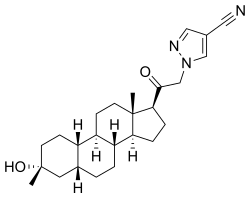
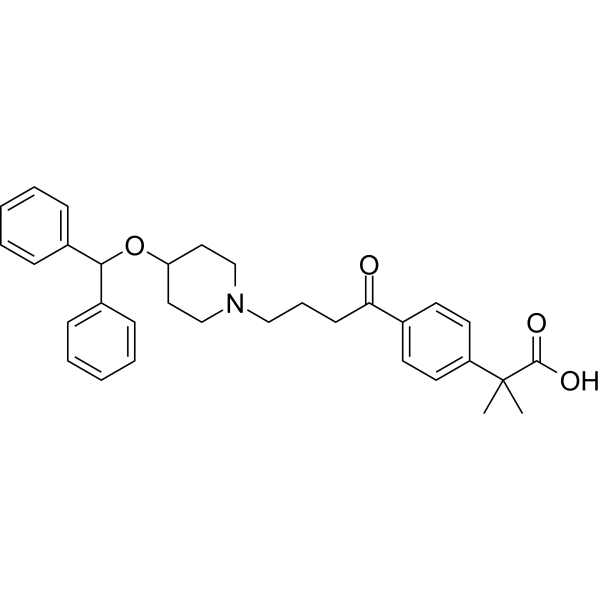
The most common side effects reported for nirsevimab are rash, pyrexia (fever) and injection site reactions (such as redness, swelling and pain where the injection is given).[6]
Nirsevimab was approved for medical use in the European Union in November 2022.[1][7]
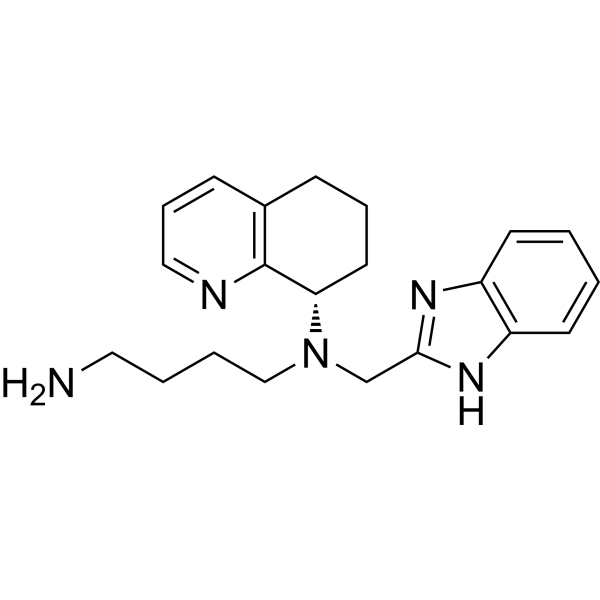
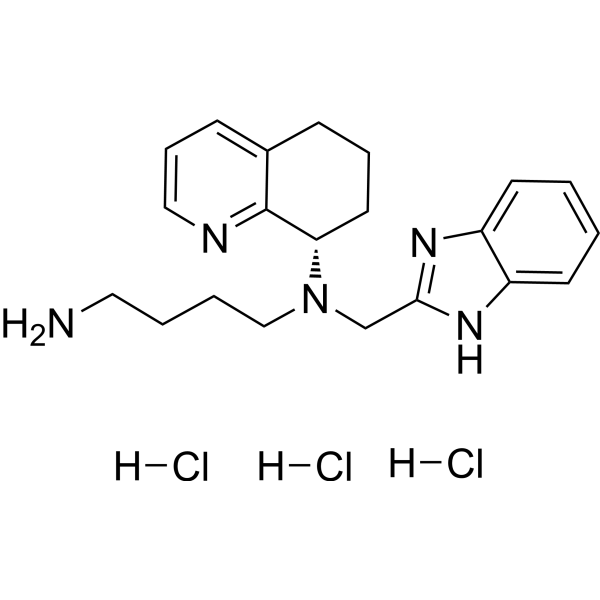
Nirsevimab (MEDI8897) is a recombinant human immunoglobulin G1 kappa (IgG1ĸ) monoclonal antibody used to prevent respiratory syncytial virus (RSV) lower respiratory tract disease in neonates and infants.6 It binds to the prefusion conformation of the RSV F protein, a glycoprotein involved in the membrane fusion step of the viral entry process, and neutralizes several RSV A and B strains.6,1 Compared to palivizumab, another anti-RSV antibody, nirsevimab shows greater potency at reducing pulmonary viral loads in animal models. In addition, nirsevimab was developed as a single-dose treatment for all infants experiencing their first RSV season, whereas palivizumab requires five monthly doses to cover an RSV season.5 This is due to a modification in the Fc region of nirsevimab that grants it a longer half-time compared to typical monoclonal antibodies.1,6
On November 2022, nirsevimab was approved by the EMA for the prevention of RSV lower respiratory tract disease in newborns and infants during their first RSV season.6
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
No major hypersensitivity reactions have been reported, and adverse events of grade 3 or higher were only reported in 8% (77 of 968) of participants in clinical trial NCT02878330.[8][4]
Pharmacology
Mechanism of action
Nirsevimab binds to the prefusion conformation of the RSV fusion protein, i.e. it binds to the site at which the virus would attach to a cell; effectively rendering it useless. It has a modified Fc region, extending the half-life of the drug in order for it to last the whole RSV season.[4]
History
The opinion by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) is based on data from two randomized, double-blind, placebo-controlled multicenter clinical trials that investigated the efficacy and safety of nirsevimab in healthy preterm (premature) and full-term infants entering their first respiratory syncytial virus (RSV) season.[6] These studies demonstrated that nirsevimab prevents lower respiratory tract infection caused by RSV requiring medical attention (such as bronchiolitis and pneumonia) in term and preterm infants during their first RSV season.[6]
The safety of nirsevimab was also evaluated in a phase II/III, randomized, double‑blind, multicenter trial in infants who were born five or more weeks prematurely (less than 35 weeks gestation) at higher risk for severe RSV disease and infants with chronic lung disease of prematurity (i.e. long-term respiratory problems faced by babies born prematurely) or congenital heart disease.[6] The results of this study showed that nirsevimab had a similar safety profile compared to palivizumab (Synagis).[6]
Society and culture
Legal status
On 15 September 2022, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Beyfortus, intended for the prevention of respiratory syncytial virus (RSV) lower respiratory tract disease in newborns and infants.[9][6] Beyfortus was reviewed under EMA’s accelerated assessment program.[9] The applicant for this medicinal product is AstraZeneca AB.[9] Nirsevimab was approved for medical use in the European Union in November 2022.[1][7]
Research
Nirsevimab is being investigated as an experimental vaccine against respiratory syncytial virus, RSV, in the general infant population.[2][3] The MELODY study is an ongoing, randomized, double-blind, placebo-controlled to evaluate the safety and efficacy of nirsevimab in late preterm and term infants. Initial results have been promising, with nirsevimab reducing LRTI (lower respiratory tract infections) by 74.5% compared to placebo in infants born at term or late preterm.[5][10][11]
