% toluene by Ή-ΝΜΡν, 0.04 wt % water content by KF). Ή-ΝΜΡν (500 MHz, CDC13) δ ppm 8.48 (s, 1 H); 6.71(s, 1 H); 3.78 (s, 3 H); 3.38 (s, 3 H).
ii): alkylation of Compound (2′) and ethyl propiolate to provide (E)-ethyl 4-(isoxazol-3-yl)-2-(methox methyl)amino)-4-oxobut-2-enoate (3′)
(2′) (3′)
[00241] Intermediate (2′) (72.2 g, 96 wt%, 444 mmoles, 1.0 equiv.), ethyl propiolate (65.7 g, 670 mmoles, 1.5 equiv.) and anhydrous THF (650 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The solution was cooled to -65 to -55 °C. Sodium bis(trimethylsilyl)amide in THF (1 M, 650 mL, 650 mmoles, 1.46 equiv.) was then charged slowly via an addition funnel while maintaining the reaction temperature -65 to -55 °C. The mixture was stirred below -55 °C over 10 min. after addition was complete. Then 1 N HC1 (650 mL, 650 mmoles, 1.46 equiv.) was charged to quench the reaction while maintaining the reaction temperature below -20 °C followed immediately with the addition of ethyl acetate (1500 mL) and water (650 mL). The top ethyl acetate layer was collected and the bottom aqueous layer was extracted with ethyl acetate (800 mL). The combined organic layers were washed with 10% citric acid (1000 mL) and saturated sodium chloride solution (650 mL). The organic layer was concentrated under reduced pressure to give a dark oil.
[00242] The dark oil was dissolved in a solution of dichloromethane/ethyl acetate/heptane
(150mL/100mL/100mL). The solution was loaded on a silica pad (410 g) and the silica pad was eluted with ethyl acetate/heptane (1/1 v/v). The filtrate (~ 3000 mL) was collected and then concentrated under reduced pressure to a volume of 150 mL to give a slurry upon standing. Heptane (200 mL) was then added to the slurry and the slurry was concentrated under reduced pressure to a volume of 150 mL. The resulting slurry was filtered, and the filter cake was washed with heptane (150 mL). The filter cake was then air dried overnight to furnish intermediate (3′) as a brown solid (63.4 g, 56% yield, >99% pure by HPLC). i-NMR (500 MHz, CDC13) δ ppm 8.42 (d, J=1.53 Hz, 1 H); 6.76 (d, J=1.53 Hz, 1 H); 6.18 (s, 1 H); 4.47 (q, J=7.07 Hz, 2H); 3.75 (s, 3 H); 3.21 (s, 3 H); 1.41 (t, J=7.17 Hz, 3 H). iii): cyclization of Compound 3′ and 2-fluorobenzylhydrazine to provide ethyl l-(2-fluorobenz l)-5-(isoxazol-3-yl)-lH-pyrazole-3-carboxylate (4′)
[00243] Intermediate (3′) (72.9 g, 287 mmoles, 1.0 equiv.) and absolute ethanol (730 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The mixture was cooled to 0 to 5 °C. 2-Fluorobenzylhydrazine (48.2 g, 344 mmoles, 1.2 equiv.) was then charged to the mixture. The mixture was stirred at 0 to 10 °C over 1 h and then warmed to 20 to 25 °C and stirred at 20 to 25 °C over 16 h. The reaction was completed by HPLC. Concentrated HCl (33.9 g, 37 wt%, 344 mmoles, 1.2 equiv.) was charged to the reaction mixture over 1 min and the batch temperature exothermed from 20 °C to 38 °C. A slurry was obtained. The mixture was cooled to 0 to 10 °C over 1 h and stirred at 0-10 °C for 1 h. The resulting slurry was filtered, and the filter cake was washed with ethanol (200 mL). The filter cake was dried under vacuum at 30 to 40 °C over 16 h to furnish intermediate (4′) as an off-white solid (81.3 g, 90% yield, >99% pure by HPLC). ¾-NMR (500 MHz, CDC13) δ ppm 8.47 (d, J=1.68 Hz, 1 H); 7.15 – 7.26 (m, 2 H); 6.94 – 7.08 (m, 2H); 6.77 – 6.87 (m, 1 H); 6.55 (d, J=1.68 Hz, 1 H); 5.95 (s, 2 H); 4.43 (q, J=7.02 Hz, 2 H); 1.41 (t, J=7.17 Hz, 3 H).
iv): amination of Compound (4′) to provide l-(2-fluorobenzyl)-5-(isoxazol-3-yl)-lH-pyrazole-3-carboximidamide hydrochloride (5’B)
[00244] Anhydrous ammonium chloride (267 g, 4991 mmoles, 5.0 equiv.) and toluene (5400 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. Trimethylaluminum in toluene (2 M, 2400 mL, 4800 mmoles, 4.8 equiv.) was charged
slowly via an addition funnel while maintaining the reaction temperature at 20 to 40 °C (Note:
Methane gas evolution was observed during addition). Then the mixture was heated to 75 to 80 °C over 30 min. and a clear white solution was obtained. Intermediate (4′) (315 g, 999 mmoles, 1.0 equiv.) was charged to reaction mixture in four equal portions over 1 h at 75 to 90 °C. The reaction was stirred at 80 to 90 °C over 30 min. and then heated to 100 to 110 °C and stirred at 100 to 110 °C over 3 h. The reaction was completed by HPLC. The reaction mixture was cooled to 10 to 20 °C and methanol (461 g, 14.4 moles, 14.4 equiv.) was charged slowly via an addition funnel while
maintaining the reaction temperature 10-40 °C. Note the quenching was very exothermic and a lot gas evolution was observed. A thick slurry was obtained. A 3N HQ (6400 mL, 3 N, 19.2 moles, 19.2 equiv.) was then charged slowly via an addition funnel while maintaining the reaction temperature at 20 to 45 °C. The mixture was heated to 80 to 85 °C and stirred at 80 to 85 °C over 10 min. to obtain a clear biphasic mixture. The mixture was cooled to 0 to 5 °C over 3 h and stirred at 0 to 5 °C over 1 h. The resulting slurry was filtered, and the filter cake was washed with water (3000 mL). The filter cake was dried under vacuum at 40 to 50 °C over 24 h to furnish intermediate (5’B) as an off-white solid (292 g, 91% yield, >99% pure by HPLC). ¾-ΝΜΡν (500 MHz, DMSO- 6) δ ppm 9.52 (s, 2 H); 9.33 (s, 2 H); 9.18 (d, J=1.53 Hz, 1 H); 7.88 (s, 1 H); 7.29 – 7.38 (m, 1 H); 7.19 – 7.25 (m, 1 H); 7.10 – 7.16 (m, 1 H); 7.03 (d, J=1.53 Hz, 1 H); 6.92 – 6.98 (m, 1 H); 5.91 (s, 2 H). M.P. 180-185 °C.
v): cyclization of Compound (5’B) and diethyl fluoromalonate to provide 5-fluoro-2-(l-(2-fluorobenz l)-5-(isoxazol-3-yl)-lH-pyrazol-3-yl)pyrimidine-4,6-diol (6′)
(5’B) (6·)
[00245] Intermediate (5’B) (224.6 g, 698 mmoles, 1.0 equiv.), methanol (2250 mL) and diethyl fluoromalonate (187 g, 1050 mmoles, 1.5 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. Then sodium methoxide in methanol solution (567 g, 30 wt %, 3149 mmoles, 4.5 equiv.) was charged via an addition funnel while maintaining the reaction temperature 20 to 35 °C. The mixture was stirred at 20 to 35 °C over 30 min. and a light suspension was obtained. The reaction was completed by HPLC. A solution of 1.5 N HQ (2300 mL, 3450 mmoles, 4.9 equiv.) was charged via an addition funnel over 1 h while maintaining the reaction temperature 20 to 30 °C. A white suspension was obtained. The pH of the reaction mixture was to be ~1 by pH paper. The slurry was stirred at 20 to 30 °C over 30 min. The resulting slurry was filtered, and the filter cake was washed with a pre-mixed solution of methanol and water (500 mL/500 mL), and then with water (1000 mL). The filter cake was dried under vacuum at 50 to 60 °C over 16 h to furnish intermediate (6′) as an off-white solid (264 g, 97% yield, >99% pure by HPLC). ¾-NMR (500 MHz,
DMSO- s) δ ppm 12.82 (br. s., 1 H); 12.31 (br. s., 1 H); 9.14 (d, J=1.53 Hz, 1 H); 7.55 (s, 1 H); 7.31 -7.37 (m, 1 H); 7.18 – 7.25 (m, 1 H); 7.10 – 7.15 (m, 2 H); 6.97 – 7.02 (t, J=7.55 Hz, 1 H); 5.88 (s, 2 H).
vi): chlorination of Compound (6′) to provide 3-(3-(4,6-dichloro-5-fluoropyrimidin-2-yl)-l-(2-fluorobenz l)-lH-pyrazol-5-yl)isoxazole (7′)
(6«) (7«)
[00246] Intermediate (6′) (264 g, 71 1 mmoles, 1.0 equiv.), acetonitrile (4000 mL) and N,N-dimethylaniline (138 g, 1 137 mmoles, 1.6 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The slurry mixture was heated to 70-80 °C. Then phosphorous oxychloride (655 g, 4270 mmoles, 6.0 equiv.) was charged via an addition funnel over 1 h while maintaining the reaction temperature 70 to 80 °C. The mixture was stirred at 75 to 80 °C over 22 h and a brown solution was obtained. The reaction was completed by HPLC. Then the mixture was cooled to between 0 and 5 °C and cotton like solids precipitated out at 25 °C. Water (3000 mL) was charged slowly via an addition funnel while maintaining the reaction temperature at 0 to 10 °C. The slurry was stirred at 0 to 10 °C over 30 min. The resulting slurry was filtered, and the filter cake was washed with a pre-mixed solution of acetonitrile and water (500 mL/500 mL). The filter cake was dried under vacuum at 35 to 45 °C over 16 h to furnish intermediate (7′) as an off-white solid (283 g, 98% yield, >99% pure by HPLC). ‘H-NMR (500 MHz, CDC13) δ ppm 8.48 (d, J=1.68 Hz, 1 H); 7.44 (s, 1 H); 7.19 – 7.25 (m, 1 H); 6.96 – 7.08 (m, 2 H); 6.81 – 6.88 (m, 1 H); 6.60 (d, J=1.68 Hz, 1 H); 6.03 (s, 2 H).
vii): substitution of Compound (7′) with meth oxide to provide 3-(3-(4-chloro-5-fluoro-6-m
(7′) (8′)
[00247] Methanol (3400 mL) and sodium methoxide in methanol (154 mL, 5.4 M, 832 mmoles,
1.2 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The reaction mixture was heated to 23 to 27 °C. Intermediate (7′) (283 g, 693 mmoles, 1.0 equiv.) was charged to the mixture in small portions (5-10 g each portion) over 40 min while maintaining the reaction temperature 23 to 27 °C. The slurry was stirred at 23 to 27 °C over 30 min. The reaction was completed by HPLC. The resulting slurry was filtered, and the filter cake was washed with methanol (850 mL) and then water (850 mL). The filter cake was dried under vacuum at 35 to 45 °C over 16 h to furnish intermediate (8′) as an off-white solid (277 g, 99% yield, 97% pure by HPLC). i-NMR (500 MHz, CDCl3) 5 ppm 8.47 (d, J=1.83 Hz, 1 H); 7.38 (s, 1 H); 7.18 – 7.25 (m, 1 H); 7.01 – 7.08 (m, 1 H); 6.94 – 7.00 (m, 1 H); 6.81 – 6.88 (m, 1 H); 6.60 (d, J=1.68 Hz, 1 H); 6.00 (s, 2 H); 4.21 (s, 3 H).
viii): hydrogenation of Compound (8′) to provide 3-(3-(5-fluoro-4-methoxypyrimidin-2-yl)-l-(2-fluorobenz l)-lH-pyrazol-5-yl)isoxazole (9′)
[00248] Intermediate (8′) (226 g, 560 mmoles, 1.0 equiv.), palladium (10% on activated carbon, nominally 50% water wet, 22.6 g, 0.01 moles, 0.018 equiv), tetrahydrofuran (3400 mL) and triethylamine (91 g, 897 mmoles, 1.6 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. Nitrogen was bubbled into the reaction mixture via teflon tubing over 10 min. at 20 to 30 °C. Then the mixture was heated to 40 to 50 °C and hydrogen gas was bubbled into the reaction mixture via teflon tubing over 6 h while maintaining the reaction temperature 40 to 50 °C. The reaction was completed by HPLC. Nitrogen was then bubbled into the reaction mixture via teflon tubing over 10 min. at 40 to 50 °C The reaction mixture was hot filtered through Hypo Supercel and the filter cake was washed with tetrahydrofuran (2000 mL). The filtrate was concentrated under reduced pressure to a volume of -1300 mL to give a slurry. Tetrahydrofuran was then solvent exchanged to methanol under reduced pressure via continuously feeding methanol (3000 mL). The final volume after solvent exchange was 1300 mL. The resulting slurry was filtered, and the filter cake was washed with methanol (500 mL). The filter cake was dried under vacuum at 20 to 25 °C over 16 h to furnish intermediate (9′) as a white solid (192 g, 93% yield, 98% pure by HPLC). ¾-NMR (500 MHz, CDC13) δ ppm 8.47 (d, J=1.68 Hz, 1 H); 8.41 (d, J=2.59 Hz, 1 H); 7.36 (s, 1 H); 7.17 – 7.24 (m, 1 H); 6.95 – 7.07 (m, 2 H); 6.83 – 6.90 (m, 1 H); 6.60 (d, J=1.68 Hz, 1 H); 5.99 (s, 2 H); 4.19 (s, 3 H).
ix: demethylation of Compound (9′) to provide 5-fluoro-2-(l-(2-fluorobenzyl)-5-(isoxazol-3-yl)-lH-pyrazol-3-yl)pyrimidin-4-ol (10′)
[00249] Intermediate (9′) (230 g, 623 mmoles, 1.0 equiv.), Me OH (3450 mL) and cone. HC1
(307 g, 37 wt%, 3117 mmoles, 5.0 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The mixture was heated to 60 to 65 °C and a solution was obtained. The mixture was then stirred at 60 to 65 °C over 17 h and a slurry was obtained. The reaction was completed by HPLC. The slurry was cooled to 20 to 25 °C over 2 h and stirred at 20 to 25 °C over 30 min. The resulting slurry was filtered, and the filter cake was washed with methanol (1000 mL). The filter cake was dried under vacuum at 35 to 45 °C over 16 h to furnish intermediate (10′) as a white solid (214 g, 97% yield, >99% pure by HPLC). ¾-NMR (500 MHz, DMSO-t/6) δ ppm 12.90 – 13.61 (br. s., 1 H); 9.11 (d, J=1.68 Hz, 1 H); 8.16 (s, 1 H); 7.64 (s, 1 H); 7.29 – 7.42 (m, 1 H); 7.17 – 7.28 (m, 2 H); 7.08 – 7.15 (m, 1 H); 6.97 (s, 1 H); 5.91 (s, 3 H).
x): chlorination of Compound (10′) to provide 3-(3-(4-chloro-5-fluoropyrimidin-2-yl)-l-(2-fluorobenzyl)-lH-pyrazol-5-yi)isoxazole (Formula IV)
Formula IV
[00250] Intermediate (10′) (214 g, 602 mmoles, 1.0 equiv.), acetonitrile (3000 mL) and NN-dimethylaniline (109 g, 899 mmoles, 1.5 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The slurry mixture was heated to 70 to 80 °C. Then phosphorous oxychloride (276 g, 1802 mmoles, 3.0 equiv.) was charged via an addition funnel over 30 min. while maintaining the reaction temperature 70-80 °C. The mixture was stirred at 75 to 80 °C over 2 h and a green solution was obtained. The reaction was completed by HPLC. Then the mixture was cooled to 0 to 5 °C. Water (1500 mL) was charged slowly via an addition funnel while maintaining the reaction temperature at 0 to 10 °C. The slurry was stirred at 0 to 10 °C over 30 min. The resulting slurry was filtered, and the filter cake was washed with a pre-mixed solution of
acetonitrile and water (500 mL/500 mL) and water (500 mL). The filter cake was dried under vacuum at 30 to 40 °C over 16 h to furnish intermediate of Formula IV as an off-white to pink solid (214 g, 95% yield, >99% pure by HPLC). 1H NMR (500 MHz, CDC13) 5 ppm 8.65 (s, 1 H); 8.48 (d, J=1.68 Hz, 1 H); 7.44 (s, 1 H); 7.21 – 7.25 (m, 1 H); 6.97 – 7.06 (m, 2 H); 6.83 – 6.87 (m, 1 H); 6.61 (d, J=1.68 Hz, 1 H); 6.03 (s, 2 H).
a): Cyanation of intermediate (15) to provide 2-(bromomethyl)-3,3,3-trifluoro-2-((trimethylsilyl)oxy)propanenitrile (16)
(15) (16)
[00251 ] Trimethylsilanecarbonitrile ( 153 g, 1.54 moles, 0.97 equiv) and triethylamine (4.44 mL,
3.22 g, 0.032 mole, 0.02 equiv) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The mixture was cooled to 5 °C. 3-Bromo-l, l, l-trifluoropropan-2-one ((15), 304 g, 1.59 moles, 1.0 equiv) was charged via an addition funnel over 35 min, while maintaining the reaction temperature between 10 to 20 °C. The mixture was stirred at 20 to 30 °C over 3 h after the addition to furnish intermediate (16) as a dense oil which was used directly in the next step. 1H-NMR (500 MHz, CDC13) δ ppm 3.68 (d, J=1 1.14 Hz, 1 H); 3.57 (d, J=11.14 Hz, 1 H), 0.34 – 0.37 (m, 9 H).
b): Conversion of nitrile Compound (16) to amide to provide 2-(bromomethyl)-3,3,3-trifluoro-2-hydroxypropanamide (17)
2
(16) (17)
[00252] Concentrated sulfuric acid (339 mL, 6.37 moles, 4.0 equiv) was stirred in a suitable reaction vessel equipped with a mechanical stirrer, digital thermometer and an addition funnel. The sulfuric acid was heated to 45 °C. The above intermediate (16) was added via an addition funnel over 50 min, while keeping the temperature below 75 °C. The reaction mixture was stirred at 75 °C for 2 h and then allowed to cool to room temperature. ¾-NMR indicated reaction complete. The reaction mixture was cooled to -15 °C and diluted with ethyl acetate (1824 mL) via an addition funnel over 45 min (very exothermic), while keeping the temperature between -15 to 5 °C. Water ( 1520 mL) was added slowly via an addition funnel for 1 h 20 min. (very exothermic) between -10 to 0 °C. The layers were separated and the organic layer was washed with 15% aqueous sodium chloride solution ( 1520
mL), 25% aqueous sodium carbonate solution (911 mL) followed by 15% aqueous sodium chloride solution (911 mL). The organic layer was filtered and concentrated under reduced pressure to get 348 g of intermediate (17) as light yellow oil. This oil was dissolved in methanol (1200 mL) and concentrated to furnish 380 g of intermediate (17). (296 g adjusted weight, 79% yield). i-NMR (500 MHz, CDC13) 5 6.61 – 6.94 (m, 1 H); 5.92 – 6.26 (m, 1 H); 3.93 – 4.00 (m, 1 H); 3.68 (d, J=l 1.14 Hz, 1 H).
c): N-Alkylation of compound (17) to provide of 2-(aminomethyl)-3,3,3-trifluoro-2-hydroxypropanamide (14)
(17) (14)
[00253] A 7 N solution of ammonia in methanol (600 mL, 4.28 moles, 10 equiv) was charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The solution was cooled to 0 to 5 °C. Then the intermediate (17) (102 g, 0.432 moles, 1 equiv) was added via an addition funnel over 30 min at 0 to 5 °C. The reaction mixture was warmed to 20 to 25 °C over 1 h and held for 72 h. The reaction was completed by HPLC. The reaction mixture was cooled to 0 to 5 °C and sodium methoxide (78 mL, 5.4 M, 0.421 moles, 0.97 equiv) was added over 2 min. The reaction mixture was then concentrated under reduced pressure to a volume of 300 mL. 2 L of ethyl acetate was added and concentration was continued under reduced pressure to a volume to 700 mL to get a slurry. 700 mL of ethyl acetate was added to the slurry to make the final volume to 1400 mL. 102 mL of water was added and stirred for 2 min to get a biphasic solution. The layers were separated. The ethyl acetate layer was concentrated under reduced pressure to a volume of 600 mL. Then the ethyl acetate layer was heated to > 60 °C and heptane (600 mL) was added slowly between 55 to 60 °C. The mixture was cooled to 15 to 20 °C to give a slurry. The slurry was stirred at 15 to 20 °C for 2 h and filtered. The solids were dried under vacuum at 25 °C for 16 h to furnish amine (14) as white solid (48 g, 64% yield). ‘H-NMR (500 MHz, MeOH-d4) δ ppm 2.94 (d, J= 13.73 Hz, 1H); 3.24 (d, J= 13.58 Hz, 1H).
d): chiral resolution of amine (14) as the 1:1 salt of (R)-2,2-dimethyl-5- (trifluoromethyl)oxazolidine-5-carboxamide (R)-2-hydroxysuccinate (18A) and (D)-malic acid.
(14) (ISA)
[00254] Amine (14) (105 g, 0.608 moles, 1.0 equiv.), (D)-Malic acid (82 g, 0.608 moles, 1.0 equiv.) and acetone (1571 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The reaction mixture was stirred at 20 to 25 °C for 16 h. The resulting slurry was filtered, and the wet cake was washed with acetone (300 mL). The wet cake was charged back to the reaction vessel, and acetone (625 mL) was charged. The slurry was heated to 53 °C and held for 6 h. The slurry was cooled to 20 to 25 °C and held at this temperature for 16 h. The slurry was filtered, and the wet cake was washed with acetone (200 mL). The wet cake was dried under vacuum at 40 °C for 4 h to furnish 82.4 g of the 1 : 1 salt of (18A) and (D)-malic acid as a white solid (82.4 g, 39% yield, 97% ee). i-NMR (500 MHz, D20) δ ppm 4.33 (br, s, 1H); 3.61 (br, d, J= 13.58 Hz, 1H); 3.40 – 3.47 (m, 1H); 2.76 (br, d, J= 15.87 Hz, 1H); 2.53 – 2.63 (m, 1H); 2.16 (br, s, 4H).
e): Coupling of the 1:1 (D)-malic acid salt of intermediate (18A) and Formula IV to provide (R)-3,3,3-trifluoro-2-(((5-fluoro-2-(l-(2-fluorobenzyl)-5-(isoxazol-3-yl)-lH-pyrazol-3-yl)pyrimidin-4-yl)amino)methyl)-2-hydroxypropanamide (Compound I)
Formula IV Compound I
[00255] The 1: 1 salt of intermediate (18A) and (D)-malic acid (74.1 g, 0.214 moles, 2.5 equiv) and water (44.8 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The reaction mixture was heated to 70 °C and stirred for 20 min. Acetone generated during the reaction was removed by blowing with nitrogen. The reaction mixture was cooled to 30 to 40 °C and Formula IV (32 g, 0.086 moles, 1.0 equiv), DMSO (448 mL) and Hunig’s base (44.7 mL, 0.257 moles, 3.0 equiv) were charged. The reaction mixture was heated to 90 °C and stirred at 90 °C over 17 h. The reaction was complete by HPLC. Then the mixture was cooled to 60 °C. Another portion of Hunig’s base (104 mL, 0.599 moles, 7.0 equiv) was charged followed by water (224 mL) at 55 to 62 °C. The reaction mixture was stirred for 15 min at 55 to 60 °C to form the seed bed. Water (320 mL) was added via addition funnel at 55 to 62 °C over the course of 30 min, and the resultant slurry was stirred for 1 h at 55 to 60 °C. The resulting slurry was filtered, and the filter cake was washed with a pre-mixed solution of methanol and water (320 mL/320 mL) followed by water (640 mL). The filter cake was then dried under vacuum at 40 °C over 16 h to furnish Compound I as an off-white solid (40 g, 92% yield, 99% pure by HPLC, 98% ee). ¾-NMR (500 MHz, DMSO-t/6) δ ppm 9.10 (s, 1 H); 8.33 (d, J=2.90 Hz, 1 H); 7.93 (s, br, 1 H); 7.90 (s, 1 H); 7.78 (s, br, 1 H); 7.69 (s, br, 1 H); 7.52 (s, 1 H); 7.33 (q, J=7.02 Hz, 1 H); 7.17 – 7.25 (m, 1 H); 7.17 – 7.25
(m, 1 H); 7.10 (t, J=7.48 Hz, l H); 6.98 (t, J=7.55 Hz, 1 H); 5.90 (s, 2 H); 3.92-4.05 (m, 2 H).
////////////OLINCIGUAT, IW-1701, phase 2, ironwood
NC(=O)[C@](O)(CNc1nc(ncc1F)c2cc(c3ccon3)n(Cc4ccccc4F)n2)C(F)(F)F
Fluoroform (CHF3) can be considered as an ideal reagent for difluoromethylation reactions. However, due to the low reactivity of fluoroform, only very few applications have been reported so far. Herein we report a continuous flow difluoromethylation protocol on sp3 carbons employing fluoroform as a reagent. The protocol is applicable for the direct Cα-difluoromethylation of protected α-amino acids, and enables a highly atom efficient synthesis of the active pharmaceutical ingredient eflornithine.
Methyl 3,3-(difluoro)-2,2-diphenylpropanoate (2a) The product mixtures were collected and the solvent removed in vacuo. The products were isolated by thin layer chromatography (dichloromethane/hexane = 3/2 (v/v)). Yield: 173 mg (0.62 mmol, 62%); 93% by 19F NMR ;light yellow viscous liquid. 1 H NMR (300 MHz, D2O): δ = 7.45 – 7.19 (m, 10H), 6.90 (t, 2 JHF = 55.0 Hz, 1H), 3.79 (s, 3H). 13C NMR (75 MHz, D2O): δ = 171.1, 136.3, 129.8, 128.3, 128.2, 115.6 (t, 1 JCF = 246.2 Hz), 64.7, 53.1.19F NMR (282 MHz, D2O):δ = -123.0 (d, 2 JHF = 55.0 Hz).



A gas–liquid continuous flow difluoromethylation protocol employing fluoroform as a reagent was reported. Fluoroform, a by-product of Teflon manufacture with little current synthetic value, is the most attractive reagent for difluoromethylation reactions. The continuous flow process allows this reaction to be performed within reaction times of 20 min with 2 equiv. of base and 3 equiv. of fluoroform. Importantly, the protocol allows the direct Cα-difluoromethylation of protected α-amino acids. These compounds are highly selective and potent inhibitors of pyridoxal phosphate-dependent decarboxylases. The starting materials are conveniently derived from the commercially available α-amino acid methyl esters, and the final products are obtained in excellent purities and yields after simple hydrolysis and precipitation. The developed process appears to be especially appealing for industrial applications, where atom economy, sustainability, reagent cost and reagent availability are important factors.
//////////
Nastorazepide (Z-360)
CAS: 209219-38-5
Chemical Formula: C29H36N4O5
Molecular Weight: 520.61994
UNII-R22TMY97SG; 209219-38-5;
Phase II, treatment of pancreatic cancer.
(R)-3-(3-(5-cyclohexyl-1-(3,3-dimethyl-2-oxobutyl)-2-oxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)ureido)benzoic acid

Nastorazepide, also known as Z-360, is a selective, orally available, 1,5-benzodiazepine-derivative gastrin/cholecystokinin 2 (CCK-2) receptor antagonist with potential antineoplastic activity. Z-360 binds to the gastrin/CCK-2 receptor, thereby preventing receptor activation by gastrin, a peptide hormone frequently associated with the proliferation of gastrointestinal and pancreatic tumor cells.
In January 2018, Zeria is developing nastorazepide calcium (phase II clinical trial), a CCK2 receptor antagonist, for the treatment of pancreatic cancer.
Zeria is developing nastorazepide calcium (Z-360), an oral CCK2 receptor (gastrin receptor) antagonist, for the potential treatment of pancreatic cancer. In September 2005, a phase Ib/IIa trial began in the UK for pancreatic cancer , in February 2008, the trial was completed ; in June 2008, data were presented . In March 2010, the drug was listed as being in phase II preparation in Europe ; in August 2011, this was still the case . In April 2014, a phase II trial began in patients with metastatic pancreatic adenocarcinoma in Japan, Korea and Taiwan. In November 2015, the drug was listed as being in phase II development

Nastorazepide (calcium salt)
CAS No. : 343326-69-2
M.Wt:540.62Formula:C29H36N4O5Ca0.5
Cholecystokinin (CK) is a digestive hormone produced and released in the duodenum, jejunal membrane and is known to have actions such as secretion of secretion, constriction of the gallbladder, stimulation of insulin secretion and the like. C CK is also known to exist in high concentrations in the cerebral cortex, hypothalamus and hippocampus, and it is also known that it has actions such as suppression of food intake, memory enhancement, anxiety action and the like. On the other hand, gastrin is a gastrointestinal hormone produced and released in G cells distributed in the pyloric region of the stomach, and it is known that it has gastric acid secretion action, contraction action of the gastric pyloric part and gallbladder, and the like. These C CK and gastrin have the same 5 amino acids at the C-terminus, and all express the action through the receptor. C CK receptors are classified into peripheral type C CK – A distributed in the ile, gall bladder and intestinal tract and central type C CK – B distributed in the brain. The gastrin receptor and the CKK – B receptor show similar properties in receptor binding experiments and sometimes called C CK 1 B / gastrin receptor due to high homology. These receptors, such as gastrin or a CCK-B receptor antagonist compound, are useful in the treatment of gastric ulcers, duodenal ulcers, gastritis, reflux esophagitis, splenitis, Zollinger-EUison syndrome, cavitary G cell hyperplasia, basal hyperplasia, Choleditis, gallstone stroke, gastrointestinal motility disorder, sensitive bowel syndrome, certain tumors, eating disorders, anxiety, panic disorder, depression, schizophrenia, Parkinson’s disease, late onset dyskinesia, It is expected to be useful for treatment and prevention of La Tourette’s syndrome, addiction due to drug ingestion, and withdrawal symptoms. It is also expected that the induction of analgesia or the enhancement of induction of analgesia by opioid drugs is expected (Journal of Pharmacology, Vol. 106, 171-180 (1995), Drugs of the Future, Vol. 18, 919-931 (1993), American Journal of Physiology, Vol.
As a gastrin receptor antagonist already, prolumide is known as a therapeutic agent for gastric ulcer and gastritis. However, proglumide has considerably low affinity for gastrin or CKK-B receptor and its therapeutic effect is weak. In addition, L – 3 6 4, 7 1 8 (Dibazepide, Japanese Unexamined Patent Publication No. 616366), L -3 6 5, 2 6 0 (Japanese Patent Laid-Open No. 6 3- 9), and the like, have been reported to exhibit either CKK-A receptor antagonism or CKK-B receptor antagonism. Furthermore, it is disclosed that a compound having a strong C 4 C – – B receptor antagonistic effect suppresses gastric acid secretion by pentagastrin stimulation (International Patent Publication WO 94/438, International Patent Publication WO 95/18110) , It is not always satisfactory and clinically applicable gastrin or CKK-B receptor antagonist has not yet been provided.
Compounds capable of strongly binding to gastrin or cholecystokinin receptors are expected for the prevention and treatment of diseases involving their respective receptors in the digestive tract and the central nervous system.
PRODUCT PATENT WO1998025911
| Inventors | Katsuo Shinozaki, Tomoyuki Yoneta, Masakazu Murata, Naoyoshi Miura, Kiyoto Maeda, Less « |
| Applicant | Zeria Pharmaceutical Co., Ltd. |
SYNTHESIS WO 2017030859

PATENT
WO 9825911
https://www.google.co.in/patents/WO1998025911A1?cl=und
PATENT
WO2017175854
PATENT
WO-2018008569
Process for producing a calcium salt of a 1,5-benzodiazepine compound – nastorazepide calcium – a cholecystokinin CCK2 receptor antagonist. Useful for the treatment of gastritis, reflux esophagitis, Zollinger-Ellison syndrome.
1: Kato H, Seto K, Kobayashi N, Yoshinaga K, Meyer T, Takei M. CCK-2/gastrin receptor signaling pathway is significant for gemcitabine-induced gene expression of VEGF in pancreatic carcinoma cells. Life Sci. 2011 Oct 24;89(17-18):603-8. doi: 10.1016/j.lfs.2011.07.019. Epub 2011 Aug 3. PubMed PMID: 21839751.
////////////NASTORAZEPIDE, phase II, treatment of pancreatic cancer,
O=C(O)C1=CC=CC(NC(N[C@@H]2CN(C3CCCCC3)C4=CC=CC=C4N(CC(C(C)(C)C)=O)C2=O)=O)=C1

LASMIDITAN, COL-144 , LY-573144
613677-28-4 HYDROCHLORIDE
439239-90-4 (free base)
2,4,6-Trifluoro-N-[6-(1-methylpiperidin-4-ylcarbonyl)pyridin-2-yl]benzamide
2,4,6-trifluoro-N-{6-[(1-methylpiperidin-4-yl)carbonyl]pyridin-2-yl}benzamide
CoLucid Pharmaceuticals, PHASE 3, MIGRAINE
UNII:760I9WM792
| Lasmiditan succinate; UNII-W64YBJ346B; Lasmiditan succinate [USAN]; W64YBJ346B; 439239-92-6; Lasmiditan succinate (USAN)
|
|
| Molecular Formula: | C42H42F6N6O8 |
|---|---|
| Molecular Weight: | 872.822 g/mol |
Lasmiditan (COL-144) is an investigational drug for the treatment of acute migraine. It is being developed by Eli Lilly and is in phase III clinical trials. It is a first-in-class “neurally acting anti-migraine agent” ditan.
WO-2018010345, from Solipharma and the inventor on this API. Eli Lilly , following its acquisition of CoLucid Pharmaceuticals , is developing lasmiditan, a 5-HT 1f agonist, for treating acute migraine.
WATCH THIS SPACE, SYNTHESIS COMING………..

SYN 2

Lasmiditan is a serotonin receptor agonist that, like the unsuccessful LY-334,370, selectively binds to the 5-HT1F receptor subtype. A number of triptans have been shown to act on this subtype as well, but only after their affinity for 5-HT1B and 5-HT1D has been made responsible for their anti-migraine activity. The lack of affinity for these receptors might result in fewer side effects related to vasoconstriction compared to triptans in susceptible patients, such as those with ischemic heart disease, Raynaud’s phenomenon or after a myocardial infarction,[1] although a 1998 review has found such side-effects to rarely occur in patients taking triptans.[2][3]
Lasmiditan was discovered by Eli Lilly and Company and was out-licensed to CoLucid Pharmaceuticals in 2006, until CoLucid was bought by Eli Lilly in 2017 to reacquire the drug.[4] The drug is protected by patents until 2031.[5]
Phase II clinical trials for dose finding purposes were completed in 2007 for an intravenous form[6] and in early 2010 for an oral form.[7]Two separate Phase III clinical trials for the oral version are currently ongoing under special protocol agreements with the US Food and Drug Administration (FDA). Eli Lilly has stated that they intend to submit a new drug application to the FDA in early 2018.[5]
As of 2017, three phase III clinical trials have been completed or are in progress. The SPARTAN trial compares placebo with 50, 100, and 200 mg of lasmiditan.[8] SAMURAI compared placebo with 100 and 200 mg doses of lasmidatin. In 2016, CoLucid announced that the trial had met its primary and secondary endpoints of patients being pain-free two hours after dosing.[5] GLADIATOR is an open-labelstudy comparing 100 and 200 mg doses of lasmidatin in patients that received the drug as part of a prior trial.[9] In August 2017 topline results from the SPARTAN trial showed that the drug induced met its primary and secondary endpoints in the trial. The primary result showed a statistically significant improvement in pain relief relative to placebo 2 hours after the first dose. The secondary result showed a statistically significantly greater percentage of patients were free of their most bothersome symptom (MBS) compared with placebo at two hours following the first dose. [10]
Novel crystalline forms of a 5-HT1F receptor agonist, particularly lasmiditan – designated as Forms 1-3 and A-D – processes for their preparation and compositions comprising them are claimed. Also claim is their use for treating anxiety, fatigue, depression, premenstrual syndrome, trauma syndrome, memory loss, dementia (including Alzheimer’s), autism, schizophrenia, attention deficit hyperactivity disorder, obsessive-compulsive disorder, epilepsy, anorexia nervosa, alcoholism, tobacco abuse, mutism and trichotillomania.
Lasmiditan (also known as COL-144 and LY573144) is a high-affinity, highly selective serotonin (5-HT) 5-HT(1F) receptor agonist.
In vitro binding studies show a K(i) value of 2.21 nM at the 5-HT(1F) receptor, compared with K(i) values of 1043 nM and 1357 nM at the 5-HT(1B) and 5-HT(1D) receptors, respectively, a selectivity ratio greater than 470-fold. Lasmiditan showed higher selectivity for the 5-HT(1F) receptor relative to other 5-HT(1) receptor subtypes than the first generation 5-HT(1F) receptor agonist LY334370.
In two rodent models of migraine, oral administration of lasmiditan potently inhibited markers associated with electrical stimulation of the trigeminal ganglion (dural plasma protein extravasation, and induction of the immediate early gene c-Fos in the trigeminal nucleus caudalis).
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.
PATENT
WO 03084949
https://www.google.co.in/patents/WO2003084949A1?cl=en
8. 2,4,6-Trifluoro-N-[6-(l -methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide mono-hydrochloride salt
Combine 2-amino-6-(l-methylpiperidin-4-ylcarbonyl)pyridine (0.20 g, 0.92 mmol), 2,4,6-Trifluorobenzoyl chloride (0.357 g, 1.84 mmol), and 1 ,4-Dioxane (10 mL), and stir while heating at reflux. After 3 hr., cool the reaction mixture to ambient temperature and concentrate. Load the concentrated mixture onto an SCX column (lOg), wash with methanol, and elute with 2M ammonia in methanol. Concentrate the eluent to obtain the free base of the title compound as an oil (0.365 g (>100%)). Dissolve the oil in methanol (5 mL) and treat with ammonium chloride (0.05 g, 0.92 mmol). Concentrate the mixture and dry under vacuum to obtain the title compound. HRMS Obs. m/z 378.1435, Calc. m/z 378.1429; m.p. 255°C (dec).
Examples
21. 2,4,6-Trifluoro-N-[6-(l-methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide
Add triethylamine (10.67 mL, 76.70 mmol, 2.4 eq) to a solution of 2-amino-(6-(l- methylpiperidin-4-ylcarbonyl)-pyridine (7g, 31.96 mmol, 1 eq) in anhydrous THF (100 mL) under a nitrogen atmosphere. Add 2,4,6-triflubenzoylchloride (7.46g, 5 mL, 38.35 mmol, 1.20 eq) dropwise at room temperature. After 2 hrs., add additional 2,4,6- triflubenzoylchloride (0.75 mL, 0.15 eq) and triethylamine (1.32 mL, 0.3 eq) to the reaction mixture and agitate the mixture for an additional 3 hrs. Quench the reaction with distilled water (10 mL) and 30%o NaOH (15 mL). Stir the resulting biphasic system for 1 hour and then separate the phases. Extract the organic fraction by adding H2O (75 mL) and acetic acid (12 mL), followed by cyclohexane (70 mL). Wash the organic fraction with H2O (50 mL) containing acetic acid (1 mL). Combine all the aqueous fractions and washes and neutralize the mixture with 30% NaOH (15 mL). Extract with methyl-tert- butyl ether (MTBE) (3×50 mL). Combine the organic fractions and dry with MgSO4, filter, concentrate under reduce pressure, and vacuum dry at room temperature, to obtain the title compound as a light-brown solid (11.031 g, 91 % yield).
Mass spectrum, (Electrospray) m/z = 378 (M+l); Η NMR (250 MHz, Chloroform-D) ppm 1.54 (m, 2 H) 2.02 (m, 2 H) 2.13 (t, J=l 1.48 Hz, 2 H) 2.29 (s, 3 H) 2.80 (m, J=l 1.96 Hz, 1 H) 3.56 (m, 1 H) 4.26 (d, J=7.87 Hz, 1 H) 6.17 (d, J=8.50 Hz, 1 H) 6.75 (m, 2 H) 7.45 (t, J=7.87 Hz, 1 H) 7.53 (m, 1 H) 7.95 (s, 1 H); 13C-NMR: (62.90 MHz, Chloroform-D) ppm 202.78; 162.6 (dm C-F-couplings); 162.0 (m C-F-couplings); 160.1 (m C-F-couplings); 158.1 ; 150.0; 139.7; 1 19.3; 1 17.9; 1 10.2 (m C-F-couplings); 100.9 (m C-F-couplings); 55.2; 46.5; 41.9; 28.1
22. 2,4,6-Trifluoro-N-[6-(l-methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide mono-hydrochloride salt
Dissolve 2,4,6-trifluoro-N-[6-(l-methylpiperidin-4-ylcarbonyl)-pyridin-2-yl]- benzamide – free base (5g, 23.26mmol) in isopropanol (50 mL) at room temperature and add a solution of 3.3 M diethylether/HCl (8 mL). Heat the reaction mixture under reflux for 30 minutes. Cool the reaction mixture to room temperature and agitate for 2 hrs. Filter the resulting white precipitate and rinse with isopropanol (5 mL). Dry the residual solid under reduce pressure at 40°C overnight to obtain the title compound (5.12 g, 93% yield). M.p. 223-224°C (sublimation); Η NMR (400 MHz, d6-DMSO) d ppm 1.94 (m, 2 H) 2.14 (m, J=11.15 Hz, 2 H) 2.74 (s, 3 H) 2.99 (m, J=9.19 Hz, 2 H) 3.49 (m, J=1 1.15 Hz, 2 H) 3.77 (m, 1 H) 7.41 (t, J=8.71 Hz, 2 H) 7.78 (d, J=7.43 Hz, 1 H) 8.10 (t, J=7.92 Hz, 1 H) 8.37 (d, J=6.85 Hz, 1 H) 10.50 (s, 1 H) 1 1.51 (s, 1 H); 13C-NMR: (100.61 MHz, Chloroform-D) ppm 200.7; 130.6-158.0 (m, C-F-couplings); 150.4; 150.1; 140.2; 118.5; 1 18.2; 11 1.9; 101.3 (t, C-F couplings); 52.8; 42.6; 25.2
23. 2,4,6-Trifluoro-N-[6-(l-methyl-piperidine-4-carbonyl)-pyridin-2-yl]- benzamide hemi-succinate salt
Add succinic acid (0.25g, 2.148 mmol, 0.5eq) to a solution of 2,4,6-trifluoro-N-[6-
(l-methyl-piperidin-4-ylcarbonyl)-pyridin-2-yl]-benzamide – free base (1.62g, 4.297 mmol, leq) in acetone (16.2 mL), at room temperature. Warm the solution under reflux for 30 minutes. Cool the solution to room temperature and filter off the resulting white precipitate. Rinse the precipitate with acetone (0.2 mL) and dry under vacuum at 50°C for 16 hours to provide the title compound (1.5g, 80% yield). M.p. 198.5°C; mass spectrum (Electrospray) m/z = 495.45
The following examples are prepared by combinatorial chemistry techniques as follows:
Examples 24-54
Combine R-acid (300 μL of 0.5M solution in dimethylformamide (DMF)), HATU (57 mg, 0.15 mmol), collidine (19 μL, 0.15 mmol), 2-amino-(6-(l-methylpiperidin-4- ylcarbonyl)-pyridine and DMF (1.5 mL), and agitate for 48 hr. Dilute the reaction mixture with 10% acetic acid in methanol (0.5 L). Load the resulting reaction mixture onto a 2 g SCX column. Wash the column thoroughly with methanol and then elute with 1 M ammonia in methanol. Concentrate the eluent and further purify the product by high- throughput mass guided chromatography. This procedure is repeated in parallel for examples 24-54.
Examples 55-58
Heat R-acid chloride (300 μL of 0.5M solution in pyridine) to 55°C, add 2-amino- (6-(l-methylpiperidin-4-ylcarbonyl)-pyridine (200 μL of 0.5M solution in pyridine), and continue heating the reaction mixture for 24 hr. Concentrate the reaction mixture and then dilute with 10% Acetic acid in methanol (0.5 mL) and methanol (0.5 mL). Load the resulting reaction mixture directly onto a 2 g SCX column. Thoroughly wash the column with methanol and then elute the column with 1 M ammonia in methanol. Concentrate the eluent and then further purify the product by high- throughput mass guided chromatography. This procedure is repeated in parallel for examples 55-58.
Examples 59-71
Heat 2-amino-(6-(l-methylpiperidin-4-ylcarbonyl)-pyridine (200 μL of 0.5M solution in pyridine) to 55°C then add R-acid chloride (0.10 mmol), heat for 2 hr. Concentrate the reaction mixture and then dilute with 10% Acetic acid in methanol (0.5 mL) and methanol (0.5 mL). Load the resulting reaction mixture directly onto a 2 g SCX column. Thoroughly wash the column with methanol and then elute the column with 1 M ammonia in methanol. Concentrate the eluent and then further purify the product by high-throughput mass guided chromatography. This procedure is repeated in parallel for examples 59-71.
PATENT
|
|
| Clinical data | |
|---|---|
| Routes of administration |
By mouth, intravenous |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C19H18F3N3O2 |
| Molar mass | 377.36 g/mol |
| 3D model (JSmol) | |
/////////////LASMIDITAN, phase III, LILY, COL-144 , LY-573144, CoLucid Pharmaceuticals, PHASE 3, MIGRAINE
CN1CCC(CC1)C(=O)C2=NC(=CC=C2)NC(=O)C3=C(C=C(C=C3F)F)F.CN1CCC(CC1)C(=O)C2=NC(=CC=C2)NC(=O)C3=C(C=C(C=C3F)F)F.C(CC(=O)O)C(=O)O
A series of (−)-nornuciferidine derivatives was synthesized and the non-natural enantiomer of the aporphine alkaloid was discovered to be a potent β1– and β2-adrenergic receptor ligand that antagonized isoproterenol and procaterol induced cyclic AMP increases from adenylyl cyclase, respectively. Progressive deconstruction of the tetracyclic scaffold to less complex cyclic and acyclic analogues revealed that the conformationally restricted (6a-R,7-R)-7-hydroxyaporphine 2 (AK-2-202) was necessary for efficient receptor binding and antagonism.