^ Jump up to:abc“Beyfortus: Pending EC decision”. European Medicines Agency (EMA). 15 September 2022. Archived from the original on 19 September 2022. Retrieved 18 September 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
The most common adverse reactions include diarrhea, nausea, fatigue, vomiting, musculoskeletal pain, hepatotoxicity, renal impairment, dyspnea, edema, decreased appetite, cough, pneumonia, dizziness, constipation, abdominal pain, and QTc interval prolongation.[2] The most common laboratory abnormalities include decreased lymphocytes, increased aspartate aminotransferase, decreased sodium, decreased hemoglobin, increased creatinine, decreased albumin, increased alanine aminotransferase, increased lipase, decreased platelets, decreased magnesium, and decreased potassium.[2]
It was approved for medical use in the United States in December 2022.[1][3]
Synthesis Reference
Fell, Jay B et al. “Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer.” Journal of medicinal chemistry vol. 63,13 (2020): 6679-6693. doi:10.1021/acs.jmedchem.9b02052
Journal of Medicinal Chemistry (2020), 63(13), 6679-6693
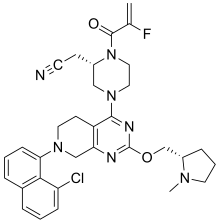
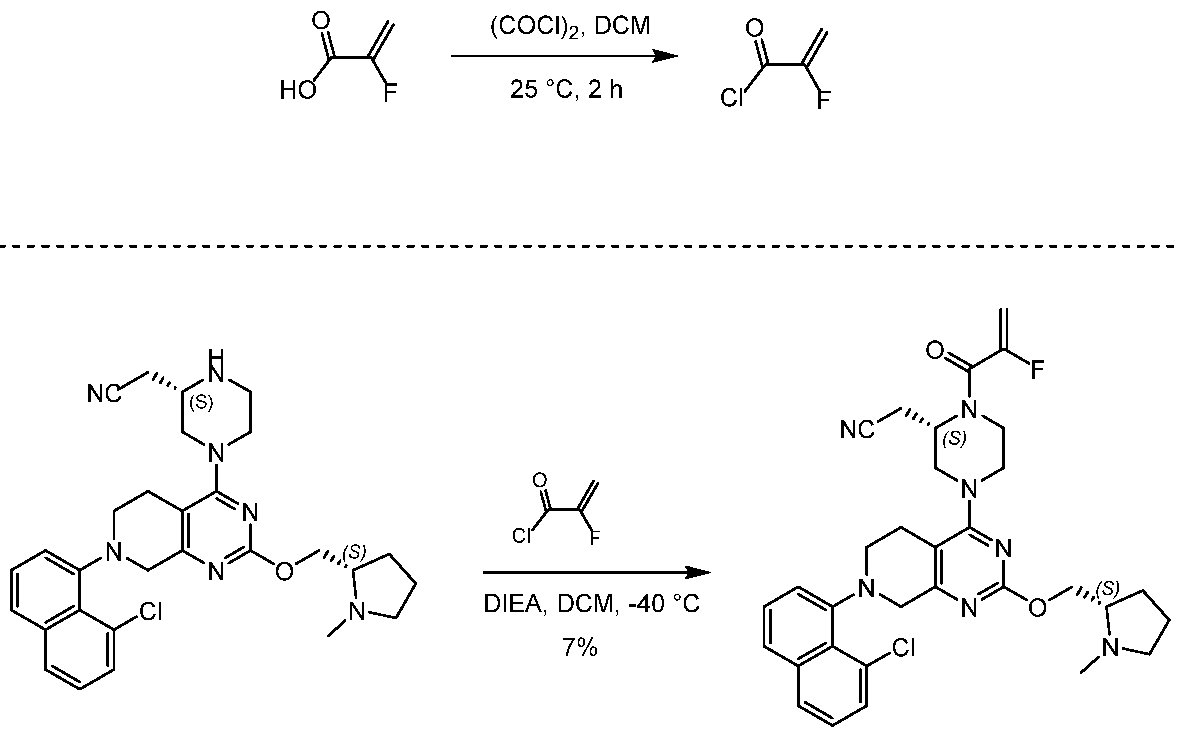
[0432] 2-fluoroprop-2-enoyl chloride. To a solution of 2-fluoroprop-2-enoic acid (400 mg, 4.44 mmol, 1 eq) in DCM (4 mL) was added (COCl)2 (846 mg, 6.66 mmol, 583 µL, 1.5 eq) and DMF (32.5 mg, 444 umol, 34.2 µL, 0.1 eq). The mixture was stirred at 25 °C for 2 hrs. The reaction mixture was concentrated under reduced pressure to remove a part of solvent and give a residue in DCM. Compound 2-fluoroprop-2-enoyl chloride (400 mg, crude) was obtained as a yellow liquid and used into the next step without further purification. [0433] Step A: 2-[(2S)-4-[7-(8-chloro-1-naphthyl)-2-[[(2S)-1- methylpyrrolidin-2-yl]methoxy]- 6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2- yl]acetonitrile. To a solution of 2-[(2S)-4-[7-(8-chloro-1-naphthyl)-2-[[(2S)- 1-methylpyrrolidin- 2-yl]methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-2-yl]acetonitrile (300 mg, 528 umol, 1 eq, HCl) in DCM (5 mL) was added DIEA (1.73 g, 13.4 mmol, 2.33 mL, 25.4 eq) and 2-fluoroprop-2-enoyl chloride (286 mg, 2.64 mmol, 5 eq) in DCM (5 mL). The mixture was stirred at 0 °C for 1 hour. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (Al2O3, Dichloromethane/Methanol = 10/1 to 10/1). The residue was purified by prep-HPLC (column: Gemini 150 * 25 5u; mobile phase: [water (0.05% ammonia hydroxide v / v) – ACN]; B%: 55% – 85%, 12min). The residue was purified by prep-HPLC (column: Phenomenex Synergi C18 150 * 30mm * 4um; mobile phase: [water (0.225% FA) – ACN]; B%: 20% – 50%, 10.5min). The residue was concentrated under reduced pressure to remove ACN, and then lyophlization. Title compound 2-[(2S)-4-[7-(8-chloro- 1-naphthyl)-2-[[(2S)-1- methylpyrrolidin-2-yl]methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin- 4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile (EXAMPLE 7, 24.1 mg, 36.7 umol, 7% yield, 99.1% purity, FA) was obtained as a brown solid. [0434] SFC condition: “AD – 3S_3_5_40_3ML Column: Chiralpak AD – 3 100 × 4.6mm I.D., 3um Mobile phase: methanol (0.05% DEA) in CO2 from 5% to 40% Flow rate: 3mL/min Wavelength: 220nm”. [0435] 1H NMR (400 MHz, Acetic) d = 7.82 (d, J = 8.0 Hz, 1H), 7.69 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 7.6 Hz, 1H), 7.49 (t, J = 7.6 Hz, 1H), 7.41 – 7.30 (m, 2H), 5.58 – 5.25 (m, 2H), 5.17 – 4.59 (m, 4H), 4.57 – 4.28 (m, 3H), 4.24 – 3.78 (m, 4H), 3.67 – 3.13 (m, 7H), 3.08 (br d, J = 2.4 Hz, 3H), 2.98 (br d, J = 6.4 Hz, 1H), 2.83 – 2.61 (m, 1H), 2.45 – 2.29 (m, 1H), 2.24 – 2.08 (m, 3H).