(6aR,7R)-1,2-Dimethoxy-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-7-ol (2) To a solution of S2 (10 mg, 0.031 mmol) in THF (2 mL) was added 2 N NaOH(aq) (1 mL), and the mixture was stirred at 70 oC for 2 days. After being quenched with H2O (10 mL), the aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (CH3OH/CH2Cl2, 5:95 to 10:90) to afford 2 (7.6 mg, 82%) as a pale yellow solid; mp 89−91 oC; [] 24 D +78 (c 0.58, CHCl3); 1H NMR (CDCl3, 500 MHz) 8.37−8.35 (1 H, m), 7.73−7.72 (1 H, m), 7.38−7.33 (2 H, m), 6.65 (1 H, s), 4.55 (1 H, d, J = 11.5 Hz), 3.88 (3 H, s), 3.67 (1 H, d, J = 11.5 Hz), 3.64 (3 H, s), 3.40−3.37 (1 H, m), 3.10−3.03 (1 H, m), 2.98 (1 H, td, J = 11.5, 3.5 Hz), 2.73 (1 H, d, J = 16.0 Hz); 13C NMR (CDCl3, 125 MHz) 152.5, 145.1, 139.0, 130.2, 129.4, 128.1, 127.8, 127.4, 125.9, 124.3, 123.1, 111.8, 72.0, 60.3, 59.0, 55.9, 42.0, 28.9; HRMS (ESI/Q-TOF) m/z [M + H]+ calculated for C18H20NO3 298.1438; found 298.1440
SIMILAR IN LIT
7-Oxygenated aporphines 1–6 possessing anti-configurations have previously been reported. In order to explore their bioactivities, a synthesis was established by utilizing a diastereoselective reductive acid-mediated cyclization followed by palladium-catalyzed ortho-arylations. Moderate XPhos precatalyst loading (10 mol %) and short reaction times (30 min) were sufficient to mediate the arylations. Alkaloids 1–5 were successfully prepared, while (−)-artabonatine A was revised to syn-isomer 30. Consequently, (−)-artabonatine E likely also has a syn-configuration (31).
///////////AK-2-202,


| CAS Number |
|---|
FDA APPROVED 2017
Naldemedine (INN, USAN; S-297,995, Symproic) is a peripherally–selective μ-opioid receptor antagonist developed by Shionogi which is approved for the treatment of opioid-induced constipation in adult patients with chronic non-cancer pain.[1] Clinical studies have thus far found it to possess statistically significant effectiveness for these indications and to be generally well-tolerated with predominantly mild to moderate gastrointestinal side effects.[2][3] No effects indicative of central opioid withdrawal or impact on the analgesic or mydriatic effects of co-administered opioids have been observed.[2]


ナルデメジントシル酸塩
Naldemedine is manufactured by Shionogi Inc., a U.S. based subsidiary of Shionogi & Co., Ltd. Shionogi & Co., Ltd. (SGIOF) is a Japanese pharmaceutical company founded in 1878 based in Osaka, Japan. Shionogi Inc. is fully funded by its parent company, Shionogi & Co., Ltd. The parent company specializes in pharmaceuticals, diagnostic reagents and medical devices in Japan and internationally. Naldemedine is their only gastroenterology product in the United States.
In the US market, Shionogi Inc. has partnered with Purdue Pharma in a joint venture for US commercialization of Symproic.[4] Purdue Pharma LP is a privately held pharmaceutical company based in the United States that specializes in chronic pain disorders.[5]
Purdue Pharma appealed to remove the Class II scheduling of Symproic as accordant to the Controlled Substances Act. The appeal was posted to the Federal Register on July 12, 2017.[6] The Drug Enforcement Administration officially removed the Class II scheduling in September 2017.[7]
SYN
US 8084460

WO 2012063933

Since 2015, Shionogi & Co., Ltd. has produced increasing net income. At the end of fiscal year 2016, Shionogi & Co., Ltd. had a net income of $66,687,000. At the end of fiscal year 2017, they increased their net income to $83,879,000.[8] How much of this is attributed to sales of Symproic is unknown. Shionogi & Co., Ltd. ends their fiscal year on March 31 of each year. Considering the drug was only FDA approved on March 23 of 2017, the true valuation of the drug is yet to be seen. Purdue Pharma has begun advertising for the medication to be available by October 2017.[9]
There are currently three patents issued for naldemedine tosylate by the United States Patent and Trademark Office. All patents are owned by Shionogi Inc. and will expire from 2026-2031.[10] Naldemedine tosylate has 46 other patents in 18 different countries.[11]
12 Phase I clinical trials were reported for the use of naldemedine in healthy volunteers.[12] In a single ascending dose study, subjects received one dose of naldemedine (0.1–100 mg) or one dose of a placebo. In a multiple ascending dose study, subjects received once daily naldemedine (3–30 mg) or placebo for 10 days. Maximum plasma concentrations were reached within 0.5-0.75 hours. There were no reported major safety concerns, even at doses 150-500 times the available dose of 0.2 mg. In both studies, gastrointestinal events occurred more frequently with naldemedine, but researchers concluded these to be treatment related.[13]
The approval of naldemedine came from the results of the COMPOSE program, a phase three clinical studies program conducted in adults 18–80 years of age with chronic non-cancer pain opioid induced constipation. COMPOSE-I and COMPOSE-II were 12-week double blind randomized controlled trials comparing the use of naldemedine to placebo in the patient population. COMPOSE-I began in August 2013 until January 2015 in 68 outpatient clinic in seven countries. COMPOSE-II began in November 2013 until June 2015 taking place in 69 outpatient clinics in six countries. In both trials, patients were randomly assigned to receive either naldemedine 0.2 mg or placebo once daily for 12 weeks. A responder had at least three spontaneous bowel movements per week with an increase of one spontaneous bowel movement for nine of the 12 weeks, including three of the final four weeks of the study. In COMPOSE-I and COMPOSE-II, the proportion of responders were significantly higher in the naldemedine group than the placebo group. Adverse events were similar in both trials, however, patients in the naldemedine group had slightly higher rates of adverse events.[14]
COMPOSE-III was a 52 week clinical trial examining the long term safety with naldemedine in patients with non cancer chronic pain. Results from this trial showed statistical significance for increased weekly bowel movements and no opioid withdrawal symptoms. The study also concluded adverse effects were more similar between two groups.[12]
All trials were conducted following Good Clinical Practice guidelines.[12]
| FDA Orange Book Patents: 1 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 9108975 |
| Expiration | Nov 11, 2031 |
| Applicant | SHIONOGI INC |
| Drug Application | N208854 (Prescription Drug: SYMPROIC. Ingredients: NALDEMEDINE TOSYLATE) |
| FDA Orange Book Patents: 2 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | RE46375 |
| Expiration | Oct 5, 2026 |
| Applicant | SHIONOGI INC |
| Drug Application | N208854 (Prescription Drug: SYMPROIC. Ingredients: NALDEMEDINE TOSYLATE) |
| FDA Orange Book Patents: 3 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | RE46365 |
| Expiration | Jan 11, 2028 |
| Applicant | SHIONOGI INC |
| Drug Application | N208854 (Prescription Drug: SYMPROIC. Ingredients: NALDEMEDINE TOSYLATE) |
|
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| KEGG | |
| Chemical and physical data | |
| Formula | C32H34N4O6 |
| Molar mass | 570.63556 g/mol |
| 3D model (JSmol) | |
//////////S-297995, Naldemedine, FDA 2017, ナルデメジントシル酸塩 , Symproic
CC(C)(C1=NC(=NO1)C2=CC=CC=C2)NC(=O)C3=C(C4C56CCN(C(C5(C3)O)CC7=C6C(=C(C=C7)O)O4)CC8CC8)O
Delafloxacin, ABT-492, RX-3341, WQ-3034, A-319492
Delafloxacin is a Fluoroquinolone Antibacterial. The chemical classification of delafloxacin is Fluoroquinolones.
![]()
Delafloxacin (INN) (trade name Baxdela) is a fluoroquinolone antibiotic used to treat acute bacterial skin and skin structure infections.[1] It was developed and marketed by Melinta Therapeutics (formerly Rib-X Pharmaceuticals),[1] which subsequently merged with Cempra.[2]

CN 104876911

Delafloxacin is used to treat acute bacterial skin and skin structure infections caused by designated susceptible bacteria.[1]
Susceptible bacteria are:[1]
It has not been tested in pregnant women.[1]
Like other drugs in the fluoroquinolone class, delafloxacin contains a black box warning about the risk of tendinitis, tendon rupture, peripheral neuropathy, central nervous system effects, and exacerbation of myasthenia gravis. The label also warns against the risk of hypersensitivity reactions and Clostridium difficile-associated diarrhea.[1]
Adverse effects occurring in more than 2% of clinical trial subjects included nausea, diarrhea, headache, elevated transaminases, and vomiting.[1]
![]()
Like other fluoroquinolones, delafloxacin chelates metals including aluminum, magnesium, sucralfate, iron, zinc, and divalent and trivalent cations like didanosine; using this drugs with antacids, some dietary supplements, or drugs buffered with any of these ions will interfere with available amounds of delafloxacin.[1]
The half-life varies in around 8 hours at normal doses. Excretion is 65% through urine, mostly in unmetabolized form, and 28% via feces. Clearance is reduced in people with severe kidney disease.[3]
Delafloxacin is more active (lower MIC90) than other quinolones against Gram-positive bacteria such as methicillin-resistant Staphylococcus aureus (MRSA). In contrast to most approved fluoroquinolones, which are zwitterionic, delafloxacin has an anionic character, which results in a 10-fold increase in delafloxacin accumulation in both bacteria and cells at acidic pH. This property is believed to confer to delafloxacin an advantage for the eradication of Staphylococcus aureus in acidic environments, including intracellular infections.[3]
The chemical name is 1-Deoxy-1 (methylamino)-D-glucitol, 1-(6-amino-3,5-difluoropyridin-2-yl)-8-chloro-6-fluoro-7-(3-hydroxyazetidin-1-yl) 4-oxo-1,4-dihydroquinoline-3-carboxylate (salt).[1]
The injectable form of delafloxacin is sold as the meglumine salt of the active ingredient and its United States Adopted Name, delafloxacin meglumine, reflects that; the injection formulation also includes EDTA and sulfobutylether-β-cyclodextrin. The tablet is made of delafloxacin, citric acid anhydrous, crospovidone, magnesium stearate, microcrystalline cellulose, povidone, sodium bicarbonate, and sodium phosphate monobasic monohydrate.[1]
Delafloxacin was known as ABT-492, RX-3341, and WQ-3034 while it was under development.[4]
Rib-X Pharmaceuticals acquired delafloxacin from Wakunaga Pharmaceutical in 2006.[5] Rib-X was renamed to Melinta Therapeutics in 2013.[6]
Key clinical trials for delafloxacin have been performed by Melinta regarding indications for skin and skin structure infections as well as complicated bacterial infections and uncomplicated gonorrhea. The trial on gonorrhea was terminated before data was released.[7]
Delafloxacin was approved by the FDA in June 2017, after it was noninferior to vancomycin plus aztreonam in two trials on 1042 patients with acute bacterial skin and skin structure infection.[8] New Drug Applications (NDA) for delafloxacin (Baxdela) 450 mg tablets and 300 mg injections were approved by the FDA in June 2017.[9]
The FDA obligated Melinta to conduct further studies as follows:[9]
Melinta merged with Cempra in August, 2017.[2]
Melinta has entered into commercialization and distribution agreements with both Menarini Therapeutics (March 2017) and Eurofarma Laboratórios (January 2015) for international commercialization of delafloxacin. The agreement with Menarini allows them to commercialize and distribute in 68 countries, including Europe, China, and South Korea among others. A similar agreement with Eurofarma allows for commercialization in Brazil.[7]
PATENT
de Iaf Ioxacin Preparation
[0101] was added to the S-neck flask resultant product of Example 11 (3.5 Yap, dirty 〇1 0.76) implemented 01. (35 blood) milky white suspension, was added glacial acetic acid (3. OmL), stirred at room temperature to embrace completely clear solution was added dropwise distilled water 70 fed blood, filter, wash coating, evaporated to dryness to give a pale yellow powder 3. Og, purity 99.8% (HPLC), m / z (MH + M41.03, IH NMR (400MHz, DMSO) S4.20 (m, 2H), 4.45 (m, lH), 4.61 (m, 2H), 5.63 (d, lH), 6.69 (s, 2H), 7.81 (d, lH), 7.95 (dd, lH), 8.69 (d, lH), 14.34 (brs, lH).
PAPER
Org. Process Res. Dev. 2006, 10, 803-807.
The total synthesis of quinolone antibiotic ABT-492 has been achieved in 67% yield over nine steps from 2,4,5-trifluorobenzoic acid. The highlights of this synthesis include a novel chemoselective chlorination at the 8-position of a highly elaborated quinolone core. In addition, a Lewis acid promoted cyclization reaction to form the quinolone heterocycle was developed which was incorporated into a one-pot, three-step cyclization/coupling/protection sequence that proceeds in 93% yield.
1-(6-Amino-3,5-difluoropyridin-2-yl)-8-chloro-6-fluoro-7-(3-hydroxyazetidin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (ABT-492), NCS Process: . Mp: 238−241 °C. 1H NMR (CDCl3) δ 14.63 (brs, 1H), 8.70 (d, J = 0.7 Hz, 1H), 7.95 (dd, J = 9.9, 0.7 Hz, 1H), 7.83 (d, J = 13.6 Hz, 1H), 6.75 (s, 2H), 5.75 (d, J = 5.8 Hz, 1H), 4.61 (m, 12H), 4.47 (m, 1H), 4.18 (m, 2H). Anal. Calcd for C18H12ClF3N4O4: C, 49.05; H, 2.74; N, 12.71. Found: C, 48.90; H, 2.48; N, 12.62.
PATENT
EXAMPLE 5
A solution of 2,4,5-trifluorobenzoic acid (139.5Kg) in DMF (8.4Kg) and toluene (613Kg) was treated with thionyl chloride (139.4Kg), stirred at 60°C for 3.5 hours, cooled to 250C, concentrated to 20% of its original volume, treated with toluene (600Kg), distilled and stored at ambient temperature.
EXAMPLE 6
A suspension of potassium ethyl malonate (50.8Kg) and magnesium chloride
(34.5Kg) in toluene (130Kg) below 00C was treated with THF (265L), cooled to 0°C, treated with triethylamine (75Kg), warmed to 5O0C, stirred for 1-5 hours, cooled to 00C, treated with 22% (w/w) of EXAMPLE 5 in toluene (163Kg), warmed to ambient temperature, stirred for 2 hours, added to 2M HCl (407Kg), stirred for 30 minutes, separated from the water layer and washed with water. This procedure was repeated, and the organic layers were combined, concentrated with an ethanol (150L) azeotrope, treated with water (30% by weight of the organic layer), stirred for 3 hours at 00C, and filtered. The andfiltrant was washed with 3:1 ethanol/water and dried under vacuum at 35-45°C to provide 86Kg of product. H NMR (CDCl3) (keto) δ 7.75 (ddd, J=10.8, 10.8, 6.0Hz, IH), 7.02 (ddd, IH), 4.27 (q, J=7.2Hz, 2H), 3.95 (d, 4.2Hz, 2H), 1.35 (t, J=7.3Hz, 3H); (enol) δ 12.72 (s, IH), 7.85 (ddd, J=10.5, 9.6, 6.6Hz, IH), 6.96 (ddd, J=10.5, 10.5, 6.6Hz, IH), 5.84 (s, IH), 4.23 (q, J=7.2Hz, 2H), 1.27 (t, J=7.4Hz, 3H).
EXAMPLE 7A
A solution of EXAMPLE 6 (83.2Kg) in triethyl orthoformate (80.1Kg) at reflux was stirred for 0.5-1 hour, treated with acetic anhydride (103.5Kg), stirred for 12 hours and cooled to ambient temperature to provide a solution that was used immediately.
EXAMPLE 7B
The solution of EXAMPLE 7A was treated with N-methylpyrrolidinone (210Kg), acetonitrile (161Kg) and water (3Kg), added to a suspension of EXAMPLE 4 (57.4Kg) in 1 : 1 N-methylpyrrolidinone (210Kg) and acetonitrile (161Kg), stirred for 2 hours, added to water (662Kg) and filtered. The fϊltrant was washed with (2:1) acetonitrile/water and water and dried under vacuum at 600C to provide 119.5Kg of product. Mp 157-16O0C; 1H NMR (CDCl3, 300 MHz) (E) δ 1.15 (t, 3H), 4.16 (q, 2H), 4.64 (br s, 2H), 6.90 (m, IH), 7.22 (t, IH), 7.32 (m, IH), 9.03 (d, IH), 12.44 (bd, IH); (Z) δ 1.03 (t, 3H), 4.11 (q, 2H), 4.60 (br s, 2H), 6.90 (m, IH), 7.20 (t, IH), 7.48 (m, IH), 8.90 (d, IH), 11.17 (bd, IH).
EXAMPLE 8A
A mixture of EXAMPLE 7 (115Kg) and lithium chloride (24.3Kg) in
N-methylpyrrolidinone (769Kg) below 350C was treated with DBU (946.1Kg) and stirred for 2 hours to provide a solution of EXAMPLE 8 A that was used immediately.
EXAMPLE 8B
The solution of EXAMPLE 8A below 4O0C was treated with EXAMPLE 2 (33.9Kg) and DBU (109Kg) and stirred for 2-5 hours to provide a solution of EXAMPLE 8B that was used immediately.
EXAMPLE 8C
The solution of EXAMPLE 8B was treated with isobutyric anhydride (99.7Kg), stirred at 350C for 1-2 hours, cooled to 20-300C, treated with ethyl acetate (104Kg) and 10% aqueous citric acid (570Kg) and filtered. The filtrant was washed with water and dried under vacuum at 500C to provide 136Kg of product. 1H NMR (DMSO-d6, 400 MHz) δ 8.49 (s, IH), 8.00 (dd, J=9.0, 9.3 Hz, IH), 7.75 (d, J=12.8 Hz, IH), 6.79 (br s, 2H), 5.95 (dd, J=I.5, 7.6 Hz, IH), 5.21 (m, IH), 4.36 (t, J=7.4 Hz, 2H), 4.02 (q, J=7.0 Hz, 2H), 3.95 (dd, J=3.7, 9.2 Hz, 2H), 2.58 (hept, J=7.0 Hz, IH), 1.26 (t, J=7.0 Hz, 3H), 1.11 (d, J=7.0 Hz, 6H).
EXAMPLE 10
A solution of N-chlorosuccinimide (25.3Kg) in methyl acetate (419Kg) at 170C was treated with sulfuric acid (560 g), transferred to a slurry of EXAMPLE 8 (92.7Kg) in ethyl acetate (244Kg) at 17°C while maintaining the reaction temperature at 17°C,
quenched/washed with 1.5% aqueous sodium bicarbonate (370Kg), washed with
10% aqueous sodium sulfite (200Kg) and concentrated. The concentrate was dissolved in isopropanol, treated with 4% (w/w) aqueous potassium hydroxide (750Kg), stirred at 5O0C until hydrolysis was complete, passed through a polishing filter, treated with 12% aqueous acetic acid (410Kg) and filtered. The filtrant was washed with water and dried at 5O0C to provide 73Kg of product. 1H NMR (CDCl3) δ 14.63 (brs, IH), 8.70 (d, J=0.7Hz, IH), 7.95 (dd, J=9.9, 0.7Hz, IH), 7.83 (d, J=13.6Hz, IH), 6.75 (s, 2H), 5.75 (d, J=5.8Hz, IH), 4.61 (m, 12H), 4.47 (m, IH), 4.18 (m, 2H).
PATENT
https://www.google.com/patents/CN104876911A?cl=en

Currently, 德拉沙 star for the synthesis mainly in the following two ways:
[0004] 1, Chinese patent CN1201459A _2,4,5_ trifluorobenzoyl from 3-chloro-ethyl ester synthesis De Lasha star. Used in this reaction is N, N- dimethylformamide high temperature and potassium carbonate cyclization, prone to impurities, after cyclization is hydrolyzed required, increase the reaction step, a low yield. Reaction scheme is as follows:
[0005]
[0006] 2, published in the Journal of Organic Chemistry (Org Process Res & Dev2006,4, 751) provides a new synthesis method 德拉沙 star from 2,4,5_ trifluoroacetic acid as the starting material, synthetic Germany Lassa star. This reaction because of the need in eight selective chlorination, so 7-hydroxy need protection, reaction step increase. And when eight were chlorinated 7 substituent easily broken, harsh reaction conditions, the reaction yield is low, is not suitable for mass production. Reaction scheme is as follows:
[0007]
Example: 8_-Chloro-6-fluoro-1- (6-amino-3,5-difluoro-2-yl) -7- (3-hydroxy-1-azetidinyl) – 1,4-dihydro-4-oxo-3-quinolinecarboxylic acid (Dela Sha star) Synthesis of
[0025] 3-chloro-2,4,5-trifluoro-benzoyl acetate (78,0.025111〇1) in 501,111 flask, triethylorthoformate (5. 9g, 0. 04mol) and vinegar anhydride, heated at reflux for 3h ~ 5h, evaporated under reduced pressure excess triethyl orthoformate and acetic anhydride, was added N- methylpyrrolidone was diluted, and then 2,6-diamino-3,5-difluoro-pyridine was suspended ( 3. 8g, 0. 026mol) and N- methylpyrrolidone were suspended, was added dropwise to the above solution, after completion of the reaction was added anhydrous lithium chloride (2. 6g) and DBU (4.6g, 0.03mol) (1 1,8-diazabicyclo [5.4.0] undec-ene _7_) was heated with stirring, HPLC monitored the reaction was complete. Then 3-hydroxy-azetidine hydrochloride (3. 52g) was added to the above solution was added dropwise DBU, the reaction was continued to completion. In the aqueous solution of isopropanol and potassium hydroxide, heating the hydrolysis, the hydrolysis is completed after adjusting PH = 3 solid precipitated. Filtering, washing, to give a yellow solid (7. 82g), yield 71%.
[0026] MP: 238-241 ° C
[0027] Tuen bandit 1 (square)?! (: 13) 14.32 0 ^ 8,1 1), 8.51 ((1, J = 0.7Hz, lH), 7.96 (dd, J = 9 · 9,0 · 7Ηζ , 1H), 7 · 64 (d, J = 13. 6Hz, 1H), 6 · 92 (s, 2H), 5 · 86 (d, J = 5. 8Hz, 1H), 4 · 89 (m, 12H ), 4 · 32 (m, 1H), 4 · 18 (m, 2H).
|url=value (help). Drugs. 77: 1481–1486 – via Springer.| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN1201459A * | Sep 20, 1996 | Dec 9, 1998 | 涌永制药株式会社 | Novel pyridonecarboxylic acid derivatives or their salts and antibacterial agent comprising same as active ingredient |
| JP2005097116A * | Title not available | |||
| WO2006015194A2 * | Jul 29, 2005 | Feb 9, 2006 | Abbott Laboratories | Preparation of pyridonecarboxylic acid antibacterials |
| FDA Orange Book Patents: 1 of 8 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8871938 |
| Expiration | Sep 23, 2029 |
| Applicant | MELINTA |
| Drug Application | N208610 (Prescription Drug: BAXDELA. Ingredients: DELAFLOXACIN MEGLUMINE) |
| FDA Orange Book Patents: 2 of 8 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 9539250 |
| Expiration | Oct 7, 2025 |
| Applicant | MELINTA |
| Drug Application | N208611 (Prescription Drug: BAXDELA. Ingredients: DELAFLOXACIN MEGLUMINE) |
| FDA Orange Book Patents: 3 of 8 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7728143 |
| Expiration | Nov 20, 2027 |
| Applicant | MELINTA |
| Drug Application | N208611 (Prescription Drug: BAXDELA. Ingredients: DELAFLOXACIN MEGLUMINE) |
|
|
| Clinical data | |
|---|---|
| Trade names | Baxdela |
| Synonyms | ABT-492; RX-3341; WQ-3034 |
| Routes of administration |
Oral, intravenous injection |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C18H12ClF3N4O4 |
| Molar mass | 440.76 g/mol |
| 3D model (JSmol) | |
/////////////Delafloxacin, ABT-492, RX-3341, WQ-3034, FDA 2017, A-319492
C1C(CN1C2=C(C=C3C(=C2Cl)N(C=C(C3=O)C(=O)O)C4=NC(=C(C=C4F)F)N)F)O

lutetium Lu 177 dotatate
FDA approves new treatment for certain digestive tract cancers
The U.S. Food and Drug Administration today approved Lutathera (lutetium Lu 177 dotatate) for the treatment of a type of cancer that affects the pancreas or gastrointestinal tract called gastroenteropancreatic neuroendocrine tumors (GEP-NETs). This is the first time a radioactive drug, or radiopharmaceutical, has been approved for the treatment of GEP-NETs. Lutathera is indicated for adult patients with somatostatin receptor-positive GEP-NETs. Continue reading.\
January 26, 2018
The U.S. Food and Drug Administration today approved Lutathera (lutetium Lu 177 dotatate) for the treatment of a type of cancer that affects the pancreas or gastrointestinal tract called gastroenteropancreatic neuroendocrine tumors (GEP-NETs). This is the first time a radioactive drug, or radiopharmaceutical, has been approved for the treatment of GEP-NETs. Lutathera is indicated for adult patients with somatostatin receptor-positive GEP-NETs.
“GEP-NETs are a rare group of cancers with limited treatment options after initial therapy fails to keep the cancer from growing,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This approval provides another treatment choice for patients with these rare cancers. It also demonstrates how the FDA may consider data from therapies that are used in an expanded access program to support approval for a new treatment.”
GEP-NETs can be present in the pancreas and in different parts of the gastrointestinal tract such as the stomach, intestines, colon and rectum. It is estimated that approximately one out of 27,000 people are diagnosed with GEP-NETs per year.
Lutathera is a radioactive drug that works by binding to a part of a cell called a somatostatin receptor, which may be present on certain tumors. After binding to the receptor, the drug enters the cell allowing radiation to cause damage to the tumor cells.
The approval of Lutathera was supported by two studies. The first was a randomized clinical trial in 229 patients with a certain type of advanced somatostatin receptor-positive GEP-NET. Patients in the trial either received Lutathera in combination with the drug octreotide or octreotide alone. The study measured the length of time the tumors did not grow after treatment (progression-free survival). Progression-free survival was longer for patients taking Lutathera with octreotide compared to patients who received octreotide alone. This means the risk of tumor growth or patient death was lower for patients who received Lutathera with octreotide compared to that of patients who received only octreotide.
The second study was based on data from 1,214 patients with somatostatin receptor-positive tumors, including GEP-NETS, who received Lutathera at a single site in the Netherlands. Complete or partial tumor shrinkage was reported in 16 percent of a subset of 360 patients with GEP-NETs who were evaluated for response by the FDA. Patients initially enrolled in the study received Lutathera as part of an expanded access program. Expanded access is a way for patients with serious or immediately life-threatening diseases or conditions who lack therapeutic alternatives to gain access to investigational drugs for treatment use.
Common side effects of Lutathera include low levels of white blood cells (lymphopenia), high levels of enzymes in certain organs (increased GGT, AST and/or ALT), vomiting, nausea, high levels of blood sugar (hyperglycemia) and low levels of potassium in the blood (hypokalemia).
Serious side effects of Lutathera include low levels of blood cells (myelosuppression), development of certain blood or bone marrow cancers (secondary myelodysplastic syndrome and leukemia), kidney damage (renal toxicity), liver damage (hepatotoxicity), abnormal levels of hormones in the body (neuroendocrine hormonal crises) and infertility. Lutathera can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception. Patients taking Lutathera are exposed to radiation. Exposure of other patients, medical personnel, and household members should be limited in accordance with radiation safety practices.
Lutathera was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition. Lutathera also received Orphan Drugdesignation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Lutathera to Advanced Accelerator Applications.
MORE……………..
Dotatate lutenium Lu-177; 437608-50-9; DTXSID20195927
2-[4-[2-[[(2R)-1-[[(4R,7S,10S,13R,16S,19R)-10-(4-aminobutyl)-4-[[(1S,2R)-1-carboxy-2-hydroxypropyl]carbamoyl]-7-[(1R)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1H-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicos-19-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-2-oxoethyl]-7,10-bis(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;lutetium(3+)

 Lutetium-177 has been quite a late addition as an isotope of significance to the nuclear medicine yet it is making big strides especially as a therapeutic radiopharmaceutical for neuroendocrine tumours in the form of 177Lu-DOTA-TATE on regular basis as described by Das & Pillai (2013). Lutetium-177 a lanthanide is an f block element that has a half-life of 6.7 days and decays mainly by beta emission to Hf-177, is accompanied by two gamma ray emissions. These radionuclide properties are very similar to those of I-131 which has long served as a therapeutic radionuclide, it was therefore not surprising that Lu-177 also emerged as a highly valuable radionuclide for similar applications,
There are several other upcoming applications especially for bone pain palliatiion. As a result of its convenient production logistics Lu-177 as discussed by Pillai et al (2003) is fast emerging a radionuclide of choice in radionuclide therapy (RNT).
Lu-177 can be prepared in a nuclear reactor by one of the two reactions given below :
176Lu(n,gamma)177Lu or
176Yb(n,gamma)177Yb –beta–> 177Lu
The former reaction has a high thermal neutron capture cross section and is presently the method adopted at our reactors in spite of the formation of long lived Lu-177m whose yield is very much low and is considered insignificant to cause any great concern.
Lutetium-177 Impact
 Recently there has been a rush of several research reviews and articles where Lu-177 holds the centre stage, for example, Banerjee et al (2015) have reviewed the chemistry and applications of Lu-177; Dash et al (2015) reviewed its production and available options; Knapp & Pillai (2015) highlighted its usefulness in cancer treatment and chronic diseases and Pillai and Knapp (2015) have discussed the evolving role of Lu-177 in nuclear medicine with this ready availability of Lu-177. Peptide receptor radionuclide therapy is one of the upcoming field of investigation where Lu-177 holds much promise among few other radionuclides. Indeed Lutetium-177 has covered a good distance especially for Therapeutic and as a palliative radiopharmaceutical.
Chemistry
Das et al (2014) have described the preparation of Lu-177 EDTMP kit.
Parus et al (2015) have discussed chemistry of bifunctional chelating agents for binding Lu-177.
Gupta et al (2014) have compiled methods of labelleing antibdoies with radioiodine and radiometals.
Applications
Limouris (2012) has reviewed applications in neuroendocrine tumors with focus on Liver metastasis. Das and Banerjee (2015) described the potential theranostic applications with Lu-177.
Anderson et al (1960) were among the first to use Lutetium (as chloride and citrate) in a clinical trial which were not so successful and did not encourage much promise. Keeling et al (1988) published their results with in vitro uptake of Lutetium hydroxylapatite particles. Lu-EDTMP as bone palliating agent by Ando et al (1998) soon followed, However the greatest impact was seen with the advent of a somatostatin analogue Lu-DOTATATE for targetting neuroendocrine tumors reported by Kwekkeboom et al (2001) and reviewed recently by Bodei et al (2013).
PRRNT – IAEA (2013) has brought out a human health series booklet on the subject with emphasis on neuroendocrine tumors.
177Lu Labelled Peptides in NET Kam et al (2012).
177Lu- DOTATATE – PRRNT – Bakker et al (2006)
177Lu-EDTMP – Bone Pain Palliation – Bahrami-Samani et al (2012)
177Lu-EDTMP – Pharmacokinetics, dosimetry and Therapeutic efficacy – Chakraborty S et al (2015)
177Lu-Hydroxylapatite – Radiosynovectomy – Kamalleshwaran et al. (2014) Shinto et al. (2015)
117Lu- Radioimmunotherapy – Kameshwaran et al (2015)
177Lu – Pretargeted Radioimmunotherapy (PRIT) Frost et al (2015).
More specific applications and additional information about the highly valuable therapeutic isotope would soon be added.
References and Notes
Anderson J, Farmer FT, Haggith JW, Hill M. (1960). The treatment of myelomatosis with Lutetium. Br J Radiol. 33:374-378.
Ando A, Ando L, Tonami N, Kinuya S, Kazuma K, Kataiwa A, Nakagawa M, Fujita N. (1998). 177Lu-EDTMP: a potential therapeutic bone agent. Nucl Med Commun. 19: 587-591.
Bahrami-Samani A, Anvari A, Jalilian AR, Shirvani-Arani S, Yousefnia H, Aghamiri MR, Ghannadi-Maragheh M. (2012). Production, Quality Control and Pharmacokinetic Studies of 177Lu-EDTMP for Human Bone Pain Palliation Therapy Trials. Iran J Pharm Res. 11:137-44.
Bakker WH, Breeman WAP, Kwekkeboom DJ, De Jong LC, Krenning EP. ((2006) Practical aspects of peptide receptor radionuclide therapy with [177Lu][DOTA0, Tyr3]octreotate. Q J Nucl Med Mol Imaging 50: 265-271.
Banerjee S, Pillai MR, Knapp FF (2015). Lutetium-177 Therapeutic Radiopharmaceuticals: Linking Chemistry, Radiochemistry, and Practical Applications. Chem Rev. 115: 2934-2974.
Bodei L, Mueller-Brand J, Baum RP, Pavel ME, Hörsch D, O’Dorisio MS, O’Dorisio TM, Howe JR, Cremonesi M, Kwekkeboom DJ, Zaknun JJ. (2013).The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2013 40:800-16.
Chakraborty S, Balogh L, Das T, Polyák A, Andócs G, Máthé D, Király R, Thuróczy J, Chaudhari PR, Jánoki GA, Jánoki G, Banerjee S, Pillai MR (2015). Evaluation of 177Lu-EDTMP in dogs with spontaneous tumor involving bone: Pharmacokinetics, dosimetry and therapeutic efficacy. Curr Radiopharm (ahead of Pub)
Das T, Banerjee S. (2015). Theranostic Applications of Lutetium-177 in Radionuclide Therapy. Curr Radiopharm. (ahead of print).
Das T , Sarma HD, Shinto A, Kamaleshwaran KK, Banerjee S. (2014). Formulation, Preclinical Evaluation, and Preliminary Clinical Investigation of an In-House Freeze-Dried EDTMP Kit Suitable for the Preparation of Lu-177-EDTMP. Cancer Biotherap Radiopharm. 29: (ahead of publication).
Das T, Pillai M.R.A. (2013).Options to meet the future global demand of radionuclides for radionuclide therapy. Nucl Med Biol. 40: 23-32.
Dash A, Pillai MR, Knapp FF Jr. (2015). Production of 177Lu for targeted radionuclide therapy : Available options. Nucl Med Mol Imaging. 49: 85-107.
Frost SH, Frayo SL, Miller BW, Orozco JJ, Booth GC, Hylarides MD, Lin Y, Green DJ, Gopal AK, Pagel JM, Bäck TA, Fisher DR, Press OW. (2015) Comparative efficacy of 177Lu and 90Y for anti-CD20 pretargeted radioimmunotherapy in murine lymphoma xenograft models. PLoS One. 2015 Mar 18;10(3):e0120561. Gupta S, Batra S, Jain M (2014) Antibody labeling with radioiodine and radiometals. Methods Mol Biol. 2014;1141:147-57.
IAEA (2013). Peptide receptor radionuclide therapy (PRRNT) for neuroendocrine tumors. IAEA Human Health Series No. 20., IAEA, Vienna.
Kam BLR, Teunissen JJM, Krenning EP, de Herder WW, Khan S, van Vliet EI, Kwekkeboom DJ. (2012). Lutetium-labelled peptides for therapy of neuroendocrine tumours. Eur J Nucl Med Mol Imaging 39 (Suppl 1):S103–S112.
Kamaleshwaran KK, Rajamani V, Thirumalaisamy SG, Chakraborty S, Kalarikal R, Mohanan V, Shinto AS.(2014).
Radiosynovectomy of the elbow joint synovitis in rheumatoid arthritis treated with Lutetium – 177 labeled hydroxylapatite (Lu-177HA) particulates; first case report and image of Lu -177 HA in the elbow joint. Indian J Nucl Med. 29:270-2.
Kameshwaran M, Pandey U, Dhakan C, Pathak K, Gota V, Vimalnath KV, Dash A, Samuel G. (2015) .Synthesis and Preclinical Evaluation of (177)Lu-CHX-A”-DTPA-Rituximab as a Radioimmunotherapeutic Agent for Non-Hodgkin’s Lymphoma. Cancer Biother Radiopharm. 2015 Aug;30(6):240-6Kwekkeboom DJ, Bakker WH, Kooij PP, Konijnenberg MW, Srinivasan A, Erion JL, Schmidt MA, Bugaj JL, de Jong M, Krenning EP.. (2001). [177Lu-DOTAOTyr3]octreotate: comparison with [111In-DTPAo]octreotide in patients.Eur J Nucl Med. 28: 1319-1325.
(Russ) Knapp FF, Pillai MR.(2015). Lutetium-177 Labeled Therapeutics: Emerging Importance for Cancer Treatment and Therapy of Chronic Disease. Curr Radiopharm. (ahead of Pub)
Parus JL, Pawlak D, Mikolajczak R, Duatti A. (2015) Chemistry and bifunctional chelating agents for binding 177Lu Curr Radiopharm (Ahead of Pub)
Limouris G. (2012) Neuroendocrine tumors: a focus on liver metastatic lesions. Front Oncol. 2:20 (Ahead of Pub) PMC article
Pillai MR, (Russ) Knapp FF. (2015). Evolving Important Role of Lutetium-177 for Therapeutic Nuclear Medicine Curr Radiopharm (ahead of print).
Pillai MR, Chakraborty S, Das T, Venkatesh M, Ramamoorthy N. (2003). Production logistics of 177Lu for radionuclide therapy. Appl Radiat Isot. 59: 109-118.
Shinto AS, Kamaleshwaran KK, Vyshakh K, Thirumalaisamy SG, Karthik S, Nagaprabhu VN, Vimalnath KV, Das T, Chakraborty S, Banerjee S. (2015) Radiosynovectomy of Painful Synovitis of Knee Joints Due to Rheumatoid Arthritis by Intra‑Articular Administration of 177Lu‑Labeled Hydroxyapatite Particulates: First Human Study and Initial Indian Experience. World J Nucl Med. 14: (ahead of print).
Videos
|
 |
|
| Names | |
|---|---|
| Other names
DOTA-(Tyr3)-octreotate
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| Properties | |
| C65H90N14O19S2 | |
| Molar mass | 1,435.63 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
DOTA-TATE, DOTATATE or DOTA-octreotate is a substance which, when bound to various radionuclides, has been tested for the treatment and diagnosis of certain types of cancer, mainly neuroendocrine tumours.
DOTA-TATE is an amide of the acid DOTA (top left in the image), which acts as a chelator for a radionuclide, and (Tyr3)-octreotate, a derivative of octreotide. The latter binds to somatostatin receptors, which are found on the cell surfaces of a number of neuroendocrine tumours, and thus directs the radioactivity into the tumour.
Gallium (68Ga) DOTA-TATE (GaTate[1]) is used for tumour diagnosis in positron emission tomography (PET).[2] DOTA-TATE PET/CT has a much higher sensitivitycompared to In-111 octreotide imaging.[1]
Lutetium (177Lu) DOTA-TATE[3] has been tested for the treatment of tumors such as carcinoid and endocrine pancreatic tumor. It is also known as Lutathera.[4]
Patients are typically treated with an intravenous infusion of 7.5 GBq of lutetium-177 octreotate. After about four to six hours, the exposure rate of the patient has fallen to less than 25 microsieverts per hour at one metre and the patients can be discharged from hospital.
A course of therapy consists of four infusions at three monthly intervals.[5]
Lu177 octreotate therapy is currently available under research protocols in five different medical centers in North America: Los Angeles (CA), Quebec City, (Qc), Birmingham, AL, Edmonton, (Ab), London, (On) as Houston (Tx) on clinical trial.[6] Medical centers in Europe also offer this treatment. For instance: Cerrahpasa Hospital in Turkey, Uppsala Centre of Excellence in Neuroendocrine Tumors in Sweden and Erasmus University in the Netherlands.[7] In Israel, treatment is available at Hadassah Ein Kerem Medical Center. In Australia, treatment is available at St George Hospital and Royal North Shore Hospital, Sydney;[8] the Royal Brisbane and Women’s Hospital in Brisbane [9], the Peter MacCallum Cancer Centre [1] and at the Department of Nuclear Medicine at Fremantle Hospital in Western Australia.[10] In Aarhus universitet hospital in Denmark. In the coming years such therapy will also become commercially available in Latvia, Riga – “Clinic of nuclear medicine”.
//////////////Lutathera, lutetium Lu 177 dotatate, fda 2018, PRIORITY REVIEW, ORPHAN DRUG
CC(C1C(=O)NC(CSSCC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)N1)CCCCN)CC2=CNC3=CC=CC=C32)CC4=CC=C(C=C4)O)NC(=O)C(CC5=CC=CC=C5)NC(=O)CN6CCN(CCN(CCN(CC6)CC(=O)[O-])CC(=O)[O-])CC(=O)[O-])C(=O)NC(C(C)O)C(=O)O)O.[Lu+3]
Netarsudil
| Molecular Formula: | C28H27N3O3 |
|---|---|
| Molecular Weight: | 453.542 g/mol |
Netarsudil; UNII-W6I5QDT7QI; W6I5QDT7QI; 1254032-66-0; Netarsudil [USAN]; AR-11324 free base
1422144-42-0 (mesylate) 1254032-66-0 (free base) 1253952-02-1 (HCl)
[4-[(2S)-3-amino-1-(isoquinolin-6-ylamino)-1-oxopropan-2-yl]phenyl]methyl 2,4-dimethylbenzoate

Netarsudil Mesylate
CAS: 1422144-42-0 (mesylate)
Chemical Formula: C30H35N3O9S2
Molecular Weight: 645.742
Netarsudil dimesylate is a light yellow-to-white powder that is freely soluble in water, soluble in methanol, sparingly soluble in dimethyl formamide, and practically insoluble in dichloromethane and heptane.
Netarsudil ophthalmic solution 0.02% is supplied as a sterile, isotonic, buffered aqueous solution of netarsudil dimesylate with a pH of approximately 5 and an osmolality of approximately 295 mOsmol/kg. It is intended for topical application in the eye. Each mL of netarsudil contains 0.2 mg of netarsudil (equivalent to 0.28 mg of netarsudil dimesylate). Benzalkonium chloride, 0.015%, is added as a preservative. The inactive ingredients are: boric acid, mannitol, sodium hydroxide to adjust pH, and water for injection
Netarsudil, also known as AR-11324, is a Rho-associated protein kinase inhibitor. Netarsudil is potential useful for treating glaucoma and/or reducing intraocular pressure. Netarsudil Increases Outflow Facility in Human Eyes Through Multiple Mechanisms. Netarsudil inhibited kinases ROCK1 and ROCK2 with a Ki of 1 nM each, disrupted actin stress fibers and focal adhesions in TM cells with IC50s of 79 and 16 nM, respectively, and blocked the profibrotic effects of TGF-β2 in HTM cells. Netarsudil produced large reductions in IOP in rabbits and monkeys that were sustained for at least 24 h after once daily dosing, with transient, mild hyperemia observed as the only adverse effect.
Netarsudil (trade name Rhopressa) is a drug for the treatment of glaucoma. In the United States, the Food and Drug Administrationhas approved a 0.02% ophthalmic solution for the lowering of elevated intraocular pressure in patients with open-angle glaucoma or ocular hypertension.[1]
Rho-associated protein kinase (ROCK) is a kinase belonging to the AGC (PKA/ PKG/PKC) family of serine-threonine kinases. It is involved mainly in regulating the shape and movement of cells by acting on the cytoskeleton. ROCK signaling plays an important role in many diseases including diabetes, neurodegenerative diseases such as Parkinson´s disease and amyotrophic lateral sclerosis, pulmonary hypertension and cancer. It has been shown to be involved in causing tissue thickening and stiffening around tumours in a mouse model of skin cancer, principally by increasing the amount of collagen in the tissue around the tumour.
WO 2014144781
WO2010127329

CONTINUED………..