Adagrasib (MRTX849) is an oral, small-molecule KRAS inhibitor developed by Mirati Therapeutics. KRAS mutations are highly common in cancer and account for approximately 85% of all RAS family mutations.5 However, the development of KRAS inhibitors has been challenging due to their high affinity for guanosine triphosphate (GTP) and guanosine diphosphate (GDP), as well as the lack of a clear binding pocket.1 Adagrasib targets KRASG12C, one of the most common KRAS mutations, at the cysteine 12 residue and inhibits KRAS-dependent signalling.2 In a phase I/IB clinical study that included patients with KRASG12C-mutated advanced solid tumors (NCT03785249), adagrasib exhibited anti-tumor activity. The phase II of the same study showed that in patients with KRASG12C-mutated non-small-cell lung cancer (NSCLC), adagrasib was efficient without new safety signals.2,3,6
In February 2022, the FDA accepted a new drug application (NDA) for adagrasib for the treatment of patients with previously treated KRASG12C–positive NSCLC.7 In December 2022, the FDA granted accelerated approval to adagrasib for the treatment of KRASG12C-mutated locally advanced or metastatic NSCLC who have received at least one prior systemic therapy.8,9 Adagrasib joins sotorasib as another KRASG12C inhibitor approved by the FDA.4
Medical uses
Adagrasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA approved test, who have received at least one prior systemic therapy.[1][2][4]
History
Approval by the US Food and Drug Administration (FDA) was based on KRYSTAL-1, a multicenter, single-arm, open-label clinical trial (NCT03785249) which included participants with locally advanced or metastatic non-small cell lung cancer with KRAS G12C mutations.[2] Efficacy was evaluated in 112 participants whose disease has progressed on or after platinum-based chemotherapy and an immune checkpoint inhibitor, given either concurrently or sequentially.[2]
Forasartan, a specific angiotensin II antagonist, is used alone or with other antihypertensive agents to treat hypertension. Forasartan competes with angiotensin II for binding at the AT1 receptor subtype. As angiotensin II is a vasoconstrictor which also stimulates the synthesis and release of aldosterone, blockage of its effects results in a decreases in systemic vascular resistance.
Indications
Forasartan is indicated for the treatment of hypertension[6] and, similar to other ARBs, it protects the kidneys from kidney blood vessel damage caused by increased kidney blood pressure by blocking renin–angiotensin system activation.[7]
Administration
Forasartan is administered in the active oral form [6] which means that it must go through first pass metabolism in the liver. The dose administered ranges between 150 mg-200 mg daily.[6] Increasing to more than 200 mg daily does not offer significantly greater AT1 receptor inhibition.[6] Forasartan is absorbed quickly in the GI, and within an hour it becomes significantly biologically active.[6] Peak plasma concentrations of the drug are reached within one hour.[6]
A solution of 1500 mL (14 mol) of 48% hydrobromic acid was cooled to 10 °C and 300 g (2.8 mol) of 2-amino-5-picoline (Aldrich) was added slowly. The solution was maintained at or below 0 °C while 450 mL (8.8 mol) of bromime was added dropwise. After the bromine addition was complete, a solution of 500 g (7.3 mol) of sodium nitrite in 1000 mL of water was added slowly over 6 h. The reaction pH was adjusted by the careful addition of 1500 mL (56 mol) of 50% sodium hydroxide at such a rate that the temperature was maintained below 30 °C. The product precipitated from the nearly colorless reaction mixture; filtration gave 450 g (94%) of 2-bromo-5-picoline as a yellow powder: mp 38-40 °C; NMR 7.27 (s, 1H), 7.28 (s, 1H), 7.12 (br s, 1H).
Step 2 : Preparation of N-methyl-N-tertbutylbenzamide.
Under nitrogen, 96.7 g (1.1 mol) of N-methyl-N-tertbutylamine and 111 g (1.1 mol) of triethylamine was dissolved in 1050 mL of anhydrous tetrahydrofuran (THF).
The solution was cooled to 0 °C and treated with 140.6 σ (1.0 mol) of benzoyl chloride. The reaction was allowed to slowly warm to ambient temperature and stir overnight. Filtration and subsequent concentration in vacuo of the filtrate gave the crude product which was purified by sublimation (65 °, 0.2 torr) to give 184 g (96%) of colorless N-methyl-N-tertbutybenzamide: mp 80.5-82.0 °C; NMR (CDCI3) δ1.52 (s, 9H), 2.87 (s, 3H), 7.34-7.40 (m, 3H), 7.40-7.46 (m, 2H).
Step 3 : Preparation of 2-(N-methyl-N-tertbutylcarboxamido)phenyIboronic acid.
Under nitrogen, a solution of 50.0 g (262 mmol) of N-methyl-N-tertbutylbenzamide from step 2 and 44 ml (2S2 mmol) of tetramethylethylenediamine (TMEDA) in 3350 mL of anhydrous THF was cooled to -78 °C and slowly treated with 262 mmol of sec-butyllithium in cyclohexane. After 1 h at -78 °C, the reaction was treated with 45 mL (393 mmol) of trimethyl borate and allowed to slowly warm to ambient temperature overnight with stirring. The reaction was concentrated in vacuo; the residue was dissolved in IK sodium hydroxide and extracted with methylene chloride. The pH of the aqueous phase was adjusted to six with dilute hydrochloric acid and extracted with methylene chloride; the organic layer was dried (MgSO4) and concentrated in vacuo to give 55.7 g (90%) of a 80:20 mixture of syn/anti isomers of 2-(N-methyl-N-tertbutylcarboxamido)phenyIboronic acid as a pale yellow glass: NMR (CDCI3) δ 1.30 (s, syn C(CH3)3, 7.3H), 1.54 (s, anti 0(0.3)3, 1.7H), 2.81 (s, anti CH3, 0.6H), 2.94 (s, syn CH3, 2.4H), 7.29-7.46 (m, 3H), 7.95-8.01 (m, 1H).
step 4 : Preparation of N-methyl-N-tertbwtyl-2-(5-methyl-2-pyridinyl)benzamide.
Under nitrogen, 4.44 g (25.8 mmcl) cf 2-bromo-5-picoline from step 1 in 60 mL of toluene was treated with 6.75 g (29 mmol) of 2- (N-methyl-N- tertbutylcarboxamido)phenyIboronic acid from step 3, 1.0 g of tetrakis (triphenylphosphine)palladium zero, 26 mL of ethanol, and 29 mL of 2M sodium carbonate; this mixture was heated to reflux and vigorously stirred for 24 h. The reaction was partitioned between water and ether; the organic layer was separated, dried (MgSθ4), and concentrated in vacuo. Purification by silica gel chromatography (Waters Prep-500A) using ethyl acetate/hexane (1:2) gave 6.51 g (90%) of N-methyl-N- tertbutyl-2-(5-methyl-2-pyridinyl)benzamide as an oil : NMR (CDCI3) δ 1.40 (s, 9H), 2.33 (s, 3H), 2.61 (s, 3H), 7.27- 7.33 (m, 1H), 7.35-7.41 (m, 2H), 7.47-7.51 (m, 2H), 7.60- 7.66 (m, 1H), 8.43 (br s, 1H).
Step 5 : Preparation of sodium 2-(5-methyl-2- pyridinyl)benzoate.
Under nitrogen, 6.5 g (23 mmol) of N-methyl-N- tertbutyl-2-(6-methyl-3-pyridinyl)benzamide from step 4 was treated with 65 mL of anhydrous trifluoroacetic acid (TFA) at reflux for 6 h. The reaction was concentrated in vacuo and the residue dissolved in water. The pH was adjusted to 10 with aqueous sodium hydroxide and lyophilized to give the sodium salt of 2- (5-methyl-2-pyridinyl)benzoic acid as a colorless solid: NMR [CDCI3/CF3CO2H (97:3)] δ 2.62 (s, 3H), 7.42-7.48 (m, 1H), 7.67-7.80 (m, 3H), 8.18-8.24 (m, 1H), 8.28 (dd, J=8 and 2 HZ, 1H), 7.67-7.80 (m, 3H), 8.18-8.24 (m, 1H), 8.28 (dd, J=8 and 2 Hz, 1H), 8.61 (s, 1H) ; MS (FAB) m/e (rel intensity) 214 (20), 196 (100); HRMS. Calc’d for M+H: 214.0868. Found: 214.0846.
step 6 : Preparation of ethyl 2-(5-methyl-2-pyridinyl)benzoate.