https://www.google.com/patents/CN107434780A?cl=en










Synthesis of Compound 12
[0091] The 2,4-dimethyl benzoic acid (1.5g, IOmmol) and a catalytic amount of DMF was added to the toluene and cooled to 2-5 ° C, was added dropwise oxalyl chloride (I.64g, 13_〇1 ), warmed to room temperature after dropwise, stirred overnight, during which a solid gradually dissolved to give a clear solution, evaporated to dryness under reduced pressure to give a yellow oil with dichloromethane (IOml) was dissolved in dichloromethane to give the acid chloride ;
[0092] Compound 11 (3.2g, 7.7mmo 1) and triethylamine (2ml) were added 20ml of dichloromethane, nitrogen, the above prepared acid chloride solution in dichloromethane dropwise at 0-5 ° C the increases after mixing, overnight; TLC (dichloromethane: methanol = 20: 1) to monitor the reaction, completion of the reaction, evaporated to dryness under reduced pressure, and then stirred with saturated sodium carbonate solution, filtered, the filter cake was washed with water 3 times, dried to give 3.9g white solid, i.e. compound 12; purity: 991%, optical purity: 100% (CHIRALPAK AS-H, 0.46cm IDX15cm L, Me0H + 0.1DEA) / C02 = 20/80 (V / V, 2.0ml / min), R-type, Rt = 3 · 253min; S type Rt = 4.3min).
Compound 12 (3.9g) in DCM was added, with stirring to obtain clear solution, was then added dropwise I, a solution of hydrogen chloride in dioxane 15ml 4_ (concentration 4mol / L, 4mol HCl gas dissolved in two IL oxygen six ring), and then stirred for 4 hours at room temperature, rotary evaporated under reduced pressure, and filtered to give 3.65g product as a white solid, was obtained HNMR detectable substance is the AR-13324 hydrochloride, which IHNMR spectrum Referring to FIG. 1 , MS, purity, 99.4%, lHNMR (400MHz, DMS0,300) S (Ppm) c3Il .773 (s, 1H), 9.702 (s, lH), 8.740 (d, lH), 8.560 (d, 1H), 8.469 (d, 1H), 8.360 (d, 1H), 8.280 (s, 3H), 8.158 (dd, lH), 7.777 (d, lH), 7.577 (d, 2H), 7.496 (d, 2H), 7.134 (s, lH), 7.111 (d, lH), 5.281 (s, 2H), 4.504 (q, lH), 3.609 (q, lH), 3.139 (q, lH), 2.483 (s, 3H), 2.302 ( s, 3H).
Example 2
[0097] In this embodiment, the same processing steps except that Compound 12, the other the same as in Example 1.
[0098] Compound 12 processing steps are as follows: The compound is dissolved in 12 (3.9g) 40ml of dichloromethane, followed by dropwise addition of methanesulfonic acid (2g, 21.6mmol), stirred at room temperature overnight, rotary evaporated under reduced pressure, IOOml diethyl ether was added thereto, followed by stirring, a large amount of white solid was filtered, dried to give a white solid (4.54 g of), yield 97.8%, purity 98.2%, the resulting substance was detected IHNMR AR-13324 is the mesylate salt.
 |
|
| Clinical data | |
|---|---|
| Trade names | Rhopressa |
| Synonyms | AR-11324 |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| UNII | |
| Chemical and physical data | |
| Formula | C28H27N3O3 |
| Molar mass | 453.54 g·mol−1 |
1: Sturdivant JM, Royalty SM, Lin CW, Moore LA, Yingling JD, Laethem CL, Sherman B, Heintzelman GR, Kopczynski CC, deLong MA. Discovery of the ROCK inhibitor netarsudil for the treatment of open-angle glaucoma. Bioorg Med Chem Lett. 2016 May 15;26(10):2475-80. doi: 10.1016/j.bmcl.2016.03.104. Epub 2016 Apr 1. PubMed PMID: 27072905.
2: Ren R, Li G, Le TD, Kopczynski C, Stamer WD, Gong H. Netarsudil Increases Outflow Facility in Human Eyes Through Multiple Mechanisms. Invest Ophthalmol Vis Sci. 2016 Nov 1;57(14):6197-6209. doi: 10.1167/iovs.16-20189. PubMed PMID: 27842161; PubMed Central PMCID: PMC5114035.
3: Li G, Mukherjee D, Navarro I, Ashpole NE, Sherwood JM, Chang J, Overby DR, Yuan F, Gonzalez P, Kopczynski CC, Farsiu S, Stamer WD. Visualization of conventional outflow tissue responses to netarsudil in living mouse eyes. Eur J Pharmacol. 2016 Sep 15;787:20-31. doi: 10.1016/j.ejphar.2016.04.002. Epub 2016 Apr 13. PubMed PMID: 27085895; PubMed Central PMCID: PMC5014700.
4: Lin CW, Sherman B, Moore LA, Laethem CL, Lu DW, Pattabiraman PP, Rao PV, deLong MA, Kopczynski CC. Discovery and Preclinical Development of Netarsudil, a Novel Ocular Hypotensive Agent for the Treatment of Glaucoma. J Ocul Pharmacol Ther. 2017 Jun 13. doi: 10.1089/jop.2017.0023. [Epub ahead of print] PubMed PMID: 28609185.
5: Lu LJ, Tsai JC, Liu J. Novel Pharmacologic Candidates for Treatment of Primary Open-Angle Glaucoma. Yale J Biol Med. 2017 Mar 29;90(1):111-118. eCollection 2017 Mar. Review. PubMed PMID: 28356898; PubMed Central PMCID: PMC5369028.
/////////////Netarsudil, fda 2017, Rhopressa, AR-11324, AR 11324
CC1=CC(=C(C=C1)C(=O)OCC2=CC=C(C=C2)C(CN)C(=O)NC3=CC4=C(C=C3)C=NC=C4)C
GSK2248761A , IDX899, Fosdevirine,
Fosdevirine; IDX899; IDX-899; GSK2248761; cas 1018450-26-4; GSK-2248761, IDX 12899
| Molecular Formula: | C20H17ClN3O3P |
|---|---|
| Molecular Weight: | 413.798 g/mol |
[R(P)]-(2-Carbamoyl-5-chloro-1H-indol-3-yl)[3-(2-cyanovinyl)-5-methylphenyl]phosphinic acid methyl ester
Phase II clinical trials for the treatment of HIV infection
Idenix (Originator)
Fosdevirine, also known as GSK2248761 and IDX899, a Highly Potent Anti-HIV Non-nucleoside Reverse Transcriptase Inhibitor having an EC50 of 11 nM against the Y181C/K103N double mutant. GSK2248761 is a novel, once-daily (QD), next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI) with activity against efavirenz-resistant strains. GSK2248761 at 100 to 800 mg QD for 7 days was well tolerated, demonstrated potent antiviral activity in treatment-naive HIV-infected subjects, and had favorable PK and resistance profiles. GSK2248761 is no longer in clinical development.
IDX-12899 is a non-nucleoside reverse transcriptase inhibitors (NNRTI) originated by Idenix (acquired by Merck & Co.). It had been in phase II clinical trials for the treatment of HIV infection. However, in 2010, the compound was placed on clinical hold by the FDA. In 2009, the compound was licensed by Idenix to GlaxoSmithKline for the treatment of HIV infection on a worldwide basis.
PATENT
WO2008/042240 A2, 2008, Compound III
compound 66a: racemic form
5-chloro-3-[ methyl 3-((Zζ)-2-cyanovinyl)-5-methylphenyl] phosphinoyl-l//-indole-2- carboxamide.
[00258] Compound 66a was synthesized according to method AL. White solid, 1H NMR (CDCl3, 300 MHz) δ 2.40 (s, 3H), 3.88 (d, J= 11.7 Hz, 3H), 5.89 (d, J= 16.5 Hz, IH), 5.97 (brs, IH), 7.33-7.67 (m, 7H), 10.46 (s, IH), 10.89 (brs, IH), 31P NMR (CDCl3, 121.49 MHz) δ 31.54. MS (ES+) m/z = 414 (MH+).
Example 8
Preparation of Compound HI
305
1 (-)cιnchonιdιne, Acetone
2 1N HCI1 EtOAc
Compound 302
[00348] A suitable reactor was charged Compound 301 (10Og, 0.23mol) and tetrahydrofuran (IL). The resulting solution was chilled between -90° to -100°C under nitrogen using a LN2 / IPA slush bath, then was treated with n-butyl lithium (2.5M in Hexanes, 99ml, 0.25mol) added over 10 minutes. To this was added diethyl chlorophosphite (37.1g, 0.24mol) over 10 minutes. HPLC (Method 001, RT = 18.9 min) showed no starting material and ca. 85% product. The reaction was then diluted with ethyl acetate (IL) and was allowed to warm to -4O0C. The mix was then treated with hydrochloric acid (0.5M, 590ml) and was allowed to warm to ambient temperature and stir for 30 minutes. The resulting layers were separated and the aqueous extracted with ethyl acetate (500ml). The organics were combined and washed with brine (500ml) dried over sodium sulfate, filtered and concentrated to an oil. 88% HPLC AUC (Method 20, RT = 5.8 min) 115g, >100% yield due to impurities and solvent. Used as is in the next step. Compound 303
[00349] A suitable reactor was charged with Compound 302 (111 g, estimated 0.18mol), iodocinnamonitrile (47.1g, 0.175mol), triethylamine (29.3ml, 0.21mol) and toluene (800ml). The resulting mix was degassed by sparging with a stream of nitrogen for 10 minutes at ambient temperature, after which time tetrakis(triphenylphosphine) palladium(O) (10. Ig, 0.0088mol) was added. The mix was sparged for an additional 5 minutes, then was heated to 80°C for 2 hours. HPLC (Method 20, RT = 6.5 min) showed a complete reaction. The mix was cooled to ambient and was filtered through celite and washed with ethyl acetate (400ml). The combined organics were washed with brine (2 x 500ml) then dried over sodium sulfate, filtered and concentrated to a volume of 350ml. The concentrate was cooled to O0C and was stirred for 1 hour, during which time the product crystallized. The solids were filtered and washed with hexane:toluene (2:1, 150ml). Dried to leave 95g, 90% yield, HPLC AUC 98% (Method 20). Used as is in the next reaction. [00350] 303: C29H26ClN2O6PS 597.02gmol“‘ m/z (ESI+): 597.0 (MH+, 100%), 599.0 (MH+, 35%) 1H NMR δH (400 MHz, CDCl3): 1.38, 1.48 (2 x 3H, 2 x t, COOCH2CH3, POOCH2CH3), 2.41 (3Η, s, Ar-CH3), 4.09-4.16 (2Η, m, POOCH2CH3), 4.52 (2H, q, COOCH2CH3), 5.93 (IH, d, CH=CHCN), 7.33-7.38 (3Η, m, CH=CHCN, 2 x Ar-H), 7.52 (2Η, t, 2 x Ar-H), 7.64 (1Η, t, Ar-H), 7.74, 7.77 (2 x 1Η, 2 x d, 2 x Ar-H), 7.85 (1Η, d, Ar- H), 7.94 (1Η, dd, Ar-H), 8.08 (2Η, d, 2 x Ar-H) 1H NMR δH (400 MHz, d6-DMSO): 1.26, 1.33 (2 x 3H, 2 x t, COOCH2CH3, POOCH2CH3), 2.34 (3Η, s, Ar-CH3), 3.95-4.10 (2Η, m, POOCH2CH3), 4.40 (2H, q, COOCH2CH3), 6.52 (IH, d, CH=CHCN), 7.52 (1Η, dd, Ar-H), 7.60-7.84 (8Η, m, CH=CHCN, 7 x Ar-H), 8.07 (3 x 1Η, m, 3 x Ar-H)
Compound 304
[003511 A suitable reactor was charged with Compound 303 (537g, 0.90mol) and methylene chloride (2.0L). The resulting solution was cooled to O0C, and was treated with bromotrimethylsilane (45Og, 2.9mol) added over 15 minutes. The reaction was then warmed to 400C for 1.5 hours. ΗPLC (Method 20, RT = 4.4 min) indicated a complete reaction. The excess TMSBr was stripped under vacuum (40 – 45°C) and the resulting sticky solid was resuspended in DCM (2.5L) and chilled to 00C. Oxalyl chloride (156ml, 1.8mol) was added over 15 minutes, followed by N,N-dimethylformamide (13.7ml, 0.18mol) both added at O0C. Gas evolution was observed during the DMF addition. After 1 hour, ΗPLC (Method 20, RT = 6.2 min, sample quenched with anhydrous methanol prior to injection) showed a complete reaction. The solvents were stripped again to remove residual oxalyl chloride and the mix resuspended in chilled methanol (3.0L) at 0° – 5°C, and then was allowed to warm to ambient. After two hours, HPLC indicated a complete reaction (HPLC Method 20, RT = 6.2 min). The solution was concentrated to a volume of 1.5L, and the resulting thin slurry was cooled to 0°C, and was diluted with an aqueous solution of sodium bicarbonate (126g, 3L water). After 2 hours at 50C, the product was filtered and washed with cold water / methanol (2:1, 1.5L) then dried to leave 50Og Compound 304. HPLC (Method 20) purity 92% used as is.
Compound 305
[00352] A suitable reactor was charged with Compound 304 (ca. 28Og, 0.48mol) and tetrahydrofuran (2.8L). The resulting solution was then cooled to 5°C and was treated with lithium hydroxide monohydrate (45g, 1.07mol) added in one portion. The reaction was allowed to warm to ambient, during which time the color lightened and a white precipitate formed. After overnight stirring, HPLC indicated an incomplete reaction (Method 20, product RT = 4.3, partially deprotected RT = 5.1, major impurity RT = 3.8). An additional 10% LiOH-H2O was added, but after 10 hours, the partially deprotected intermediate remained at 5%, and the impurity peak at 3.8 minutes had increased to ca. 25%. The reaction was cooled to 50C and was acidified with hydrochloric acid (5N, 280ml) then was diluted with ethyl acetate (2L). The layers were separated and the aqueous extracted with ethyl acetate (500ml). The combined organics were washed with brine (IL) and dried with sodium sulfate, then concentrated to leave a crude oily solid, Compound 305. Ca. 300g, HPLC AUC 57%.
[00353] The crude product was taken up in acetonitrile (1.2L) at 4O0C, and the product triturated w/ water (1.2L). The resulting slurry was cooled to 50C and was allowed to granulate for 30 minutes, after which time the product was filtered and washed with ACN:H2O (1 :1, 100 ml). Ca. 103g, 88% by HPLC. The product was then recrystallized from 360ml ACN at 400C and 360ml water as before. Filtered, washed and dried to leave 75g Compound 305. HPLC AUC 97%. Used as is in the next step.
Compound 306 (chiral resolution)
[00354] A suitable reactor was charged with Compound 305 (28Og, 0.66mol) and acetone (4.2L). The resulting thin slurry was then treated with (-)-cinchonidine (199g, 0.66mol) added in one portion. After one hour, a solution had formed, and after an additional hour, a white solid precipitated, and the mix was left to stir for an additional two hours (four hours total) after which time the solids were filtered, washed with acetone (200ml) and dried to leave 199g Crude Compound 306 cinchonidine salt. HPLC showed an isomer ratio of 96:4.
[00355] The crude salt was then slurried in ethyl acetate (3L) and hydrochloric acid (IN, 3L). The two phase solution was vigorously stirred for 2 hours at ambient temperature. The layers were separated, and the aqueous extracted with ethyl acetate (3L). The organics were combined, dried with sodium sulfate, and concentrated to leave the free base Compound 306, 107g, 95:5 by chiral HPLC.
[00356] The crude Compound 306 was then suspended in acetone (1.07L) and treated with (-)-cinchonidine (76g, 0.26 mol.) After 4 hours total stir time (as above) the solids were filtered, washed with acetone (200ml) and dried to leave 199g of the salt. HPLC 98.6:1.4.
[00357] The salt was broken by dissolving in ethyl acetate (3L) and hydrochloric acid (IN, 3L). The two phase solution was stirred for 2 hours at ambient temperature. The layers were separated, and the aqueous extracted with ethyl acetate (2L). The organics were combined, dried with sodium sulfate, and concentrated to leave the free base Compound 306, 98g, 98.6:1.4 by chiral HPLC. 70% recovery of the desired isomer, 35% yield from the racemic Compound 306. #6: C20H16ClN2O4P 414.78gmol“‘ m/z (ESI+): 415.1 (MH+, 100%), 417.0 (MH+, 35%) [α]D 25 : -47.51 (c, 10.66mgml“‘ in EtOAc) [Opposite enantiomer [α]D 25 : +47.26 (c, 9.60mgml“‘ in EtOAc)] 1H NMR δH (400 MHz, d6-DMSO): 2.33 (3 H, s, Ar-CH3), 3.71 (3H, d, CH3OP), 6.50 (1Η, d, CH=CHCN), 7.36 (1Η, dd, H-6), 7.57 (1Η, d, H-I), 7.66-7.71 (2Η, m, H-4, Ar-Hortho), 7.67 (1Η, d, CH=CHCN), 7.84 (IH, d, Ar-Hortho), 7.98 (1Η, s, Ar-Hpara), 12.97 (1Η, s, N-H), 14.38 (1Η, br-s, COOH) Multiple δc values indicate splitting of carbon signal due to P. 13C NMR δc (100 MHz, d6-DMSO): 20.68 (Ar-CH3), 51.70 (CH3OP), 98.15 (CH=CHCN), 102.33, 103.85, 1 14.98, 120.91 (3 x Q, 118.47 (CN), 125.39 (C), 126.78 (Q, 127.74, 127.86 (C- Hortho), 129.78, 129.88 (Q, 131.25 (Q, 132.06 (Q, 133.44, 133.55 (Q, 133.89, 134.05 (Q, 134.62, 134.75 (Q, 135.47, 135.66 (Q, 138.78, 138.91 (Q, 149.62 (CH=CHCN), 160.40 (C=O) 31P NMR δP (162 MHz, d6-DMSO): 33.50 (IP, s)
Compound HI
[00358] A suitable reactor was charged with Compound 306 (0.63g, O.OOHmol) and 1 ,2-dimethoxyethane (10ml.) The mix was treated with 1,1-carbonyldiimidazole (0.47g, 0.0028mol) added in one portion, and the mix was allowed to stir at ambient temperature until gas evolution ceased (ca. 1.5 hours.) The solution was then cooled to 50C, and was sparged with ammonia gas for 5 minutes. HPLC (Method 20, product RT=5.0 min) showed a complete reaction after one hour at ambient. The reaction was quenched by the addition of 1Og crushed ice, and was concentrated under reduced pressure to remove the DME. The resulting slurry was stirred for one hour at 50C to granulate the product. The solids were filtered and dried to leave pure Compound III ((2-Carbamoyl-5-chloro-4-fluoro-lH-indol-3- yl)-[3-((E)-2-cyano-vinyl)-5-methyl-phenyl]-(S)-phosphinic acid methyl ester) as a white solid 0.56g, 89% yield. HPLC (Method 20) chemical purity 98.5%. Chiral purity 97%. [00359] A suitable reactor was charged with Compound 306 (1Og, 0.024mol) and 1,2- dimethoxyethane (150ml). The mix was treated with 1,1-carbonyldiimidazole (7.8g, 0.048mol) added in one portion, and the mix was allowed to stir at ambient temperature until gas evolution ceased. The solution was then cooled to 5°C, and was sparged with ammonia gas for 5 minutes. HPLC (Method 20, product RT=5.0 min) showed a complete reaction after one hour. The reaction was quenched by the addition of lOOg crushed ice, and was concentrated under reduced pressure to remove the DME. The resulting oily solid (in water) was diluted with methanol (20ml) and stirred for one hour at 50C to granulate the product. The solids were filtered and dried to leave pure Compound III ((2-Carbamoyl-5- chloro-4-fluoro-lH-indol-3-yl)-[3-((E)-2-cyano-vinyl)-5-methyl-phenyI]-(S)-phosphinic acid methyl ester). 9.8g, 98% yield. HPLC (Method 20) chemical purity 99.5%. Chiral purity 94.3%.
Compound III: C20Hi7ClN3O3P 413.79gmol“‘ m/z (ESI+): 414.1 (MH+, 100%), 416.1 (MH+, 35%)
vmax (KBr disc) (cm“1) 1620.0 (amide I), 1670.6 (amide II), 2218.7 (CN), 3125.5, 3291.9 (N-H)
[α]D 20 : -75.08 (c, 9.04mgmr’ in CHCl3)
m.p.: 144- 1480C transition to opaque semi-solid, 209-2100C melts
Elemental analysis: C20H17ClN3O3P calculated C 58.05%, H 4.14%, N 10.15%, Cl 8.57%, P 7.49%. Found C 58.13%, H 4.08%, N 10.16%, Cl 8.69%, P 7.44%
1H NMR δH (400 MHz, d6-DMSO): 2.32 (3H, s, Ar-CH3), 3.74 (3Η, d, CH3OP), 6.52 (1Η, d, CH=CHCN), 7.30 (1Η, dd, H-6), 7.53-7.58 (3Η, m, H-4, H-7, H-6′), 7.68 (1Η, d, CH=CHCN), 7.73 (IH, s, H-4′), 7.75 (1Η, d, H-2′), 8.02, 10.15 (2 x 1Η, 2 x s, NH2), 12.80 (1Η, s, N-H) Multiple δc values indicate splitting of carbon signal due to P.
13C NMR δc(100 MHz, d6-DMSO): 20.77 (Ar-CH3), 51.75, 51.81 (CH3OP), 98.39, 98.91 (C-3), 98.44 (CH=CHCN), 1 15.05 (C-7), 1 18.53 (CN), 119.96 (C-4), 124.73 (C-6), 126.68 (C-5), 127.15, 127.26 (C-2′), 129.25, 129.35 (C-9), 131.37 (C-4′), 132.45, 134.04 (C-I ‘), 132.69, 132.80 (C-6′), 133.92 (C-8), 134.30, 134.44 (C-3′), 139.33, 139.46 (C-5’), 139.96, 140.17 (C-2), 149.55 (CH=CHCN), 160.65 (C=O)
31P NMR δP (162 MHz, d6-DMSO): 33.72 (IP, s)
PATENT
http://www.google.ch/patents/WO2009120914A1?cl=en&hl=de

(2-carbamoyl-5-chloro-lH-indol-3-yl)-[3-((E)-2-cyano-vinyl)-5-methyl-phenyl]- (7?)-phosphinic acid methyl ester (I):
| WO2008042240A2 * | 28. Sept. 2007 | 10. Apr. 2008 | Idenix Pharmaceuticals, Inc. | Enantiomerically pure phosphoindoles as hiv inhibitors |
| US20060074054 * | 16. Sept. 2005 | 6. Apr. 2006 | Richard Storer | Phospho-indoles as HIV inhibitors |
Figure 7 provides an infrared spectrum of Form I.
Paper
Amidation of indole 2-carboxylate 1 with ammonia gas via the imidazolide 2 gave GSK2248761A API 3, which was in development for the treatment of HIV. Three significant impurities, namely the phosphinic acid 4, the N-acyl urea 8, and the indoloyl carboxamide 6, were formed during the reaction, and the original process was unable to produce API within clinical specification when run at scale. Investigation into the origin, fate, and control of these impurities led to a new process which was able to produce API within clinical specification.
A new and improved synthetic route to an intermediate in the synthesis of the phosphinate ester GSK2248761A is described. In the key step, we describe the first process-scale example of a palladium-catalyzed phosphorus–carbon coupling to give the entire backbone of GSK2248761A in one telescoped stage in 65% average yield on a 68 kg scale. This unusual chemistry enabled the route to be reduced from six chemistry stages to four and eliminated a number of environmentally unfriendly reagents and solvents.
1: Dousson C, Alexandre FR, Amador A, Bonaric S, Bot S, Caillet C, Convard T, da Costa D, Lioure MP, Roland A, Rosinovsky E, Maldonado S, Parsy C, Trochet C, Storer R, Stewart A, Wang J, Mayes BA, Musiu C, Poddesu B, Vargiu L, Liuzzi M, Moussa A, Jakubik J, Hubbard L, Seifer M, Standring D. Discovery of the Aryl-phospho-indole IDX899, a Highly Potent Anti-HIV Non-nucleoside Reverse Transcriptase Inhibitor. J Med Chem. 2016 Feb 3. [Epub ahead of print] PubMed PMID: 26804933.
2: Margolis DA, Eron JJ, DeJesus E, White S, Wannamaker P, Stancil B, Johnson M. Unexpected finding of delayed-onset seizures in HIV-positive, treatment-experienced subjects in the Phase IIb evaluation of fosdevirine (GSK2248761). Antivir Ther. 2014;19(1):69-78. doi: 10.3851/IMP2689. Epub 2013 Oct 24. PubMed PMID: 24158593.
3: Ölgen S. Recent development of new substituted indole and azaindole derivatives as anti-HIV agents. Mini Rev Med Chem. 2013 Oct;13(12):1700-8. Review. PubMed PMID: 23895189.
4: Castellino S, Groseclose MR, Sigafoos J, Wagner D, de Serres M, Polli JW, Romach E, Myer J, Hamilton B. Central nervous system disposition and metabolism of Fosdevirine (GSK2248761), a non-nucleoside reverse transcriptase inhibitor: an LC-MS and Matrix-assisted laser desorption/ionization imaging MS investigation into central nervous system toxicity. Chem Res Toxicol. 2013 Feb 18;26(2):241-51. doi: 10.1021/tx3004196. Epub 2012 Dec 20. PubMed PMID: 23227887.
5: Zala C, St Clair M, Dudas K, Kim J, Lou Y, White S, Piscitelli S, Dumont E, Pietropaolo K, Zhou XJ, Mayers D. Safety and efficacy of GSK2248761, a next-generation nonnucleoside reverse transcriptase inhibitor, in treatment-naive HIV-1-infected subjects. Antimicrob Agents Chemother. 2012 May;56(5):2570-5. doi: 10.1128/AAC.05597-11. Epub 2012 Feb 6. PubMed PMID: 22314532; PubMed Central PMCID: PMC3346662.
6: Piscitelli S, Kim J, Gould E, Lou Y, White S, de Serres M, Johnson M, Zhou XJ, Pietropaolo K, Mayers D. Drug interaction profile for GSK2248761, a next generation non-nucleoside reverse transcriptase inhibitor. Br J Clin Pharmacol. 2012 Aug;74(2):336-45. doi: 10.1111/j.1365-2125.2012.04194.x. PubMed PMID: 22288567; PubMed Central PMCID: PMC3630753.
7: La Regina G, Coluccia A, Silvestri R. Looking for an active conformation of the future HIV type-1 non-nucleoside reverse transcriptase inhibitors. Antivir Chem Chemother. 2010 Aug 11;20(6):213-37. doi: 10.3851/IMP1607. Review. PubMed PMID: 20710063.
8: Klibanov OM, Kaczor RL. IDX-899, an aryl phosphinate-indole non-nucleoside reverse transcriptase inhibitor for the potential treatment of HIV infection. Curr Opin Investig Drugs. 2010 Feb;11(2):237-45. Review. PubMed PMID: 20112173.
9: Zhou XJ, Garner RC, Nicholson S, Kissling CJ, Mayers D. Microdose pharmacokinetics of IDX899 and IDX989, candidate HIV-1 non-nucleoside reverse transcriptase inhibitors, following oral and intravenous administration in healthy male subjects. J Clin Pharmacol. 2009 Dec;49(12):1408-16. doi: 10.1177/0091270009343698. Epub 2009 Sep 23. PubMed PMID: 19776293.
10: Zhou XJ, Pietropaolo K, Damphousse D, Belanger B, Chen J, Sullivan-Bólyai J, Mayers D. Single-dose escalation and multiple-dose safety, tolerability, and pharmacokinetics of IDX899, a candidate human immunodeficiency virus type 1 nonnucleoside reverse transcriptase inhibitor, in healthy subjects. Antimicrob Agents Chemother. 2009 May;53(5):1739-46. doi: 10.1128/AAC.01479-08. Epub 2009 Feb 17. PubMed PMID: 19223643; PubMed Central PMCID: PMC2681571.
11: Mascolini M, Larder BA, Boucher CA, Richman DD, Mellors JW. Broad advances in understanding HIV resistance to antiretrovirals: report on the XVII International HIV Drug Resistance Workshop. Antivir Ther. 2008;13(8):1097-113. PubMed PMID: 19195337.
12: Dalton P. Two new NNRTIs enter the pipeline. Proj Inf Perspect. 2008 Sep;(46):13. PubMed PMID: 19048672.
13: Sweeney ZK, Klumpp K. Improving non-nucleoside reverse transcriptase inhibitors for first-line treatment of HIV infection: the development pipeline and recent clinical data. Curr Opin Drug Discov Devel. 2008 Jul;11(4):458-70. Review. PubMed PMID: 18600563.
/////////////GSK2248761A , IDX899, Fosdevirine, PHASE 2
CC1=CC(=CC(=C1)C=CC#N)P(=O)(C2=C(NC3=C2C=C(C=C3)Cl)C(=O)N)OC

Besifovir
Besifovir (INN) is an investigational medication to treat hepatitis B virus (HBV) infection. It is a novel and potent acyclic nucleotide phosphonate with a similar chemical structure to adefovir and tenofovir.[2]
Besifovir dipivoxil maleate
CAS:1039623-01-2, Propanoic acid, 2,2-dimethyl-, 1,1′-[[[[[1-[(2-amino-9H-purin-9-yl)methyl]cyclopropyl]oxy]methyl]phosphinylidene]bis(oxymethylene)] ester, (2Z)-2-butenedioate (1:1)
| Molecular Formula | C22 H34 N5 O8 P . C4 H4 O4 |
| Molecular Weight | 643.58 |
| Highest Phase | Launched – 2017, korea
Besifovir dipivoxil maleate |
Besifovir, also known as ANA-380; LB-80380; PMCDG dipivoxil, is a reverse transcriptase inhibitor potentially for treatment of hepatitis B infection. LB80380 is a prodrug and an oral nucleotide analogue that inhibits viral replication by incorporation into the viral DNA. Antiviral activity against wild-type virus and virus with drug-resistant mutations was demonstrated in Phase II trials, with significant reduction of viral load in patients treated with LB80380. LB80380 was also shown to be safe and well tolerated.

CAS 441785-26-8
Chemical Formula: C22H34N5O7P
Molecular Weight: 511.5158
Besifovir; ANA-380; AN-380; ANA 380; LB-80380; LB 80380; LB80380; PMCDG dipivoxil.
IUPAC/Chemical Name: ((((1-((2-amino-9H-purin-9-yl)methyl)cyclopropoxy)methyl)phosphanediyl)bis(oxy))bis(methylene) bis(2,2-dimethylpropanoate)
|
Ildong Pharmaceutical will release its first chronic hepatitis B therapy of nucleotide series “Besivo” as an insurance benefit drug next month, the company said Thursday. Besivo is a treatment for chronic hepatitis B based on the nucleotide sequence, which is composed of besifovir. The price of the insurance is 3,403 won ($3.02) per tablet, which was recently confirmed by the Ministry of Health and Welfare보건복지부. Insurance benefits also cover El-carnitine medications used in combination, and the insurance price for one tablet (330mg) is 111 won. According to the results of clinical trials, Besivo has demonstrated comparable levels of therapeutic efficacy in a randomized, double-blind trial compared with traditional therapies such as Entecavir (trade name: Baraclude) and Tenofovir (trade name: Viread). Besivo improved the prospects as a valid option for the treatment of chronic hepatitis B by improving the side effects found in the existing medications. In particular, further analysis of clinical trials has shown that typical side effects such as decreased renal function and decreased bone density, which was a problem in the existing Tenofovir. Knodell necro-inflammatory score, was also superior to the control group regarding the histological improvement of the liver.
For the deterioration of renal function, the rate of increase in serum creatinine — a test that measures kidney function – was significantly lower than that of Tenofovir. With the case of measuring bone mineral density, the proportion of patients showing bone turnover increased and the percentage of patients showing average bone mineral density decreased in the case of Tenofovir. With Besivo, the rate of patients with bone loss decreased, and the percentage of patients with average bone mineral density increased. Ildong Pharma 일동제약 (CEO: Yun Woong-sup윤웅섭) plans to go on marketing with the idea that Besivo is a domestic drug that secures safety by improving side effects of existing medicines as well as treatment effects that are comparable to that of foreign pharmaceutical companies. In particular, the company expects the cost of pharmaceuticals to be 25 percent lower than that of Viread, the leading drug in the market. “Due to the nature of chronic hepatitis B treatment for long-term use, safety is critical, and there are few side effects, so Besivo is highly valuable as a few nucleotide drugs existing in consideration of cross-resistance,” said Professor Ahn Sang-hoon안상훈 of Severance Hospital세브란스병원who participated in the clinical study. “There is a strong competitive edge regarding the advantages of Besivo in connection with the entry into the Asian market, where the demand for therapeutic drugs is increasing as a major outbreak of hepatitis B,” he added. |
Il Dong, under license from LG Life Sciences , has developed and launched Besivo (besifovir dipivoxil maleate), a phosphonate nucleoside inhibitor of HBV polymerase, for treating HBV infection. In October 2012, Il Dong was planning on seeking to outlicense the drug outside of Korea.
Besifovir dipivoxil maleate is a DNA polymerase inhibitor discovered and developed by LG Chem. The product was launched in Korea in 2017 by codeveloper ILDONG for the treatment of hepatitis B.
In April 2004, Anadys (acquired by Roche in 2011) obtained an exclusive license from LG Chem for the commercialization of LB-80380 worldwide excluding China, Korea, India and Southeast Asia. In August 2007, Anadys reported that it had discontinued development of LB-80380 and returned all rights to LG Chem in order to focus on other key compounds.
PAPER
9-[1-(Phosphonomethoxycyclopropyl)methyl]guanine (PMCG, 1), representative of a novel class of phosphonate nucleosides, blocks HBV replication with excellent potency (EC50 = 0.5 μM) in a primary culture of HepG2 2.2.15 cells. It exhibits no significant cytotoxicity in several human cell lines up to 1.0 mM. It does not inhibit replication of human immunodeficiency virus (HIV-1) or herpes simplex virus (HSV-1) at 30 μM. Many purine base analogues of 1 also exhibit inhibitory activity against HBV, but at 30 μM, pyrimidine analogues do not. 1 is 4 times more potent than 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA), which was used as a positive control (EC50 = 2.0 μM). The characteristic cyclopropyl moiety at the 2‘-position of 1 was prepared by titanium-mediated Kulinkovich cyclopropanation. 1 was modified to give the orally available drug candidate, PMCDG Dipivoxil (2). Compound 2 exhibited excellent efficacy when administered at 5 mg per kg per day in a study with woodchucks infected with woodchuck hepatitis B virus (WHBV). Drug candidate 2 has successfully completed phase I clinical trials and is currently undergoing phase II clinical studies for evaluation of efficacy.
({1-[(2-Amino-9H-purin-9-yl)methyl]cyclopropyl}oxy)methylphosphonic Acid (PMCDG, 8). 8 (89.5% yield) as yellowish solids. The compound was recrystallized from water for X-ray crystallography. 1H NMR (400 MHz, DMSO-d6): δ 0.92 (br q, 4H), 3.76 (d, J = 12.0 Hz, 2H), 4.33 (s, 2H), 8.0 (br s, 2H), 8.74 (s, 1H), 9.00 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 11.6 (2C), 45.9, 62.9 (d, J = 15.0 Hz), 63.0 (d, J = 161 Hz), 125.6, 139.1, 149.8, 154.2, 157.1. HRMS (MH+): 300.0862 calcd for C10H14N5O4P, found 300.0872. Anal. (C10H14N5O4P·H2O) C, H, N.
({1-[(2-Amino-9H-purin-9-yl)methyl]cyclopropyl}oxy)methylphosphonic Acid Dipivoxyl (PMCDG Dipivoxyl, 2). 2 (38.5% yield) as white solids. mp: 92 °C. 1H NMR (400 MHz, CDCl3): δ 0.89 (br t, 2H), 1.06 (br t, 2H), 1.21 (s, 18H), 3.97 (d, J = 10.0 Hz, 2H), 4.23 (s, 2H), 5.0 (br s, 2H), 5.62 (m, 2H), 8.01 (s, 1H), 8.68 (s, 1H). 13C NMR (100 MHz, CDCl3): δ 12.3 (2C), 26.7 (6C), 38.6 (2C), 46.0, 62.1 (d, J = 170 Hz), 64.1 (d, J = 15.0 Hz), 81.6 (d, J = 6.0 Hz, 2C), 127.6, 143.0, 149.4, 153.4, 158.9, 176.6 (2C). HRMS (MH+): 528.2223 calcd for C10H14N5O4P, found 528.2233. Anal. (C22H34N5O8P) C, H, N.
PATENT
Besifovir dipivoxil’s product PAT, WO2057288
https://encrypted.google.com/patents/WO2002057288A1?cl=en

(a) CH3CH2MgBr, Ti(Oi-Pr)4 (0.25 equiv), THF, 0 °C to 25 °C, 10 h;
(b) BrCH2P(O)(Oi-Pr)2, LiOt-Bu, LiI (cat.), DMF, THF, 60 °C, 4 h;
(c) NH4F, MeOH, reflux, 10 h;
CONTD……….

(d) MsCl, TEA, MDC, 0 °C to 25 °C; (e) 6-chloroguanine, NaH, DMF, 80 °C, 4 h; (f) H2, 5% Pd on C, THF, 1 atm, 18 h; (g) TMSBr, MDC, reflux, 18 h; (h) 2 N HCl, reflux, 6 h; (i) chloromethyl pivalate, TEA, 1-methyl-2-pyrrolidinone, 25 °C, 48 h.
WO 2002057288
PATENT
WO-2018016795
Novel crystalline polymorphic forms of 3-[({1-[(2-amino-9H-furyn-9-yl) methyl] cyclopropyl}oxy) methyl]-8,8-dimethyl-3,7-dioxo-2,4,6-trioxa-3λ5-phosphanon-1-yl-pivalate orotate (Besifovir dipivoxil), a process for its preparation, and composition comprising the salt for treating viral infections are claimed.
|
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C10H14N5O4P |
| Molar mass | 299.223022 g/mol |
| 3D model (JSmol) | |
////////////Besifovir, бесифовир , بيسيفوفير , 贝西福韦 , ANA-380, AN-380, ANA 380, LB-80380, LB 80380, LB80380, PMCDG dipivoxil
C1CC1(CN2C=NC3=CN=C(N=C32)N)OCP(=O)(O)O
NC1=NC=C2N=CN(CC3(OCP(OCOC(C(C)(C)C)=O)OCOC(C(C)(C)C)=O)CC3)C2=N1




Acalabrutinib, rINN, ACP-196,
FDA 2017 APPROVED, Lymphoma, mantle cell, ACERTA PHARMA
Orphan Drug, breakthrough therapy designation,
(S)-4-[8-Amino-3-[1-(but-2-ynoyl)pyrrolidin-2-yl]imidazo[1,5-a]pyrazin-1-yl]-N-(pyridin-2-yl)benzamide
(S)-4-(8-amino-3-n-but-2-vnoylpyrrolidin-2-vnimidazo[1 ,5-alpyrazin-1-yl)-N-(pyridin-2-yl)benzamide
Acalabrutinib (rINN,[1] ACP-196) is a novel experimental anti-cancer drug and a 2nd generation Bruton’s tyrosine kinase (BTK) inhibitor[2][3] developed by Acerta Pharma.[4] It is more potent and selective (fewer side-effects) than ibrutinib, the first-in-class BTK inhibitor.[2][3][5]
The compound was granted orphan drug designation for the treatment of chronic lymphocytic leukemia, Waldenström’s macroglobulinemia and mantle cell lymphoma in the U.S. and the E.U. in 2015 and 2016, respectively. In 2017, the product was granted breakthrough therapy designation in the U.S. for the treatment of patients with mantle cell lymphoma who have received at least one prior therapy.
Acalabrutinib is an orally available inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, acalabrutinib inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways. This leads to an inhibition of the growth of malignant B cells that overexpress BTK. BTK, a member of the src-related BTK/Tec family of cytoplasmic tyrosinekinases, is overexpressed in B-cell malignancies; it plays an important role in B lymphocyte development, activation, signaling, proliferation and survival.
Acalabrutinib is a Bruton’s Tyrosine Kinase (BTK) inhibitor developed at Acerta Pharma launched in 2017 in the U.S. for the oral treatment of adults with mantle cell lymphoma who have received at least one prior therapy.
-Facebook.png)

Relative to ibrutinib, acalabrutinib demonstrated higher selectivity and inhibition of the targeted activity of BTK, while having a much greater IC50 or otherwise virtually no inhibition on the kinase activities of ITK, EGFR, ERBB2, ERBB4, JAK3, BLK, FGR, FYN, HCK, LCK, LYN, SRC, and YES1.[3] In addition, in platelets treated with ibrutinib, thrombus formation was clearly inhibited while no impact to thrombus formation was identified relative to controls for those treated with acalabrutinib.[3] These findings strongly suggest an improved safety profile of acalabrutinib with minimized adverse effects relative to ibrutinib.[3]
As was conducted in the development of ibrutinib, pre-clinical studies of acalabrutinib included in vitro and in vivo pharmacodynamic evaluation in a canine lymphoma model.[6] A dose-dependent relationship resulting in cyto-toxicity and anti-proliferative effects was first demonstrated in a canine lymphoma cell line in vitro.[6] In vivo, the compound was found to be generally safe and well tolerated in the dosage range of 2.5–20 mg/kg every 12 or 24 hours, with clinical benefit observed in 30% of canine patients while observed adverse events consisted primarily of gastrointestinal effects such as anorexia, weight loss, vomiting, diarrhea and lethargy.[6]

The interim results of the still on-going first human phase 1/2 clinical trial (NCT02029443) with 61 patients for the treatment of relapsed chronic lymphocytic leukemia (CLL) are encouraging, with a 95% overall response rate demonstrating potential to become a best-in-class treatment for CLL.[2][7] Notably, a 100% response rate was achieved for those patients which were positive for the 17p13.1 gene deletion – a subgroup of patients that typically results in a poor response to therapy and expected outcomes.[3]
The most common adverse events were headache, diarrhea and weight gain.[3] Despite the appearance of a greater occurrence of transient headaches, the pre-clinical data suggests a preferred advantage of acalabrutinib over ibrutinib due to expected reduced adverse events of skin rash, severe diarrhea, and bleeding risk.[3] An additional clinical trial is currently in progress to directly compare the safety and efficacy performance of acalabrutinib to ibrutinib to better elucidate the differences in the therapeutic agents.[3]
While the primary indication is for CLL, as of late 2016, acalabrutinib is under evaluation for multiple indications in 20+ clinical trials (alone and in combination with other interventions) for various blood cancers, solid tumors, and rheumatoid arthritis.[7][8] Approximately 1,000 patients have been treated with acalabrutinib in clinical trials so far, including more than 600 on acalabrutinib alone and almost 400 on additional therapies in combination with acalabrutinib.[9]
As of February 2016, acalabrutinib had received orphan designation in the United States for CLL only,[10] and was similarly designated as an orphan medicinal product by the European Medicines Agency (EMA) Committee for Orphan Medicinal Products (COMP) for treatment of three indications – chronic lymphocytic leukemia (CLL)/ small lymphocytic lymphoma (SLL), mantle cell lymphoma (MCL), and lymphoplasmacytic lymphoma (Waldenström’s macroglobulinaemia, MG).[11] If the drug is ultimately approved, this designation will result in a 10-year period of market exclusivity for the stated indications within Europe.[12]
Acerta Pharma, the innovator responsible for the discovery and development of acalabrutinib, is a clinical stage biopharmaceutical company recently founded in 2012 in Oss, the Netherlands.[13] A combined $13 Million in Series A funding was secured March 14, 2013 from various investor sources including the venture capital firms of BioGeneration Ventures and OrbiMed Advisors, the Dutch State and Province of Brabant through the Brabant Development Agency, and the private US equity firm Frazier Healthcare.[14] Further undisclosed amounts of Series B funding was secured May 2015 from the mutual fund company T. Rowe Price.[15]
After the promising results for the treatment of CLL in initial clinical trials,[2] Astra Zeneca purchased a 55% stake in Acerta Pharma for $4 billion in December 2015, with an option to acquire the remaining 45% stake for an additional $3 billion, conditional on the first approval in both the US and Europe and the establishment of commercial opportunity.[16]
Several patents have been filed by Acerta Pharma through the World Intellectual Property Organization (WIPO) for the use of acalabrutinib (and structurally similar derivatives) either alone or in combination with additional therapeutic agents for the treatment of various hematological and solid tumor cancers as well as inflammatory and autoimmune diseases.[17][18][19][19][20][21][22][23][24][25][26][27]
Notably, patents filed through WIPO still need to be filed appropriately for each individual nation on the path to commercialization. For example, one related United States patent application is US2014155385, which was filed July 11, 2012 and approved June 5th, 2014 for the use of 6-5 membered fused pyridine ring compounds (including acalabrutnib and its structurally similar derivatives) in the treatment of BTK mediated disorders.[28]
SYNTHESIS
WO 2013010868

Synthesis of acalabrutinib, using 3-chloropyrazine-2-carbonitrile as the starting material, is described. The method comprises reduction of the starting material, condensation with N-Cbz-L-proline, intramolecular cyclization, bromination, Suzuki coupling with (4-(2-pyridylcarbamoyl)phenyl)boronic acid and condensation with 2-butynoic acid. WO 2013010868
Reduction of 3-chloropyrazine-2-carbonitrile with H2 over Raney-Ni in AcOH, followed by treatment with aqueous HCl in Et2O gives (3-chloro-2-pyrazinyl)methylamine hydrochloride , which upon condensation with N-Cbz-L-proline in the presence of HATU and Et3N in CH2Cl2 affords amide .
Intramolecular cyclization of intermediate by means of DMI and POCl3 in acetonitrile at 63 °C provides N-Cbz-8-chloro-3-[2(S)-pyrrolidinyl]imidazo[1,5-a]pyrazine , which is brominated with NBS in DMF to yield N-Cbz-1-bromo-8-chloro-3-[2(S)-pyrrolidinyl]imidazo[1,5-a]pyrazine .
Reaction of chloro compound with NH3 in i-PrOH at 110 °C produces N-Cbz-1-bromo-3-[2(S)-pyrrolidinyl]imidazo[1,5-a]pyrazin-8-amine , which upon Suzuki coupling with (4-(2-pyridylcarbamoyl)phenyl)boronic acid in the presence of PdCl2(dppf) and K2CO3 in dioxane at 140 °C under microwave irradiation furnishes diaryl derivative .
Removal of the benzyloxycarbonyl moiety in intermediate using HBr in AcOH generates pyrrolidine derivative , which is condensed with 2-butynoic acid in the presence of HATU and Et3N in CH2Cl2 to afford the target acalabrutinib
PATENT
WO 2013010868
https://www.google.com/patents/WO2013010868A1?cl=en
scheme I
scheme II
Intermediate 1
(S)-Benzyl 2-(8-amino-1-bromoimidazo[1 ,5-alpyrazin-3-vnpyrrolidine-1-carboxylate
(a) (3-Chloropyrazin-2-yl)methanamine. hydrochloride
To a solution of 3-chloropyrazine-2-carbonitrile (160 g, 1 .147 mol) in acetic acid (1.5 L) was added Raney Nickel (50% slurry in water, 70 g, 409 mmol). The resulting mixture was stirred under 4 bar hydrogen at room temperature overnight. Raney Nickel was removed by filtration over decalite and the filtrate was concentrated under reduced pressure and co-evaporated with toluene. The remaining brown solid was dissolved in ethyl acetate at 50°C and cooled on an ice-bath. 2M hydrogen chloride solution in diethyl ether (1 .14 L) was added in 30 min. The mixture was allowed to stir at room temperature over weekend. The crystals were collected by filtration, washed with diethyl ether and dried under reduced pressure at 40°C. The product brown solid obtained was dissolved in methanol at 60°C. The mixture was filtered and partially concentrated, cooled to room temperature and diethyl ether (1000 ml) was added. The mixture was allowed to stir at room temperature overnight. The solids formed were collected by filtration, washed with diethyl ether and dried under reduced pressure at 40°C to give 153.5 g of (3-chloropyrazin-2- yl)methanamine. hydrochloride as a brown solid (74.4 %, content 77 %).
(b) (S)-benzyl 2-((3-chloropyrazin-2-yl)methylcarbamoyl)pyrrolidine-1-carboxylate
To a solution of (3-chloropyrazin-2-yl)methanamine.HCI (9.57 g, 21.26 mmol, 40% wt) and Z-Pro-OH (5.3 g, 21 .26 mmol) in dichloromethane (250 mL) was added triethylamine (1 1.85 mL, 85 mmol) and the reaction mixture was cooled to 0°C. After 15 min stirring at 0°C, HATU (8.49 g, 22.33 mmol) was added. The mixture was stirred for 1 hour at 0°C and then overnight at room temperature. The mixture was washed with 0.1 M HCI-solution, 5% NaHC03, water and brine, dried over sodium sulfate and concentrated in vacuo. The product was purified using silica gel chromatography (heptane/ethyl acetate = 1/4 v/v%) to give 5 g of (S)-benzyl 2-((3-chloropyrazin-2-yl)methylcarbamoyl)pyrrolidine-1-carboxylate (62.7%).
(c) (S)-Benzyl 2-(8-chloroimidazo[1 ,5-alpyrazin-3-yl)pyrrolidine-1-carboxylate
(S)-Benzyl 2-((3-chloropyrazin-2-yl)methylcarbamoyl)pyrrolidine-1-carboxylate (20.94 mmol, 7.85 g) was dissolved in acetonitrile (75 ml), 1 ,3-dimethyl-2-imidazolidinone (62.8 mmol, 6.9 ml, 7.17 g) was added and the reaction mixture was cooled to 0°C before POCI3 (84 mmol, 7.81 ml, 12.84 g) was added drop wise while the temperature remained around 5°C. The reaction mixture was refluxed at 60-65°C overnight. The reaction mixture was poured carefully in ammonium hydroxide 25% in water (250 ml)/crushed ice (500 ml) to give a yellow suspension (pH -8-9) which was stirred for 15 min until no ice was present in the suspension. Ethyl acetate was added, layers were separated and the aqueous layer was extracted with ethyl acetate (3x). The organic layers were combined and washed with brine, dried over sodium sulfate, filtered and evaporated to give 7.5 g crude product. The crude product was purified using silica gel chromatography (heptane/ethyl acetate = 1/4 v/v%) to give 6.6 g of (S)-benzyl 2-(8- chloroimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1-carboxylate (88%).
(d) (S)-Benzyl 2-(1-bromo-8-chloroimidazo[1 ,5-alpyrazin-3-yl)pyrrolidine-1-carboxylate
N-Bromosuccinimide (24.69 mmol, 4.4 g) was added to a stirred solution of (S)-benzyl 2-(8- chloroimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1-carboxylate (24.94 mmol, 8.9 g) in DMF (145 mL). The reaction was stirred 3 h at rt. The mixture was poored (slowly) in a stirred mixture of water (145 mL), ethyl acetate (145 mL) and brine (145 mL). The mixture was then transferred into a separating funnel and extracted. The water layer was extracted with 2×145 mL ethyl acetate. The combined organic layers were washed with 3×300 mL water, 300 mL brine, dried over sodium sulfate, filtered and evaporated. The product was purified using silica gel chromatography (ethyl acetate/heptane = 3/1 v/v%) to give 8.95 g of (S)-benzyl 2-(1-bromo-8-chloroimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1-carboxylate (82.3%).
(e) (S)-Benzyl 2-(8-amino-1-bromoimidazo[1 ,5-alpyrazin-3-yl)pyrrolidine-1-carboxylate
(S)-Benzyl 2-(8-amino-1-bromoimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1-carboxylate (20.54 mmol, 8.95 g) was suspended in 2-propanol (1 13 ml) in a pressure vessel. 2-propanol (50 ml) was cooled to -78°C in a pre-weighed flask (with stopper and stirring bar) and ammonia gas (646 mmol, 1 1 g) was lead through for 15 minutes. The resulting solution was added to the suspension in the pressure vessel. The vessel was closed and stirred at room temperature and a slight increase in pressure was observed. Then the suspension was heated to 1 10 °C which resulted in an increased pressure to 4.5 bar. The clear solution was stirred at 1 10 °C, 4.5 bar overnight. After 18h the pressure remained 4 bar. The reaction mixture was concentrated in vacuum, the residue was suspended in ethyl acetate and subsequent washed with water. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water, saturated sodium chloride solution, dried over sodium sulfate and concentrated to give 7.35 g of (S)-benzyl 2-(8-amino-1-bromoimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1- carboxylate (86%).
Intermediate 2
(S)-4-(8-Amino-3-(pyrrolidin-2-v0im^
(a) (S)-Benzyl 2-(8-amino-1-(4-(pyridin-2-ylcarbamov0
carboxylate
(S)-benzyl 2-(8-amino-1-bromoimidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1 -carboxylate (0.237 mmol, 98.5 mg) and 4-(pyridin-2-yl-aminocarbonyl)benzeneboronic acid (0.260 mmol, 63.0 mg) were suspended in a mixture of 2N aqueous potassium carbonate solution (2.37 mmol, 1 .18 mL) and dioxane (2.96 mL). Nitrogen was bubbled through the mixture, followed by the addition of 1 , 1 ‘- bis(diphenylphosphino)ferrocene palladium (ii) chloride (0.059 mmol, 47.8 mg). The reaction mixture was heated for 20 minutes at 140°C in the microwave. Water was added to the reaction mixture, followed by an extraction with ethyl acetate (2x). The combined organic layer was washed with brine, dried over magnesium sulfate and evaporated. The product was purified using silicagel and dichloromethane/methanol = 9/1 v/v% as eluent to afford 97.1 mg of (S)-benzyl 2-(8-amino-1-(4-(pyridin- 2-ylcarbamoyl)phenyl)imidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1 -carboxylate (77%).
(b) (S)-4-(8-Amino-3-(pyrrolidin-2-yl)imidazo[1 ,5-alpyrazin-1-yl)-N-(pyridin-2-yl)benzamide
To (S)-benzyl 2-(8-amino-1-(4-(pyridin-2-ylcarbamoyl)phenyl)imidazo[1 ,5-a]pyrazin-3-yl)pyrrolidine-1- carboxylate (0.146 mmol, 78 mg) was added a 33% hydrobromic acid/acetic acid solution (1 1.26 mmol, 2 ml) and the mixture was left at room temperature for 1 hour. The mixture was diluted with water and extracted with dichloromethane. The aqueous phase was neutralized using 2N sodium hydroxide solution, and then extracted with dichloromethane. the organic layer was dried over magnesium sulfate, filtered and evaporated to give 34 mg of (S)-4-(8-Amino-3-(pyrrolidin-2-yl)imidazo[1 ,5-a]pyrazin-1-yl)-N- (pyridin-2-yl)benzamide (58%).
Example 6
(S)-4-(8-amino-3-n-but-2-vnoylpyrrolidin-2-vnimidazo[1 ,5-alpyrazin-1-yl)-N-(pyridin-2-yl)benzamide
This compound was prepared, in an analogues manner as described in Example 2, from the compound described in intermediate 2b and 2-butynoic acid, to afford the title compound (10.5 mg, 18.0%). Data: LCMS (B) Rt : 2.08 min; m/z 466.1 (M+H)+.