Under nitrogen, the crude sodium salt from step 5 was suspended in 50 mL of chloroform and treated with 9 mL (103 mmol) of oxalyl chloride. The reaction was stirred for 72 h, filtered under nitrogen, and concentrated in vacuo; the residue was dissolved in absolute ethanol. Concentration in vacuo gave 2.0 g (8 mmol) of ethyl 2-(5-methyl-2-pyridinyl)benzoate as a brown oil: NMR (CDCI3) δ 1.09 (t, J=7 Hz, 3H), 2.36 (s, 3H), 4.15 (q, J=7 Hz, 2H), 7.34 (d, J=8 Hz, 1H), 7.38-7.48 (m, 1H), 7.48-7.58 (m, 3H), 7.80 (d, J=8 Hz, 1H), 8.46 (s, 1H).
Step 7 : Preparation of ethyl 2-(5-bromomethyl-2-pyridinyl)benzoate.
Under nitrogen, the crude ethyl 2-(5-methyl-2-pyridinyl)benzoate from step 6 was treated with 1.7 g (9.5 mmol) of NBS and 160 mg (0.66 mmol) of benzoyl peroxide in 145 mL of anhydrous carbon tetrachloride at reflux for 2.5 h. The reaction was filtered under nitrogen and concentrated in vacuo to give crude ethyl 2-(5-bromomethyl-2-pyridinyl)benzoate; no purification was attempted.
Under nitrogen, 630 mg (3.5 mmol) of 3,5-dibutyl-1H-1,2,4-triazole from step 3 of Example 1 was added in small portions to 5.4 mmol of sodium hydride in 8 mL of DMF; stirring was continued until hydrogen evolution had ceased. The anion solution was cooled to 0 °C and treated with a solution of the crude ethyl 2-(5-bromomethyl-2-pyridinyl)benzoate from step 7 in 10 mL of DMF. The reaction was stirred at ambient temperature overnight, quench with 1 mL of absolute ethanol, and concentrated in vacuo; the resulting residue was redisolved in methylene chloride, filtered, and reconcentrated in vacuo to give crude ethyl 2-[5-[(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]-2-pyridinyl]benzoate.
A 1.0 g sample of the crude ethyl 2-[5-[(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]-2-pyridinyl]benzoate from step 8 in 10 mL of water was treated with 3 mL of 101 aqueous sodium hydroxide and stirred at ambient temperature overnight. The reaction mixture was washed with 30 mL of ether and the pH adjusted to six with dilute hydrochloric acid. Purification by reverse phase chromatography (Waters Deltaprep-3000) using isocratic acetonitrile/water (28:72) (0.05% TFA) gave 5 mg of 2-[5-[(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]-2-pyridinyl]benzoic acid: NMR (D2O + NaO3S(CH2)3 Si(CH3)3] δ 0.80 (t, J=7 Hz, 3H), 0.86 (t, J=7 Hz, 3H), 1.19-1.33 (m, 4H), 1.54-1.68 (m, 4H), 2.65 (t, J=7 Hz, 2H), 2.82 (t, _ϊ=7 Hz, 2H), 5.43 (s, 2H), 7.45-7.59 (m, 5H), 7.64 (dd, J=8 and 2 Hz, 1H), 8.37-8.45 (m, 1H); MS (FAB) m/e (rel intensity) 393 (80), 375 (30), 212 (40), 182 (100); HRMS. Calc’d for M+Li: 399.2373. Found: 399.2374.
Step 1 : Preparation of 2-bromo-5-bromomethylpyridine.
A solution of 296.3 g (1.72 mol) of 2-bromo-5-picoline from step 1 of Example 2 in 6000 mL of carbon tetrachloride was treated with 306.5 g (1.72 mol) of N-bromosuccinimide (NBS) and 28.3 g (173 mmol) of azobisisobutyronitrile (AIBN). The reaction was stirred at reflux under nitrogen for 3 h, filtered, and concentrated in vacuo providing 476 g of crude 2-bromo-5-bromomethylpyridine as a brownish yellow solid (NMR indicates that this material is only 60% monobromomethyl product): NMR (CDCI3 δ 4.42 (s, 2H), 7.48 (d, .J=9 Hz, 1H), 7.60 (dd, J=9 and 3 Hz, 1H), 8.37 (d, J=3 Hz, 1H).
Step 2: Preparation of 2-bromo-5-[(3.5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]pyridine.
Under nitrogen, 3.15 g (17 mmol) of solid 3,5-dibutyl-1H-1,2,4-triazole from step 3 of Example 1 was added in small portions to 33 mmol of sodium hydride in 31 ml of dimethylformamide (DMF); stirring was continued until hydrogen evolution had ceased. The anion solution was cooled to 0 °C and treated with a solution of 7.9 g (19 mmol) of crude 2-bromo-5-bromomethylpyridine from step 1 in 10 ml of dry DMF. The reaction was allowed to warm to ambient temperature and stir overnight. Methanol (10 ml) was added to destroy any unreacted sodium hydride and the
DMF was removed in vacuo. The residue was dissolved in ethyl acetate, washed with water, and dried (MgSO4). Silica gel chromatography (Waters Prep-500A) using ethyl acetate/hexane (60:40) gave 4.8 g (47%) of 2-bromo-5-[(3,5- dibutyl-1H-1,2,4-triazol-1-yl)methyl]pyridine as an oil: NMR (CDCI3) δ 0.88 (t, J=7 Hz, 1H), 0.92 (t, J=7 Hz, 1H), 1.27-1.44 (m, 4H), 1.59-1.76 (m, 4H), 2.60-2.71 (m, 4H), 5.18 (s, 2H), 7.35 (dd, J=8 and 3 Hz), 7.46 (d, J=8 Hz, 1H), 8.23 (d, .1=3 Hz, 1H).
Step 3: Preparation of 5-[2-[5-[(3,5-dibutyl-1H-1,2,4- triazol-1-yl)methyl]-2-pyridinyl]phenyl]-1H-tetrazole.
Under nitrogen, 1.03 g (2.9 mmol) of 2-bromo-5- [(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]pyridine from step 2 and 2.46 g (5.7 mmol) of 2-(N-triphenyImethyltetrazol-5-yl)phenyIboronic acid from step 5 of Example 1 were treated with 1.0 g (0.86 mmol) of tetrakis (triphenyl-phosphine)palladium zero, 15 mL of toluene, 10 mL of ethanol, and 6.3 mL of 2M aqueous sodium carbonate. The reaction mixture was heated to reflux and vigorously stirred overnight. The product was purified by reverse phase chromatography (Waters Deltaprep-3000) using acetonitrile/water (20-40:80-60) (0.05% TFA). The pure fractions (by analytical HPLC) were combined, the acetonitrile removed in vacuo, the pH adjusted to four with dilute sodium hydroxide, and the resulting suspension extracted 4 times with ether. The extracts were combined, dried (MgSθ4), and concentrated in vacuo to give 340 mg (28%) of 5-[2-[5-[(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]-2-pyridinyl]phenyl-1H-tetrazole as a colorless solid: mp 139-141 °C; NMR (CD3OD) δ 0.90 (t, J=7 Hz, 3H), 0.93 (t, J=7 Hz, 3H), 1.29-1.44 (m, 4H), 1.58-1.75 (m, 4H), 2.65 (t, J=7 Hz, 2H), 2.81 (t, J=7 Hz, 2H), 5.40 (s, 2H), 7.47 (d, J=8 Hz, 1H), 7.61-7.77 (m, 5H), 8.33 (d, J=2 Hz, 1H); MS (FAB) m/e (rel intensity) 417 (100), 208 (30); HRMS. Calc’d for M+H: 417.2515. Found: 417.2527.