PATENT
https://www.google.com/patents/WO2016024228A1?cl=en
PATENT
CN 107056786


Step SI:
[0029] The pressure in the reactor was added 3-chloro-2-carboxaldehyde l-yl P ratio of (II) (0.71g, 5mmol) and dioxane (20mL), under stirring ammonia gas (I. 7g, 0 . Imol), was added 4- (pyridin-2-yl – aminocarbonyl) phenylboronic acid (III) (2.42g, lOmmol), Ming dicarbonyl acetylacetonate (0.26g, lmmol), and water 4mL. The reactor was sealed, gradually warmed to 80~90 °, the reaction 16-18 hours, TLC detection, the reaction was complete. Concentrated under reduced pressure, the residue was dissolved in dichloromethane, washed with saturated sodium bicarbonate and water successively, dried over anhydrous sodium sulfate. Concentrated to give brown oil, ethyl acetate and petroleum ether (volume ratio 1: 2) column chromatography to give an off-white solid 4- [amino (3-chloro-2-pyrazinyl) methyl] -N- (2-pyridyl) benzamide (IV) 1.38g, yield 81 · 2%; ESI-MS (m / z): 340 (m + H).
[0030] Step S2:
[0031] added in the reactor [1- (1-oxo-2-butyn-1-yl)] – L- proline (1.09g, 6mmol) and thionyl chloride (IOmL), was added dropwise 4mL of triethylamine and heated to 30 to 40 degrees, after the reaction for 2-4 hours under reduced pressure to remove excess thionyl chloride, the residue that is [I- (1- oxo-2-butyn-1-yl )] – L- proline acid chloride (V). The resulting [I- (1- oxo-2-butynyl -1_ yl)] _ L_ proline acid chloride (V) dissolved in 20mL dichloromethane burning, to a solution of 4- [amino (3-chloro -2-P ratio piperazinyl) methyl] -N- (2- pyridinyl) benzamide (IV) (1.35g, 4mmol) and triethylamine (0.6g, 6mmol) in dichloromethane (30mL) solution of in. Dropwise, warmed to 30-50 °, the reaction was stirred for 6 ~ 8 hours, TLC detection, the reaction was complete. Cooled to room temperature, washed with saturated sodium bicarbonate solution, brine and water, dried over anhydrous sodium sulfate. Concentrated to give a beige solid of 4- [1- (1-acyl-2-yne-2-yl) carboxamido (3-chloro-2-pyrazinyl) methyl] -N- (2- pyridinyl) benzamide (VI) 1.8g, yield 89.6% C3ESI-MS (m / z): 503 (m + H).
[0032] Step S3:
[0033] in a reaction flask was added 4- [I- (1- but-2-yn-acyl-2-yl) carboxamido (3-chloro-2-pyrazinyl) methyl] -N- ( 2-P ratio piperidinyl) benzamide (VI) (1 · 0g, 2mmol), phosphorus oxychloride (1 · 53g, IOmmol) and acetonitrile (25 mL), warmed to 80 ~ 100 ° with stirring, maintaining the temperature reaction 6 ~ 8 h, TLC the reaction was complete. Cooled to room temperature, the reaction solution was poured into 50mL concentration of 8% aqueous ammonia was added ethyl acetate, and the organic phase was separated, the aqueous phase was extracted twice with ethyl acetate. The combined organic phases were washed with brine and water, dried over anhydrous over sodium sulfate. Concentrated and the resulting residue with ethyl acetate and petroleum ether (volume ratio 2: 1) column chromatography to give an off-white solid 4- [8-Chloro -3- [(2S) -I- (1- oxo-2 – butyn-1-yl) -2-pyrrolidinyl] imidazo [I, 5-a] pyrazin-1-yl] -N-2- pyridinyl benzamide (VII) 0.85g, yield 87.8 %; EI-MS m / z: 485 [m + H] + square
[0034] Step S4:
[0035] The pressure reactor was added to 4- [8-Chloro -3- [(2S) -I- (1- oxo-2-butyn-1-yl) -2-pyrrolidinyl] imidazo [ I, 5-a] pyrazin – Buji] -N-2- pyridinyl benzamide (VII) (0.48g, lmmol) and isopropanol (15 mL), cooled to 0 degrees, by controlling the dose into ammonia gas (0.51g, 30mmol), the reactor is closed, warmed up to room temperature for 1 hour, and then continuously increasing the reaction temperature to 110~120 °, maintained at the reaction temperature and pressure 20~24 h, TLC the reaction was complete. Cooled to room temperature, slowly vented, and concentrated under reduced pressure, the resulting residue was dissolved with ethyl acetate, water and saturated brine, dried over anhydrous sodium sulfate. Concentrated and the resulting residue with ethyl acetate and petroleum ether (volume ratio 2: 1) column chromatography to give an off-white solid Acre imatinib ⑴ 0.40g, yield 86 · 0%; 1Η bandit R (DMS0-d6) 1.63 (m, lH), 1.97 (s, 3H), 2.02 ~2.12 (m, lH), 2 · 28~2.35 (m, 2H); 3.36~3.85 (m, 2H), 5 · 47~5.49 (m , lH), 6 · 17~6.23 (m, 2H), 7.12~7.20 (m, 2H), 7 · 73~7.86 (m, 4H), 8 · 16~8.25 (m, 3H), 8 · 41 ( dd, lH), 10.86 (s, lH); EI-MS m / z: 466 [m + H] +.
[0036] 3-chloro starting material employed in the method above relates to the present invention yl pyrazin-2-carbaldehyde (II) and 4- (pyridin-2-yl – aminocarbonyl) phenylboronic acid (III), respectively, refer to methods for their preparation Document “Tetrahedron Letters, 47 (l), 31-34; 2006” international Patent W02013010868 and method for preparing the same compound. Raw [1- (1-oxo-2-butyn-1-yl)] – L- proline acid chloride (V), in one embodiment, the compound may be made [the I-(1-oxo-known -2-yn-1-yl)] – L- proline acylation.
PATENT
US 20170224688
PATENT





Example I
[0030] (1) Preparation of ⑸-2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0031] (S) -2- (8- chloro-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (10g, 28mmol) was dissolved in N- methylpyrrolidone ( SOML), the mass concentration was added 28% aqueous ammonia (168mm〇l), the reaction mixture was placed in a sealed stainless steel autoclave at 85 ° C, stirring the reaction under a pressure of 2.5 atm 6h, after the completion of the reaction, was cooled to 40 ° C and delivery system pressure, slow addition of water (50 mL), cooled to 10 ° C, crystallization 3h, filtered, and recrystallized from isopropanol to give ⑸-2- (8- amino-imidazo [I, 5-a] pyrazin – 3- yl) -1-pyrrolidine-carboxylate, an off-white solid (8.5 g of), yield 90%, reaction formula of this step is as follows:
[0033] (2) Preparation of (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [I, 5_a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0034] (S) -2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (8g, 24mmol) was dissolved in dichloromethane (IOOmL) was added tert-butyl dicarbonate (5.7g, 26mmol), reaction mixture was stirred 3h at 25 ° C, after completion of the reaction, post-treatment and purification to give ⑸-2- (8- tert-butoxycarbonyl-amino-imidazole and [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (IoG), 96% yield, this step follows the reaction formula:
[0036] (3) Preparation of (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0037] (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (IOg, 23mmol) was dissolved in tetrahydrofuran ( 80mL), was slowly added N- bromosuccinimide (4.5g, 25mmol), the reaction mixture was 25 ° C the reaction was stirred for 4h. The mixture was then slowly added water (80 mL), cooled to -10 ° C crystallization 3h, filtered, and recrystallized from isopropanol to give (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [ I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (I I. Ig), a yield of 94.5%, the reaction formula of this step is as follows:
[0039] (4) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} 1-pyrrolidine-carboxylic acid benzyl ester:
[0040] (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (I Ig, 2lmmol ), 4- (2-pyridyl-carbamoyl) phenylboronic acid (5.7g, 23.4mmol), [1, Γ – bis (diphenylphosphino) ferrocene] dichloropalladium cesium (〇.78g, the I · lmmol), potassium carbonate (4.0g, 29mmol), N, N- dimethylformamide (120 mL) and water (50mL) added to the reaction flask, the reaction mixture was heated to 90 ° C the reaction was stirred for 20 h, the reaction solution was reduced at room temperature, was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, washed with brine, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethyl acetate and n-hexane and recrystallized to give (S) -2- {8- tert butoxycarbonyl group -I- [4- (2-P of pyridine-ylcarbamoyl) phenyl] imidazole and sat Jie [I, 5_a] pyrazin-3-yl} -1-pyrrolidine-carboxylate, class as a white solid (10.3 g of), a yield of 76.5%, the reaction formula of this step is as follows:
[0042] (5) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} pyrrolidine:
[0043] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } -1- [1-carboxylic acid than the burning section slightly ester (10g, 15.8mmol) was dissolved in methanol (80mL), was added cesium charcoal (0.5g), under a hydrogen pressure into 35 ° C the reaction 8h. Concentrated suction through Celite to remove the catalyst and the filtrate was rotary evaporated to dryness to afford ⑸-2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [ I, 5-a] pyrazin-3-yl} pyrrolidine as a white solid powder (7.6 g of), 96% yield, this step follows the reaction formula:
[0045] (6) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} -1- (2-butynoyl) pyrrolidine:
[0046] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } ratio slightly burning Jie (7g, 14mmol) was dissolved in tetrahydrofuran (75 mL), with stirring, was added 2-butyne chloride (I. 7g, 16.6mmol), was added dropwise N, N- diisopropylethylamine (2.7 g, 21 mmol), the reaction mixture was 50 ° C the reaction was stirred for 8h, the reaction solution was concentrated by rotary evaporation to dryness, dilute hydrochloric acid was added was adjusted to neutral, extracted with ethyl acetate was added, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, recrystallized from methanol to give ⑸ -2_ {8-tert-butoxycarbonyl-amino -1- [4- (2-P of pyridine-ylcarbamoyl) phenyl] imidazole and sat Jie [I, 5_a] [! than 3-yl} -1 – (2_ butynoyl) pyrrolidine-white solid (7g), in 88% yield, this step follows the reaction formula:
[0048] ⑺ prepared Acalabrutinib:
[0049] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } -1- (2-butynoyl) pyrrolidine (7g, 12.4mmol) and dissolved in methanol (70 mL), trifluoroacetic acid (1.55g, 13.6mmol), 65 ° C until the reaction was complete the reaction was stirred for 6h, the reaction was added dropwise to a stirred solution of water (150 mL), cooled to 0 ° C crystallization 3h, filtered to give the treatment of chronic lymphocytic leukemia BTK inhibitors Acalabrut inib, as a white solid (5.3 g of), 92% yield, this step is the following reaction formula:
[0051] Example 2:
[0052] (1) Preparation of ⑸-2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0053] (S) -2- (8- chloro-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (15g, 42mmol) was dissolved in N- methylpyrrolidone ( 75 mL), aqueous ammonia (273_〇1) was added mass percent concentration of 28%, the reaction mixture was placed in a sealed stainless steel autoclave at 70 ° C, stirring the reaction under a pressure of 3 atm 8h, after the completion of the reaction, was cooled to 40 ° C and releasing the pressure in the system, slow addition of water (50 mL), cooled to 10 ° C, crystallization 3h, filtered, and recrystallized from isopropanol to give ⑸-2- (8- amino-imidazo [I, 5-a] pyrazine 3-yl) pyrrolidine-carboxylic acid benzyl ester, off-white solid (12.9 g of), yield 91% ,, this step reaction scheme in Example 1.
[0054] (2) Preparation of (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [I, 5_a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0055] (S) -2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (12g, 35.6mmol) was dissolved in chloroform (80mL), was added tert-butyl dicarbonate (7.8g, 35.6mmol), the reaction mixture was stirred for lh the reaction at 35 ° C, after completion of the reaction, post-treatment and purification to give ⑸-2- (8- tert-butoxycarbonyl-amino-imidazole and [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (14.8 g of), in 95% yield, this step is the same reaction scheme as in Example 1.
[0056] (3) Preparation of (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0057] (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (Hg, 32mmol) was dissolved in 1, 1,2-dichloroethane (90mL), was slowly added bromine (6g, 37.8mmol), the reaction mixture was 20 ° C the reaction was stirred for 6h. After the reaction, water was slowly added (I5mL), cooled to -5 ° C crystallization 4h, filtered and recrystallized from isopropanol to give ⑸-2- (8- tert-butoxycarbonyl-amino-1-bromo-imidazo [1, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (15.8 g), yield 95.5%, the reaction of the present step is the same formula as in Example 1.
[0058] (4) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} 1-pyrrolidine-carboxylic acid benzyl ester:
[0059] (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (15g, 29mmol) , 4- (2-pyridyl-carbamoyl) phenylboronic acid (34 · 7mmol 8 · 4g,), tetrakis (triphenylphosphine) palladium (0 · 84g, 0.73mmol), sodium carbonate (6.9g, 65mmol), tetrahydrofuran (IOOmL) and water (40 mL) was added a reaction flask, the reaction mixture was heated to 80 ° C the reaction was stirred for 24h, the reaction was cooled to room temperature, and concentrated by rotary evaporation to dryness, extracted with ethyl acetate, washed with brine, dried over magnesium sulfate, concentrated by rotary evaporation to dryness, a mixed solvent of ethyl acetate and n-hexane and recrystallized to give ⑸-2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazole and [I, 5-a] pyrazin-3-yl} -1-pyrrolidine-carboxylate, an off-white solid (14.4g), 78% yield, this step is the same reaction scheme as in Example 1.
[0060] (5) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} pyrrolidine:
[0061] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-yl _3- it is slightly burned} -1-carboxylic acid ester section (14g, 22mmol) dissolved in isopropanol (85mL), was added Raney nickel (0.5g), under a hydrogen pressure into the reaction 60 ° C 12h. Concentrated suction through Celite to remove the catalyst and the filtrate was rotary evaporated to dryness to afford ⑸-2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [ I, 5-a] pyrazin-3-yl} pyrrolidine as a white solid powder (10.4 g of), 94% yield, this step is the same reaction scheme as in Example 1.
[0062] (6) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} -1- (2-butynoyl) pyrrolidine:
[0063] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } pyrrolidine (10g, 20mmo 1) was dissolved in N, N- dimethylformamide (SOML), with stirring, was added 2-butyne chloride (3. lg, 30mmol), dropwise addition of triethylamine (2.2g, 22mmol ), the reaction mixture was 60 ° C the reaction was stirred for 4h, the reaction solution was concentrated by rotary evaporation to dryness, dilute hydrochloric acid was added was adjusted to neutral, extracted with ethyl acetate was added, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, and recrystallized from methanol to give ⑸- 2- {8-tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl} -l- (2- butynoyl) pyrrolidine-white solid (10.2 g of), a yield of 90.2%, the same reaction scheme of the present embodiment step 1〇
[0064] ⑺ prepared Acalabrutinib:
[0065] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } -1- (2-butynoyl) pyrrolidine (IOg, 17.7mmol) was dissolved in ethanol, and (IOOmL), trifluoroacetic acid (2.6g, 23mmol), 50 ° C with stirring until the reaction was complete IOh reaction, the reaction solution was added dropwise to a stirred solution of water (70 mL), cooled to 0 ° C crystallization 3h, filtered to give the treatment of chronic lymphocytic leukemia BTK inhibitors AcaIabrut inib, as a white solid (7.5 g of), yield 91%, reaction of this step formula same as in Example 1.
[0066] Example 3:
[0067] (1) Preparation of ⑸-2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0068] (S) -2- (8- chloro-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (4.5g, 12.6mmol) was dissolved in N- methyl pyrrolidinone (70 mL), was added mass percent concentration of 28% aqueous ammonia (69.4 mmol), the reaction mixture was placed in the autoclave 90 ° C, the reaction was stirred under atmospheric pressure of 4h, after the completion of the reaction, it was cooled to 35 ° C a sealed stainless steel reactor and releasing the pressure in the system, slow addition of water (50 mL), cooled to 10 ° C, crystallization 3h, filtered, and recrystallized from isopropanol to give ⑸-2- (8- amino-imidazo [I, 5-a] pyrazine 3-yl) pyrrolidine-carboxylic acid benzyl ester, off-white solid (3.9 g of), 92% yield, this step is the same reaction scheme as in Example 1.
[0069] (2) Preparation of (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0070] (S) -2- (8- amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester (3 · 5g, 10 · 4mmol) was dissolved in 1, 4- dioxane (50 mL), was added tert-butyl dicarbonate (2.7g, 12.4mmol), the reaction mixture was stirred at 10 ° C the reaction 6h, after the completion of the reaction, workup and purification, to give (S) 2- (8-tert-butoxycarbonyl-amino-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (4.3 g of), in 95% yield, according to the present step reaction scheme in Example 1.
[0071] (3) Preparation of (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylic acid benzyl ester:
[0072] (S) -2- (8- tert-butoxycarbonyl-amino-imidazo [l, 5_a] pyrazin-3-yl) -1_ pyrrolidine-carboxylate (4g, 9.6mmol) was dissolved in toluene (50 mL ), was slowly added N- bromosuccinimide (I. 8g, 10. lmmol), the reaction mixture was 35 ° C the reaction was stirred for 2h. The mixture was then slowly added water (25 mL), cooled to -10 ° C crystallization 3h, filtered, and recrystallized from isopropanol to give (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [ I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate, an off-white solid (4.7 g), 94% yield, this step is the same reaction scheme as in Example 1.
[0073] (4) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} 1-pyrrolidine-carboxylic acid benzyl ester:
[0074] (S) -2- (8- tert-butoxycarbonyl-1-bromo-imidazo [I, 5-a] pyrazin-3-yl) -1-pyrrolidine-carboxylate (4g, 7 · 7mmol), 4_ (2- piperidinyl than Jie carbamoyl) phenylboronic acid (2 · 4g, IOmmol), bis (triphenylphosphine) dichloride Leba (0.41g, 0.58mmol), potassium phosphate (I. 9g, 8.9mmol), methyl tert-butyl ether (IOOmL) and water (40 mL) was added a reaction flask, the reaction mixture was heated to 100 ° C the reaction was stirred for 12h, the reaction was cooled to room temperature, and concentrated by rotary evaporation to dryness, was added acetic acid extracted with ethyl, brine, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethyl acetate and n-hexane and recrystallized to give ⑸-2- {8- tert-butoxycarbonyl-amino-1- [4- (2 – pyridin-ylcarbamoyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl} -1-pyrrolidine-carboxylate, an off-white solid (3.9 g of), in 79% yield, this step the reaction scheme in Example 1.
[0075] (5) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5_a] pyrazin-3 -} pyrrolidine:
[0076] (S) -2- {8- tert-butoxycarbonyl group -I- [4- (2- carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } -1 Jie section than slightly burning acid ester (3.5g, 5.5mmol) was dissolved in ethanol (50mL), was added cesium charcoal (0.2g), under a hydrogen pressure into 45 ° C the reaction 6h. Concentrated suction through Celite to remove the catalyst and the filtrate was rotary evaporated to dryness to afford ⑸-2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [ I, 5-a] pyrazin-3-yl} pyrrolidine as a white solid powder (2.6 g of), in 95% yield, this step is the same reaction scheme as in Example 1.
[0077] (6) Preparation of (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [1, 5-a] pyrazine 3-yl} -1- (2-butynoyl) pyrrolidine:
[0078] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl } ratio slightly burning Jie (2.5g, 5mmol) was dissolved in toluene (50 mL), with stirring, was added 2-butyne chloride (0.62g, 6mmol), was added dropwise N, N- dimethylaniline (Ig, 8.5mmo 1), The reaction mixture was 40 ° C the reaction was stirred for 12h, the reaction solution was concentrated by rotary evaporation to dryness, dilute hydrochloric acid was added was adjusted to neutral, extracted with ethyl acetate was added, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, and recrystallized from methanol to give ⑸-2- {8-tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-3-yl} -1- (2-butyn acyl) pyrrolidine-white solid (2.5g), 88% yield, this step is the same reaction scheme as in Example 1.
[0079] ⑺ prepared Acalabrutinib:
[0080] (S) -2- {8- tert-butoxycarbonyl-amino-1- [4- (2-carbamoyl-pyridyl) phenyl] imidazo [I, 5-a] pyrazin-yl _3_ } -1- (2-block group) ratio slightly burning Jie (2.5g, 4.4mmol) was dissolved in dichloromethane and burned (IOmL), two gas was added acetic acid (0.76g, 6.6mmol), 80 ° C The reaction was stirred 4h until the reaction was complete, the reaction was added dropwise to a stirred solution of water (25 mL), cooled to 0 ° C crystallization 3h, filtered to give the treatment of chronic lymphocytic leukemia BTK inhibitors AcaIabrut inib, as a white solid (1.8 g of), the yield of 89%, this step is the same reaction scheme as in Example 1.
PATENT
US 20170035881
Acalabrutinib is a potent and selective BTK (Bruton’s tyrosine kinase) inhibitor. BTK is a cytoplasmic, non-receptor tyrosine kinase that transmits signals from a variety of cell-surface molecules, including the B-cell receptor (BCR) and tissue homing receptors. Genetic BTK deletion causes B-cell immunodeficiency in humans and mice, making this kinase an attractive therapeutic target for B-cell disorders. BTK inhibitors targeting B cell receptor signaling and other survival mechanism showed great promise for the treatment of chronic lymphocytic leukemia (CLL)s holds great promise.
As of 2015 it is in late stage clinical trials for relapsed chronic lymphocytic leukemia. Interim results are encouraging : 95% overall response rate. It is also in another 20 clinical trials (alone and in combination) for various cancers.
REFERENCES
1: Maly J, Blachly JS. Chronic Lymphocytic Leukemia: Exploiting Vulnerabilities with Targeted Agents. Curr Hematol Malig Rep. 2016 Feb 11. [Epub ahead of print] PubMed PMID: 26893063.
2: Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, Chaves J, Wierda WG, Awan FT, Brown JR, Hillmen P, Stephens DM, Ghia P, Barrientos JC, Pagel JM, Woyach J, Johnson D, Huang J, Wang X, Kaptein A, Lannutti BJ, Covey T, Fardis M, McGreivy J, Hamdy A, Rothbaum W, Izumi R, Diacovo TG, Johnson AJ, Furman RR. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016 Jan 28;374(4):323-32. doi: 10.1056/NEJMoa1509981. Epub 2015 Dec 7. PubMed PMID: 26641137.
 |
|
| Identifiers | |
|---|---|
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C26H23N7O2 |
| Molar mass | 465.507 g/mol |
| 3D model (JSmol) | |
| FDA Orange Book Patents: 1 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 9290504 |
| Expiration | Jul 11, 2032 |
| Applicant | ASTRAZENECA |
| Drug Application | N210259 (Prescription Drug: CALQUENCE. Ingredients: ACALABRUTINIB) |
| FDA Orange Book Patents: 2 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 9758524 |
| Expiration | Jul 11, 2032 |
| Applicant | ASTRAZENECA |
| Drug Application | N210259 (Prescription Drug: CALQUENCE. Ingredients: ACALABRUTINIB) |
////////////Acalabrutinib, rINN, ACP-196, fda 2017, Акалабрутиниб , أكالابروتينيب , 阿可替尼 , Orphan Drug, breakthrough therapy designation, Lymphoma, mantle cell, ACERTA PHARMA
CC#CC(=O)N1CCC[C@H]1c2nc(c3n2ccnc3N)c4ccc(cc4)C(=O)Nc5ccccn5
CC#CC(=O)N1CCCC1C2=NC(=C3N2C=CN=C3N)C4=CC=C(C=C4)C(=O)NC5=CC=CC=N5