Angiotensin II binds to AT1 receptors, increases contraction of vascular smooth muscle, and stimulates aldosterone resulting in sodium reabsorption and increase in blood volume.[9]Smooth muscle contraction occurs due to increased calcium influx through the L-type calcium channels in smooth muscle cells during the plateau component, increasing the intracellular calcium and membrane potential which sustain depolarization and contraction.[10]
Effects
Forasartan is a competitive and reversible ARB that competes with the angiotensin II binding site on AT1[11] and relaxes vascular smooth muscle,[10] resulting in decreased blood pressure. Forasartan has a high affinity for the AT1 receptor (IC50=2.9 +/- 0.1nM).[12] In dogs, it was found to block the pressor response of Angiotensin II with maximal inhibition, 91%.[10] Forasartan administration selectively inhibits L-type calcium channels in the plateau component of the smooth muscle cells, favoring relaxation of the smooth muscle.[10] Forasartan also decreases heart rate by inhibiting the positive chronotropic effect of high frequency preganglionic stimuli.[13]
Scarce use
Even though experiments have been conducted on rabbits,[6] guinea pigs,[10] dogs [14] and humans,[6][13] forasartan is not a popular drug of choice for hypertension due to its short duration of action; forasartan is less effective than losartan.[6] Research demonstrates that forasartan is also significantly less potent than losartan.[6]
^ Olins GM, Corpus VM, Chen ST, McMahon EG, Palomo MA, McGraw DE, et al. (October 1993). “Pharmacology of SC-52458, an orally active, nonpeptide angiotensin AT1 receptor antagonist”. Journal of Cardiovascular Pharmacology. 22 (4): 617–25. doi:10.1097/00005344-199310000-00016. PMID7505365. S2CID93468.
^ Jump up to:abcdefghijk Hagmann M, Nussberger J, Naudin RB, Burns TS, Karim A, Waeber B, Brunner HR (April 1997). “SC-52458, an orally active angiotensin II-receptor antagonist: inhibition of blood pressure response to angiotensin II challenges and pharmacokinetics in normal volunteers”. Journal of Cardiovascular Pharmacology. 29 (4): 444–50. doi:10.1097/00005344-199704000-00003. PMID9156352.
^ Ram CV (August 2008). “Angiotensin receptor blockers: current status and future prospects”. The American Journal of Medicine. 121 (8): 656–63. doi:10.1016/j.amjmed.2008.02.038. PMID18691475.
^ Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S (April 2007). “Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology”. Clinical Science. 112 (8): 417–28. doi:10.1042/cs20060342. PMID17346243.
^ Jump up to:abcde Usune S, Furukawa T (October 1996). “Effects of SC-52458, a new nonpeptide angiotensin II receptor antagonist, on increase in cytoplasmic Ca2+ concentrations and contraction induced by angiotensin II and K(+)-depolarization in guinea-pig taenia coli”. General Pharmacology. 27 (7): 1179–85. doi:10.1016/s0306-3623(96)00058-4. PMID8981065.
^ Olins GM, Chen ST, McMahon EG, Palomo MA, Reitz DB (January 1995). “Elucidation of the insurmountable nature of an angiotensin receptor antagonist, SC-54629”. Molecular Pharmacology. 47 (1): 115–20. PMID7838120.
‘Top 10 Prominent & Great Personalities of the year 2022’ campaign by Fame Finders. Feeling great to be selected in the ‘Top 10 Prominent & Great Personalities of the year 2022’ campaign by Fame Finders. It is an online campaign to honour inspiring personalities and feature them in the upcoming edition of the top news sites, including – Deccan Chronicle/Asian Age/Deccan Herald, ANI, Zee5, Latestly, Lokmat Times, DailyHunt, Google News, JioNews, MSN and 70+ sites.Jan 16 2023
I myself Dr Anthony Melvin Crasto Looking for a post retirement assignment as Advisor API & INT, Chem. With 36 yrs rich experience, about dozen patents, 10000plus steps covered, 200 API targets, 30 plus products commercialization in plant in full career. Hands on knowledge of Synthesis, Process, scaleup, cost reduction, DOE , softwares etc
Kindly contact me Dr Anthony Melvin Crasto +919321316780 amcrasto@gmail.com
About myself Dr Anthony Crasto click on my website to know about me
1000 lakh google hits, 100lakh blog views, 10 lakh viewers in USA alone, all in 7 continents, 226 countries, 30 Indian and International awards, helping millions across the world
Selective estrogen receptor downregulator (SERD) Treatment of breast cancer
SAR439859 (compound 43d) is an orally active, nonsteroidal and selective estrogen receptor degrader (SERD). SAR439859 is a potent ER antagonist and has ER degrading activity with an EC50 of 0.2 nM for ERα degradation. SAR439859 demonstrates robust antitumor efficacy and limited cross-resistance in ER+ breast cancer.
Amcenestrant is an orally available, nonsteroidal selective estrogen receptor degrader/downregulator (SERD), with potential antineoplastic activity. Upon oral administration, amcenestrant specifically targets and binds to the estrogen receptor (ER) and induces a conformational change that promotes ER degradation. This prevents ER-mediated signaling and inhibits both the growth and survival of ER-expressing cancer cells.
Amcenestrant is reported to be a selective estrogen receptor degrader (SERD) which has estrogen receptor antagonist properties and accelerates the proteasomal degradation of the estrogen receptor. Amcenestrant is under clinical investigation as an anticancer agent, in particular for treatment of breast cancer.
The compound and processes for preparation thereof are described in International Publication No. WO 2017/140669.
Crystalline forms are described in International Publication No. WO 2021/116074.
PAPER
Journal of Medicinal Chemistry (2020), 63(2), 512-52
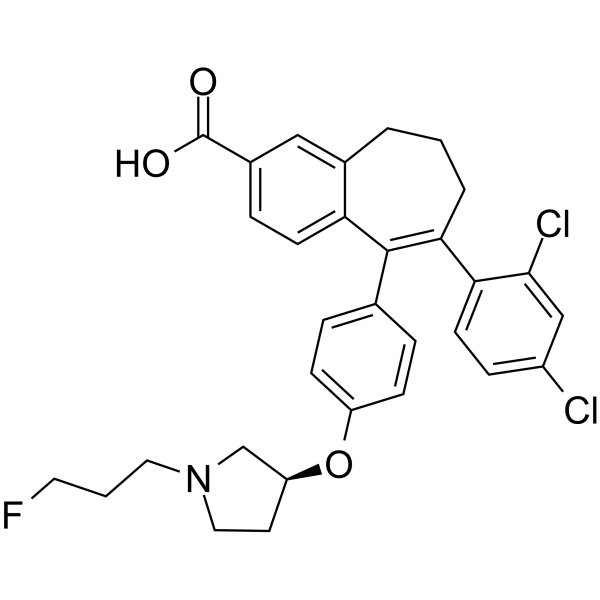
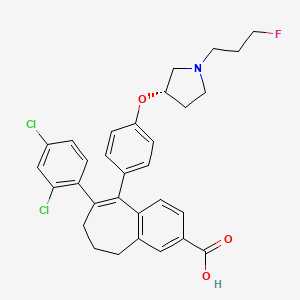
To a solution of 6-(2,4-dichloro-phenyl)-5-[4-[1-(3-fluoropropyl)-pyrrolidin-3-yloxy]-phenyl]-8,9-dihydro-7H-benzocycloheptene-2-carboxylic acid methyl ester (42d) (80 mg, 140.72 μmol) in methanol (5 mL) was added 5 N NaOH (562.88 μL), the reaction mixture was heated to 60 °C for 5 h, and the solvent was removed under reduced pressure. The residue was taken up in water (10 mL), and aqueous HCl (5 M) was added to pH 7. The slurry was extracted with dichloromethane, dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The solid was purified by column chromatography eluting with a mixture of dichloromethane, acetonitrile, and methanol (90/5/5 v/v/v) to give 60 mg (77%) of 6- (2,4-dichlorophenyl)-5-[4-[(3S)-1-(3-fluoropropyl)pyrrolidin-3-yl]- oxyphenyl]-8,9-dihydro-7H-benzo[7]annulene-2-carboxylic acid (43d). 1 H NMR (400 MHz, DMSO-d6): 1.68 (m, 1H), 1.79 (dm, J = 25.3 Hz, 2 H), 2.07 to 2.23 (m, 5H), 2.38 (m, 1H), 2.46 (t, J = 7.2 Hz, 2H), 2.52 (m, 1H), 2.62 (m, 1H), 2.55 to 2.89 (m, 3H), 4.47 (td, J = 6.2 and 47.6 Hz, 2H), 4.72 (m, 1H), 6.63 (d, J = 8.9 Hz, 2H), 6.71 (m, 3H), 7.18 (d, J = 8.4 Hz, 1H), 8.26 (dd, J = 2.0 and 8.4 Hz, 1H), 7.58 (d, J = 2.0 Hz, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.79 (s, 1H), 12.3 (m, 1H). LCMS: 554 (M + H)+ .