A study from the Johns Hopkins Bloomberg School of Public Health found the natural decline in lung function over a 10-year period was slower among former smokers with a diet high in tomatoes and fruits, especially apples, suggesting certain components in these foods might help restore lung damage caused by smoking.The researchers found that adults…
via Diet rich in tomatoes and apples may help restore lung damage caused by smoking — Med-Chemist
Glimepiride (original trade name Amaryl) is an orally available medium-to-long-acting sulfonylurea antidiabetic drug. It is sometimes classified as either the first third-generation sulfonylurea,[1] or as second-generation.[2]
Glimepiride is a Sulfonylurea. The chemical classification of glimepiride is Sulfonylurea Compounds.
Glimepiride is a long-acting, third-generation sulfonylurea with hypoglycemic activity. Compared to other generations of sulfonylurea compounds, glimepiride is very potent and has a longer duration of action. This agent is metabolized by CYP2C9 and shows peroxisome proliferator-activated receptor gamma (PPARgamma) agonistic activity.
Glimepiride is indicated to treat type 2 diabetes mellitus; its mode of action is to increase insulin production by the pancreas. It is not used for type 1 diabetes because in type 1 diabetes the pancreas is not able to produce insulin.[3]
Its use is contraindicated in patients with hypersensitivity to glimepiride or other sulfonylureas.
Side effects from taking glimepiride include gastrointestinal tract (GI) disturbances, occasional allergic reactions, and rarely blood production disorders including thrombocytopenia, leukopenia, and hemolytic anemia. In the initial weeks of treatment, the risk of hypoglycemia may be increased. Alcohol consumption and exposure to sunlight should be restricted because they can worsen side effects.[3]

Two generic oral tablets of glimepiride, 2 mg each
Gastrointestinal absorption is complete, with no interference from meals. Significant absorption can occur within one hour, and distribution is throughout the body, 99.5% bound to plasma protein. Metabolism is by oxidative biotransformation, it is hepatic and complete. First, the medication is metabolized to M1 metabolite by CYP2C9. M1possesses about 1⁄3 of pharmacological activity of glimepiride, yet it is unknown if this results in clinically meaningful effect on blood glucose. M1 is further metabolized to M2metabolite by cytosolic enzymes. M2 is pharmacologically inactive. Excretion in the urine is about 65%, and the remainder is excreted in the feces.
Like all sulfonylureas, glimepiride acts as an insulin secretagogue.[4] It lowers blood sugar by stimulating the release of insulin by pancreatic beta cells and by inducing increased activity of intracellular insulin receptors.
Not all secondary sufonylureas have the same risks of hypoglycemia. Glibenclamide (glyburide) is associated with an incidence of hypoglycemia of up to 20–30%, compared to as low as 2% to 4% with glimepiride. Glibenclamide also interferes with the normal homeostatic suppression of insulin secretion in reaction to hypoglycemia, whereas glimepiride does not. Also, glibenclamide diminishes glucagon secretion in reaction to hypoglycemia, whereas glimepiride does not.[5]

Nonsteroidal anti-inflammatory drugs (such as salicylates), sulfonamides, chloramphenicol, coumadin and probenecid may potentiate the hypoglycemic action of glimepiride. Thiazides, other diuretics, phothiazides, thyroid products, oral contraceptives, and phenytoin tend to produce hyperglycemia.
SYNTHESIS

CLIP

Following is one of the synthesis routes: 3-Ethyl-4-methyl-3-pyrrolin-2-one could be condensed (I) with 2-phenylethyl isocyanate (II) at 150 C to produce 3-ethyl-4-methyl-2-oxo-N-(2-phenylethyl)-3-pyrrolin-1-carboxamide (III), which is sulfonated with chlorosulfonic acid at 40 C to yield the corresponding benzenesulfonyl chloride (IV). The reaction of (IV) with concentrated NH4OH affords the sulfonamide (V), which is finally condensed with 4-methylcyclohexyl isocyanate (VI) in acetone.
CLIP
http://science24.com/paper/6906

https://www.sciencedirect.com/science/article/pii/S073170850500378X

https://www.google.com/patents/US20070082943
schemes I to III.

EXAMPLE 2Preparation of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV)
EXAMPLE 3APurification of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide (IV)
EXAMPLE 3BPurification of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide (IV)
EXAMPLE 4Preparation of 3-Ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methyl cyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-1 H-pyrrole-1-carboxamide (I).
EXAMPLE 5Purification of 3-Ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methyl cyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-1H-pyrrole-1-carboxamide (I)
CLIP

Magnified 1H NMR spectra of (a) glimepiride and its solid dispersions with hyperbranched polymers containing the (b) hydroxyl and (c) the tertiary amino functional groups.

Magnified 13C NMR spectra of (a) glimepiride and its solid dispersions with hyperbranched polymers containing (b) the hydroxyl and (c) the tertiary amino functional groups.

The difference spectra of the solid dispersions of glimepiride and the hyperbranched polymer containing (a) the hydroxyl groups and (b) the tertiary amino groups. The difference spectra were obtained by subtraction of the spectra of the pure hyperbranched polymers from the spectra of the solid dispersions. The ATR spectra of the pure hyperbranched polymers were recorded on samples that were prepared under the same conditions as solid dispersions, only without the presence of the glimepiride drug.
| FDA Orange Book Patents: 1 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7358366 |
| Expiration | Apr 19, 2020. 7358366*PED expiration date: Oct 19, 2020 |
| Applicant | SB PHARMCO |
| Drug Application | |
| FDA Orange Book Patents: 2 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8071130 |
| Expiration | Jun 8, 2028 |
| Applicant | TAKEDA PHARMS USA |
| Drug Application |
|
| FDA Orange Book Patents: 3 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7700128 |
| Expiration | Jan 30, 2027 |
| Applicant | TAKEDA PHARMS USA |
| Drug Application |
|
CLIP

Journal of China Pharmaceutical University 1999 , 30(3):163 ~ 165
Ethyl acetoacetate (2) Preparation of literature more, such as with ethyl iodide or ethyl bromide as ethyl reagents will produce a double ethylation or oxyethylation, it is difficult to separate. We use dimethylamine and ethyl acetoacetate reaction enamine, then diethyl sulfate as ethylating agent, you can reduce the side reactions, product purity, the yield up to 80%. Preparation of cyanohydrin (3) Hydrochloric acid anhydrous literature, toxicity, difficult to operate, we use solid sodium cyanide and sodium bisulfite in the aqueous phase reaction, get 3, easy to operate. In the literature 1-acetyl-3- Ethyl-4-methyl-3-pyrrolin-2-one (4) was purified by high vacuum distillation and then hydrolyzed to give 3-ethyl- In the distillation of the product easy to loss, after the change to the crude hydrolysis, two-step yield of 33%. Reported in the literature 5 and phenethyl isocyanate (6) without solvent direct reaction of 3-ethyl-4-methyl-2-oxo-3-pyrroline-1 – N- (2 – phenethyl) A Amide (7), the experiment found that the reaction heat when the heating easy to red material, we add toluene as a solvent, the reaction is smooth, easy to post-treatment. 6 preparation, the general method is to use phosgene, but phosgene often Temperature of gas, highly toxic, difficult to operate, we use triphosgene instead of triphosgene as a yellow solid, easy to transport, weighing, laboratory convenience. We refer to the process of domestic glyburide, 7 chlorosulfonated, ammoniated sulfonamide (9), two-step yield of 77%. The last 9 reacts with trans-4-methylcyclohexylisocyanate to form glimepiride (1). Ethyl acetoacetate as the starting material, eight-step total yield of 11.5%.
Journal of China Pharmaceutical University 1999 , 30(3):163 ~ 165
Glimepi ride (1) trade name Amary l, chemical name 1- [4- [2- (3-ethyl-4-methyl-2-oxo-3-pyrroline-1-carboxamido ) Ethyl] phenylsulfonyl] -3- (trans-4-methylcyclohexyl) urea, a new sulfonylurea hypoglycemic agent developed by Hoechst AG in Germany and listed in the Netherlands and Switzerland in 1995, In 1996 the United States FDA approval
and further investigations in the pyrrolone series.Ann Chem ,
1964 , 680 :60
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4379785 * | Dec 17, 1980 | Apr 12, 1983 | Hoechst Aktiengesellschaft | Heterocyclic substituted sulfonyl ureas, and their use |
|
|
| Clinical data | |
|---|---|
| Trade names | Amaryl |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a696016 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral (tablets) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 100% |
| Protein binding | >99.5% |
| Metabolism | Complete hepatic (1st stage through CYP2C9) |
| Biological half-life | 5–8 hours |
| Excretion | Urine (~60%), feces (~40%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.170.771 |
| Chemical and physical data | |
| Formula | C24H34N4O5S |
| Molar mass | 490.617 g/mol |
| 3D model (JSmol) | |
///////////Amaryl, glimepiride, glymepiride, HOE 490
CCC1=C(CN(C1=O)C(=O)NCCC2=CC=C(C=C2)S(=O)(=O)NC(=O)NC3CCC(CC3)C)C
AMISELIMOD
UNII-358M5150LY; CAS 942399-20-4; 358M5150LY; MT-1303; Amiselimod, MT-1303
| Molecular Formula: | C19H30F3NO3 |
|---|---|
| Molecular Weight: | 377.448 g/mol |
2-amino-2-[2-[4-heptoxy-3-(trifluoromethyl)phenyl]ethyl]propane-1,3-diol
Phase II Crohn’s disease; Multiple sclerosis; Plaque psoriasis
Amiselimod, also known as MT1303, is a potent and selective immunosuppressant and sphingosine 1 phosphate receptor modulator. Amiselimod may be potentially useful for treatment of multiple sclerosis; inflammatory diseases; autoimmune diseases; psoriasis and inflammatory bowel diseases. Amiselimod is currently being developed by Mitsubishi Tanabe Pharma Corporation
Mitsubishi Tanabe is developing amiselimod, an oral sphingosine-1-phosphate (S1P) receptor antagonist, for treating autoimmune diseases, primarily multiple sclerosis, psoriasis and inflammatory bowel diseases, including Crohn’s disease.
EU states expire 2026, and
Expire in the US in June 2030 with US154 extension.
| Inventors | Masatoshi Kiuchi, Kaoru Marukawa, Nobutaka Kobayashi, Kunio Sugahara |
| Applicant | Mitsubishi Tanabe Pharma Corporation |
In recent years, calcineurin inhibitors such as cyclosporine FK 506 have been used to suppress rejection of patients receiving organ transplantation. While doing it, certain calcineurin inhibitors like cyclosporin can cause harmful side effects such as nephrotoxicity, hepatotoxicity, neurotoxicity, etc. For this reason, in order to suppress rejection reaction in transplant patients, development of drugs with higher safety and higher effectiveness is advanced.
[0003] Patent Documents 1 to 3 are useful as inhibitors of (acute or chronic) rejection in organ or bone marrow transplantation and also useful as therapeutic agents for various autoimmune diseases such as psoriasis and Behcet’s disease and rheumatic diseases 2 aminopropane 1, 3 dioly intermediates are disclosed.
[0004] One of these compounds, 2-amino-2- [2- (4-octylphenel) propane] 1, 3 diol hydrochloride (hereinafter sometimes referred to as FTY 720) is useful for renal transplantation It is currently under clinical development as an inhibitor of rejection reaction. FTY 720 is phosphorylated by sphingosine kinase in vivo in the form of phosphorylated FTY 720 [hereinafter sometimes referred to as FTY 720-P]. For example, 2 amino-2-phosphoryloxymethyl 4- (4-octafil-el) butanol. FTY720 – P has four types of S1 P receptors (hereinafter referred to as S1 P receptors) among five kinds of sphingosine – 1 – phosphate (hereinafter sometimes referred to as S1P) receptors It acts as an aggroove on the body (other than S1P2) (Non-Patent Document 1).
[0005] It has recently been reported that S1P1 among the S1P receptors is essential for the export of mature lymphocytes with thymus and secondary lymphoid tissue forces. FTY720 – P downregulates S1P1 on lymphocytes by acting as S1P1 ghost. As a result, the transfer of mature lymphocytes from the thymus and secondary lymphatic tissues is inhibited, and the circulating adult lymphocytes in the blood are isolated in the secondary lymphatic tissue to exert an immunosuppressive effect Has been suggested (
Non-Patent Document 2).
[0006] On the other hand, conventional 2-aminopropane 1, 3 dioly compounds are concerned as transient bradycardia expression as a side effect, and in order to solve this problem, 2-aminopropane 1, 3 diiori Many new compounds have been reported by geometrically modifying compounds. Among them, as a compound having a substituent on the benzene ring possessed by FTY 720, Patent Document 4 discloses an aminopropenol derivative as a S1P receptor modulator with a phosphate group, Patent Documents 5 and 6 are both S1P Discloses an amino-propanol derivative as a receptor modulator. However, trihaloalkyl groups such as trifluoromethyl groups are not disclosed as substituents on the benzene ring among them. In any case, it is currently the case that it has not yet reached a satisfactory level of safety as a pharmaceutical.
Patent Document 1: International Publication Pamphlet WO 94 Z 08943
Patent Document 2: International Publication Pamphlet WO 96 Z 06068
Patent Document 3: International Publication Pamphlet W 0 98 z 45 429
Patent Document 4: International Publication Pamphlet WO 02 Z 076995
Patent document 5: International public non-fret WO 2004 Z 096752
Patent Document 6: International Publication Pamphlet WO 2004 Z 110979
Non-patent document 1: Science, 2002, 296, 346-349
Non-patent document 2: Nature, 2004, 427, 355-360
Reference Example 3
5 bromo 2 heptyloxybenzonitrile
(3- 1) 5 Synthesis of bromo-2 heptyloxybenzonitrile (Reference Example Compound 3- 1)
1-Heptanol (1.55 g) was dissolved in N, N dimethylformamide (24 ml) and sodium hydride (0.321 g) was added at room temperature. After stirring for 1 hour, 5 bromo-2 fluoborosyl-tolyl (2.43 g) was added and the mixture was further stirred for 50 minutes. The reaction solution was poured into water, extracted with ethyl acetate, washed with water, saturated brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. After eliminating the 5 bromo 2 fluconate benzonitrile as a raw material, the reaction was carried out again under the same conditions and purification was carried out by silica gel column chromatography (hexane: ethyl acetate = 50: 1 to 5: 1) to obtain the desired product (3.10 g ) As a colorless oil.
– NMR (CDCl 3) δ (ppm): 0.89 (3H, t, J = 6.4 Hz), 1.24-1.35 (6H, m
J = 8.8 Hz), 1.48 (2H, quint, J = 7.2 Hz), 1.84 7.59 (1 H, dd, J = 8.8, 2.4 Hz), 7.65 (1 H, d, J = 2.4 Hz).
Example 1
2 Amino 2- [2- (4-heptyloxy-3 trifluoromethylph enyl) propane-1, 3-diol hydrochloride
(1 – 1) {2, 2 Dimethyl 5- [2- (4 hydroxy 3 trifluoromethylfuethyl) ethyl] 1,3 dioxane 5 mercaptothenylboronic acid t butyl ester (synthesis compound 1 1)
Reference Example Compound 2-5 (70.3 g) was dissolved in tetrahydrofuran (500 ml), t-butoxycallium (13.Og) was added, and the mixture was stirred for 1 hour. To the mixed solution was dropwise added a solution of the compound of Reference Example 1 (15.Og) in tetrahydrofuran (100 ml) under ice cooling, followed by stirring for 2 hours under ice cooling. Water was added to the reaction solution, the mixture was extracted with ethyl acetate, washed with water, saturated brine, dried with anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane: ethyl acetate = 3: D to obtain 31. Og of a pale yellow oily matter.) The geometric isomer ratio of the obtained product was (E : Z = 1: 6).
This pale yellow oil was dissolved in ethyl acetate (200 ml), 10% palladium carbon (3.00 g) was added, and the mixture was stirred under a hydrogen atmosphere at room temperature for 7 hours. After purging the inside of the reaction vessel with nitrogen, the solution was filtered and the filtrate was concentrated. The residue was washed with diisopropyl ether to obtain the desired product (2.2 g) as a colorless powder.
1 H-NMR (CDCl 3) δ (ppm): 1. 43 (3H, s), 1.44 (3H, s), 1. 47 (9H, s), 1
(2H, m), 91- 1. 98 (2H, m), 2. 50-2.66 (2H, m), 3. 69 (2H, d, J = Il. 6 Hz), 3. 89 J = 8.2 Hz), 7. 22 (1 H, dd J = 8 Hz), 5. 02 (1 H, brs), 5. 52 . 2, 1. 7 Hz), 7. 29 (1 H, d, J = l. 7 Hz).
(1-2) {2,2 Dimethyl-5- [2- (4heptyloxy-3 trifluoromethyl) ethyl] 1,3 dioxane 5-mercaptobutyric acid t-butyl ester Synthesis (compound 1 2)
Compound 1-1 (510 mg) was dissolved in N, N dimethylformamide (10 ml), potassium carbonate (506 mg) and n-heptyl bromide (0.235 ml) were added and stirred at 80 ° C. for 2 hours. Water was added to the reaction solution, the mixture was extracted with ethyl acetate, washed with water and saturated brine, dried with anhydrous sulfuric acid
The resultant was dried with GENSCHUM and the solvent was distilled off under reduced pressure to obtain the desired product (640 mg) as a colorless oil.
– NMR (CDCl 3) δ (ppm): 0.89 (3H, t, J = 6.8 Hz), l.30-1.37 (6H, m
(2H, m), 1.91-1.98 (2H, m), 1.42-1.50 (2H, m), 1.42 (3H, s), 1.44 (3H, s), 1.47 J = 16.6 Hz), 4.00 (2H, t, J = 6.4 Hz), 4.9 8 (2H, d, J = 11.6 Hz), 3.69 1 H, brs), 6.88 (1 H, d, J = 8.5 Hz), 7.26 – 7.29 (1 H, m), 7.35 (1 H, d, J = 1.5 Hz).
(1-3) Synthesis of 2-amino-2- [2- (4heptyloxy 3 trifluoromethyl) ethyl] propane 1, 3 diol hydrochloride (Compound 1- 3)
Compound 12 (640 mg) was dissolved in ethanol (15 ml), concentrated hydrochloric acid (3 ml) was caught and stirred at 80 ° C. for 2 hours. The reaction solution was concentrated, and the residue was washed with ethyl ether to give the desired product (492 mg) as a white powder.
MS (ESI) m / z: 378 [M + H]
– NMR (DMSO-d) δ (ppm): 0.86 (3H,
6 t, J = 6.8 Hz), 1.24 – 1.39 (6
(4H, m), 3.51 (4H, d, J = 5. lHz), 4.06 (2H, m), 1.39-1.46 (2H, m), 1.68-1.78 (4H, m), 2.55-2.22 , 7.32 (2H, t, J = 5.1 Hz), 7.18 (1 H, d, J = 8.4 Hz), 7.42 – 7.45 (2 H, m), 7.76 (3 H, brs;).
PATENT
WO 2009119858
JP 2011136905
WO 2017188357
PATENT
WO-2018021517
Patent Document 1: WO2007 / 069712
[Chemical formula 3]

PATENTS
////////////AMISELIMOD, Phase II, Crohn’s disease, Multiple sclerosis, Plaque psoriasis, MT-1303, MT1303, MT 1303, Mitsubishi Tanabe Pharma Corporation, Mitsubishi , JAPAN, PHASE 2
CCCCCCCOC1=C(C=C(C=C1)CCC(CO)(CO)N)C(F)(F)F








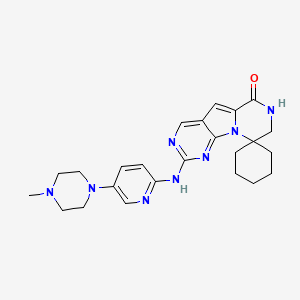











































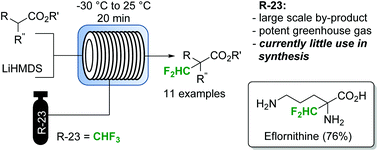
 Open Access
Open Access