Amcenestrant can be prepared according to methods known from the literature, for example U.S. Patent No. 9,714,221.
Example 1: Preparation of amorphous Amcenestrant
[00164] Amcenestrant (20 mg, prepared according to U.S. Patent No. 9,714,221) was dissolved in ethyl acetate (0.2 mL) at room temperature (25°C). Solution was left in opened flask at RT for 16 days, until all the solvent evaporated. Obtained solid was analyzed by XRPD.
To a solution of methyl 8-bromo-9-(4-{[(3S)-1-(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7-dihydro-5H-benzo[7]annulene-3-carboxylate hydrobromide (D5) (150 mg, 298.56 μιηοΙ), in dioxane (12 ml) and water (2 ml), was added 2,4-dichlorophenyl-boronic acid (62.67 mg, 328.41 μηηοΙ), Cs2C03 (204.48 mg, 626.97 μηιοΙ), and Pd(dppf)CI2 (14.63 mg, 17.91 μιηοΙ). The reaction mixture was heated at 90°C for 3 hours, and partitioned between AcOEt and water. The phases were separated and the organic phase washed with brine, dried over MgS04 and concentrated under reduced pressure. The residue was purified by column chromatography eluting with a mixture of DCM, acetonitrile and MeOH (96/2/2; V/V/V) to give 80 mg (47%) of 6-(2,4-dichloro-phenyl)-5-{4-[1-(3-fluoro-propyl)-pyrrolidin-3-yloxy]-phenyl}-8,9-dihydro-7H-benzocycloheptene-2-arboxylic acid methyl ester.
To a solution of 6-(2,4-dichloro-phenyl)-5-{4-[1-(3-fluoro-propyl)-pyrrolidin-3-yloxy]-phenyl}-8,9-dihydro-7H-benzocycloheptene-2-arboxylic acid methyl ester (80 mg, 140.72μιηο!) in MeOH (5 ml) was added a solution of NaOH (562.88 μΙ, 5 M) and the reaction mixture was heated at 60°C for 5 hours and the solvent removed under reduced pressure. The residue was taken up in water (10 ml) and aqueous HCI (5 M) added to pH
7. The slurry was extracted with DCM, dried over MgS04 and concentrated under reduced pressure. The solid was purified by column chromatography eluting with a mixture of DCM, acetonitrile and MeOH (90/5/5; V/V/V) to give 60 mg (77%) of 6-(2,4-dichlorophenyl)-5-[4-[(3S)-1-(3-fluoropropyl)pyrrolidin-3-yl]oxyphenyl]-8,9-dihydro-7H-benzo[7]annulene-2-carboxylic acid.
To a solution of commercially available 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenol (a) (82.7 g, 364.51 mmol) in THF (2 L) was added under argon (R)-1 -N-Boc-3-hydroxypyrrolidine (b) (84.43 g, 437.41 mmol) followed by Ν,Ν,Ν’,Ν’-tetramethylazodicarboxamide (99.1 g, 546.77 mmol). The clear reaction mixture turned orange and triphenylphosphine (143.41 g, 546.77 mmol) was added. The reaction mixture was stirred at room temperature for 24 hours, meanwhile a precipitate of triphenylphosphine oxide formed (Ph3P=0). The reaction mixture was poured in water (1 .5 L) and extracted with ethyl acetate (AcOEt) (3×1 .5 L). Gathered organic phases were dried over magnesium sulfate (MgS04), filtered and concentrated under reduced pressure. The residue was taken up into diisopropylether (1 .5 L) and the solid formed (Ph3P=0) was filtered. The solvent was concentrated under reduced pressure and the residue purified by column chromatography eluting with a mixture of heptane with AcOEt (90/10; v/v) to give 145 g (100%) of tert-butyl (3S)-3-[4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenoxy]pyrrolidine-1 -carboxylate (c) as a colorless oil.
To a solution of (S)-tert-butyl 3-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenoxy)pyrrolidine-1 -carboxylate (c) (80 g, 195.23 mmol) in MeOH (450 ml) was added slowly HCI 4N in dioxane (250 ml).
After 1 .5 hours, the reaction mixture was concentrated under reduced pressure and the residue was taken up into Et20 with stirring to give a solid which then was filtered and dried under vacuum to give 61.8 g (95%) of (3S)-3-[4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2yl)phenoxy]pyrrolidine, hydrochloride (d) as a white powder.
To a suspension of (S)-3-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenoxy)pyrrolidine hydrochloride (d) (20 g, 61.42 mmol) in acetonitrile (100 ml), was added K2C03 (21 .22 g, 153.54 mmol) and 1 -iodo-3-fluoropropane (12.15 g, 61.42 mmol), under argon. The reaction
mixture was stirred at 40°C for 24 hours. After cooling to room temperature, the reaction mixture was filtered and washed with acetonitrile. The filtrate was concentrated under reduced pressure and the residue was taken up in DCM and the solid formed was filtered and washed with DCM. The filtrate was concentrated to give 21.5 g (100%) of (3S)-1 -(3-fluoropropyl)-3-[4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenoxy]pyrrolidine (e) as a yellow foam.
To a solution of 2-hydroxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (A) (1 .52 g, 8.63 mmol), in acetone (60 ml), was added K2C03 (1 .19 g, 8.63 mmol) and pivaloyl chloride (1.06 ml, 8.63 mmol). The reaction mixture was stirred at room temperature for 16 hours, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with a gradient of heptane in AcOEt (100/0 to 85/15, v/v) to give 1.55 g (69%) of 5-oxo-6,7,8,9-tetrahydro-5H-benzo[7]annulen-2-yl 2,2-dimethylpropanoate (B) as a colorless oil.
To a solution of 5-oxo-6,7,8,9-tetrahydro-5H-benzo[7]annulen-2-yl 2,2-dimethylpropanoate (B) (15 g, 57.62 mmol) in DCM (500 ml) was added dropwise under argon pyridine (7.28 ml, 86.43 mmol) and trifluoromethanesulfonic anhydride (19.58 ml, 1 15.24 mmol). The reaction mixture was stirred at room temperature for 2 hours and ice (200 g) was added. The phases were separated, the aqueous phase was washed with DCM and the gathered organic phases were dried over MgS04, filtered and evaporated under reduced pressure to give 22 g (97%) of 9-(trifluoromethanesulfonyloxy)-6,7-dihydro-5H-benzo[7]annulen-3-yl 2,2-dimethylpropanoate (C) as a white solid.
To a solution of 9-(trifluoromethanesulfonyloxy)-6,7-dihydro-5H-benzo[7]annulen-3-yl-2,2-dimethylpropanoate (C) (22 g, 56.07 mmol) and (3S)-1 -(3-fluoropropyl)-3-[4-(tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenoxy]pyrrolidine (e) (20.56 g, 58.87 mmol) in dioxane (420 ml) and water (120 ml) was added under argon Pd(dppf)CI2 (2.75 g, 3.36 mmol) and Cs2C03 (36.57 g, 1 12.13 mmol). The reaction mixture was stirred for 1 hour at room temperature and was partitioned between water and DCM. The aqueous phase was washed with DCM and the gathered organic phases dried over MgS04, filtered and concentrated under reduced pressure. The residue was purified by column chromatography eluting with a gradient of MeOH in DCM (0 to 5%; V/V) to give 31 g (100 %) of 9-(4-{[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7-dihydro-5H-benzo[7]annulen-3-yl-2,2-dimethylpropanoate (D).
To a solution under argon of 9-(4-{[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7-dihydro-5H-benzo[7]annulen-3-yl-2,2-dimethylpropanoate (D) (24.8 g, 53.26 mmol) in MeOH (300 ml), was added NaOH 5M (23 ml, 1 15.00 mmol). The reaction mixture was stirred for 2 hours at room temperature. pH was then adjusted to 7 by addition of 6N aqueous HCI solution. The MeOH was concentrated under reduced pressure, then DCM was added. The organic phase was dried over MgS04, and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with a gradient of DCM/ MeOH from 100/0 to 95/05 to give 18.8 g (93%) of 9-(4-{[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7-dihydro-5H-benzo[7]annulen-3-ol (E) as a beige solid.
To a solution of 9-(4-{[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7-dihydro-5H-benzo[7]annulen-3-ol (E) (20.6 g, 54.00 mmol) in DCM (200 ml) and pyridine (6.55 ml, 81 .00 mmol), cooled to 5°C (ice bath), was added dropwise trifluoromethanesulfonic anhydride (18.93 ml, 108.00 mmol) under argon, and the reaction temperature was maintained <15°C. The ice bath was removed, and the brown suspension was stirred at room temperature for 2 hours. Ice (200 g) and DCM (200 ml) were added and the phases separated. The organic phase was dried over MgS04, and concentrated under reduced pressure. The residue was
purified by flash chromatography eluting with a gradient of DCM/MeOH from 100/0 to 95/05 to give 24.7 g (89.1 %) of 9-(4-{[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7-dihydro-5H-benzo[7]annulen-3-yl trifluoromethanesulfonate (F) as a brown oil.
To a solution of 9-(4-{[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7-dihydro-5H-benzo[7]annulen-3-yl trifluoromethanesulfonate (F) (10.1 g, 19.67 mmol) in DMF (66 ml) and MeOH (33 ml), were added Pd(dppf)CI2 (909 mg, 1.18 mmol) and diisopropylethylamine (7.21 ml). The black suspension was carbonylated in an autoclave at 70°C under 5 bars of CO for 5 hours. The reaction mixture was filtered, then the filtrate was partially concentrated under reduced pressure. The residue was partitioned between AcOEt and water. The organic phase was washed with water (2x 100 ml), dried over MgS04, and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with a gradient of DCIW MeOH from 100/0 to 95/05 to give 7.13 g (86%) of methyl 9-(4-{[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7-dihydro-5H-benzo[7]annulene-3-carboxylate (G) as a brown gum.
To a solution of commercially available 2-hydroxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (A) (18.5 g, 105 mmol) in DCM (185 ml) and lutidine (13.35 ml, 1 13.505 mmol), cooled at 5°C under argon, was added dropwise trifluoromethanesulfonic anhydride (20.22 ml,
123.29 mmol) while keeping temperature between 10 and 20°C. The reaction mixture was stirred for 1 hour at 5°C then at room temperature for 1 hour.
Then, ice (200 g) was added and the slurry partitioned between water and DCM. The organic phase was washed with aqueous NaHC03 solution, dried over MgS04, filtered off and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with a gradient of heptane/AcOEt from 100 to 90/10 to give 28.2 g (87%) of 5-oxo-6,7,8,9-tetrahydro-5H-benzo[7]annulen-2-yl trifluoromethanesulfonate (A1 ) as an orange oil. LC/MS (m/z, MH+): 309
To a solution of 5-oxo-6,7,8,9-tetrahydro-5H-benzo[7]annulen-2-yl trifluoromethanesulfonate (A1 ) (5.03 g, 16.32 mmol) in DMF (24 ml) and MeOH (12 ml), were added Pd(dppf)CI2 (754 mg, 0.98 mmol) and diisopropylethylamine (6 ml). The black suspension was carbonylated in an autoclave at 70°C under 5 bars of CO for 2.5 hours. The reaction mixture was filtered, then the filtrate was partially concentrated under reduced pressure, and the residue, was partitioned between AcOEt and water. The organic phase was washed with water (2x 75 ml) and aqueous HCI 0.5 N, dried over MgS04 and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with a gradient of heptane/AcOEt from 100/0 to 90/10 to give 3.4 g (95%) of methyl 5-oxo-6,7,8,9-tetrahydro-5H-benzo[7]annulene-2-carboxylate (B1 ) as a colorless oil.
To a solution of methyl 5-oxo-6,7,8,9-tetrahydro-5H-benzo[7]annulene-2-carboxylate (B1 ) (18,19 g, 83,34 mmol) in DCM (500 ml) and anhydrous pyridine (1 1 ml, 130,56 mmol), cooled at 5°C under argon, was added dropwise trifluoromethanesulfonic anhydride (30 ml, 176,54 mmol). The reaction mixture, a thick suspension, was stirred at room temperature for 24 hours, then ice was added and partitioned between water and DCM. The organic phase was dried over MgS04, filtered off and concentrated under reduced pressure to give 29 g (100%) of methyl 9-(trifluoromethanesulfonyloxy)-6,7-dihydro-5H-benzo[7]annulene-3-carboxylate (C1 ) as a yellow gum.
To a solution of methyl 9-(trifluoromethanesulfonyloxy)-6,7-dihydro-5H-benzo[7]annulene-3-carboxylate (C1 ) (29 g, 82.9 mmol), (3S)-1 -(3-fluoropropyl)-3-[4-(tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenoxy]pyrrolidine (e) (28.9 g, 82.9 mmol), in dioxane (225 ml) were added Pd(dppf)CI2 under argon, complex with DCM (3.73 g, 4.57 mmol) and Cs2C03 1 .5 M aqueous solution (1 1 1.12 ml, 166.68 mmol). The reaction mixture was stirred at 60°C for 1 hour.
After cooling to room temperature, the reaction mixture was poured into a mixture of water (500 ml) and AcOEt (400ml). The organic phase was washed with brine, dried over MgS04, filtered on celite and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with a gradient of DCM/MeOH from 100/0 to 95/05 to give 23 g (65%) of methyl 9-(4-{[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7-dihydro-5H-benzo[7]annulene-3-carboxylate (G) as a brown gum.
To a solution of methyl 9-(4-{[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7-dihydro- 5H-benzo[7]annulene-3-carboxylate (G) (13.93 g, 32.89 mmol), in DCM (150 ml) was added under argon pyridinium tribromide (15.78 g, 44.41 mmol). The reaction mixture was stirred for 1 hour at room temperature. Water (200 ml) was added, organic phase was then dried over MgS04, and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with a gradient of DCM/MeOH from 100/0 to 95/05 to give 16.4 g (85%) of methyl 8-bromo-9-(4-{[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7-dihydro- 5H-benzo[7]annulene-3-carboxylate hydrobromide (H) as a yellow meringue.
To a solution of methyl 8-bromo-9-(4-{[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxy}phenyl)-6,7- dihydro-5H-benzo[7]annulene-3-carboxylate hydrobromide (H) (150 mg, 298.56 μηηοΙ), in dioxane (12 ml) and water (2 ml), was added 2,4-dichlorophenyl-boronic acid (62.67 mg, 328.41 μηιοΙ), Cs2C03 (204.48 mg, 626.97 μπιοΙ), and Pd(dppf)CI2 (14.63 mg, 17.91 mol). The reaction mixture was heated at 90°C for 3 hours, and partitioned between AcOEt and water. The phases were separated and the organic phase washed with brine, dried over MgS04 and concentrated under reduced pressure. The residue was purified by column
chromatography eluting with a mixture of DCM, acetonitrile and MeOH (96/2/2; V/V/V) to give 80 mg (47%) of 6-(2,4-dichloro-phenyl)-5-{4-[1 -(3-fluoro-propyl)-pyrrolidin-3-yloxy]-phenyl}-8,9-dihydro-7H-benzocycloheptene-2-arboxylic acid methyl ester (I).
To a solution of 6-(2,4-dichloro-phenyl)-5-{4-[1 -(3-fluoro-propyl)-pyrrolidin-3-yloxy]-phenyl}-8,9-dihydro-7H-benzocycloheptene-2-arboxylic acid methyl ester (I) (80 mg, 140.72 μηηοΙ) in MeOH (5 ml) was added a solution of NaOH (562.88 μΙ, 5 M) and the reaction mixture was heated at 60°C for 5 hours and the solvent removed under reduced pressure. The residue was taken up in water (10 ml) and aqueous HCI (5 M) added to pH 7. The slurry was extracted with DCM, dried over MgS04 and concentrated under reduced pressure. The solid was purified by column chromatography eluting with a mixture of DCM, acetonitrile and MeOH (90/5/5; V/V/V) to give 60 mg (77%) of 6-(2,4-dichlorophenyl)-5-[4-[(3S)-1 -(3-fluoropropyl)pyrrolidin-3-yl]oxyphenyl]-8,9-dihydro-7H-benzo[7]annulene-2-carboxylic acid. 1H NMR (400 MHz, DMSO-d6, δ ppm): 1 .68 (m, 1 H); 1 ,79 (dm, J=25.3 Hz, 2 H); 2.07 to 2.23 (m, 5 H); 2.38 (m, 1 H); 2.46 (t, J=7.2 Hz, 2 H); 2.52 (m, 1 H); 2.62 (m, 1 H); 2.55 to 2.89 (m, 3 H); 4.47 (td, J=6.2 and 47.6 Hz, 2 H); 4.72 (m, 1 H); 6.63 (d, J=8.9 Hz, 2 H); 6.71 (m, 3 H); 7.18 (d, J=8.4 Hz, 1 H); 8.26 (dd, J=2.0 and 8.4 Hz, 1 H); 7.58 (d, J=2,0 Hz, 1 H); 7.63 (d, J=8.4 Hz, 1 H); 7.79 (s, 1 H); 12.3 (m, 1 H)
LC/MS (m/z, MH+): 554
//////////////
Admin note for myself . I am up for Grabs
I myself Dr Anthony Melvin Crasto Looking for a post retirement assignment as Advisor API & INT, Chem. With 36 yrs rich experience, about dozen patents, 10000plus steps covered, 200 API targets, 30 plus products commercialization in plant in full career. Hands on knowledge of Synthesis, Process, scaleup, cost reduction, DOE , softwares etc
Kindly contact me Dr Anthony Melvin Crasto +919321316780 amcrasto@gmail.com
About myself Dr Anthony Crasto click on my website to know about me
1000 lakh google hits, 100lakh blog views, 10 lakh viewers in USA alone, all in 7 continents, 226 countries, 30 Indian and International awards, helping millions across the world
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Are you aware of any Chemical Database which offers one stop solution to the Sourcing, R&D and Business Development department? Explore Smartchem to Quickly find Suppliers (Procurement), Customers (BD) & Synthetic pathways (R&D)
Is this the information you looking for? Evaluate SmartChem, lets schedule a demo. Try us once. You will use us for life.
Mavorixafor (AMD-070) is a potent, selective and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively.
AMD-070 is a small molecule drug candidate that belongs to a new investigational class of anti-HIV drugs known as entry (fusion) inhibitors. Approximately 76% of HIV-patients with measurable viral load are infected with a strain of virus that is resistant to one or more classes of antiretroviral agents, thus reducing treatment options. Unlike many existing HIV drugs that target the virus after it has infected a healthy cell, AMD-070 blocks the virus from entering a healthy cell, thus preventing the replication process. AMD-070 targets the CXCR4 receptor on HIV and prevents the virus from entering and infecting healthy cells. AMD-070 is specific for the CXCR4 receptor and does not interact with any other chemokine receptors in vitro. AMD-070 strongly inhibits viral infection by all CXCR4 using virus (including virus using CXCR4 alone and/or virus using CXCR4 and CCR5) in vitro. AMD-070 is orally bioavailable in animals, it has suitable PK and toxicity profile for oral dosing. AMD-070 shows additive or synergistic effects in vitro in combination with other known anti-HIV agents. AMD-070 is active against CXCR4 using HIV strains that are resistant to existing antiretroviral therapies in vitro, reveals potent anti-HIV activity against CXCR4-using laboratory strains and clinical isolates. MD-070 had been in phase II clinical trials by Genzyme for the treatment of HIV infection. However, this research has been discontinued. AMD-070 has been studied in Phase I/II clinical trials for the treatment of Renal cell carcinoma and Phase I clinical trials for the treatment of malignant melanoma and solid tumours.
A novel and practical synthesis of mavorixafor (1) is reported. The novelty of this synthetic route is the use of 8-chloro-5,6,7,8-tetrahydroquinoline (9) and 1,4-diaminobutane as the materials, instead of 8-amino-5,6,7,8-tetrahydroquinoline (4) and N,N-diprotected aminobutyraldehyde (6a or 6b). The preparation of (S)-8-(4-aminobutylamino)-5,6,7,8-tetrahydroquinoline (13) by resolution with N-acetyl-l-leucine was first achieved. Then the one-pot synthesis of 1 from 13 involving protection, condensation, and subsequent hydrolysis was successfully developed. In addition, the final product with a satisfactory purity (>99.5%, detected by both achiral and chiral HPLC) was obtained by a simple operation (salification) without column chromatographic purification.
Charge diethyl-4-aminobutyl acetal (E) (1.00 wt, 1.00 eq) to vessel A. Charge acetonitrile (10.0 vol, 7.8 wt) and adjust temperature to 20°C. Heat the mixture to 40°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C.
[0098] Acetonitrile filler (5.0 vol, 3.9wt) at 35 to 45°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure 35 to 45°C. This step is repeated once as described below.
[0099] Acetonitrile filler (5.0 vol, 3.9wt) at 35 to 45°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C. Cool to 20°C.
[00100] Charge di-tert-butyl dicarbonate (1.1 eq, 1.5 wt) to a drum, followed by acetonitrile (0.4 vol, 0.3 wt) and agitate until fully dissolved. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C.
[00101] Charge this di-tert-butyl dicarbonate solution in acetonitrile to vessel A maintaining 20°C. Charge acetonitrile (1.5 vol, 1.1 wt) to the solution as a line rinse and stir at 20°C for 30 to 60 min..
[00102] Charge 4-dimethylaminopyridine (0.076 wt, 0.10 eq) to the vessel A at 20°C. Heat the solution to 40°C. Concentrate the reaction mixture to 5.0 vol under reduced pressure. Charge acetonitrile (5.0 vol, 3.9 wt) to the solution. Concentrate the reaction mixture to 5.0 vol under reduced pressure.
[00103] Take the resulting solution of Dl into next reaction without isolation.
Step IB: Preparation of Cl
[00104] Charge acetonitrile (2.0 vol, 1.6 wt) at 35 to 45°C to vessel A containing solution of D-1 from Step 1A.
[00105] Charge di-tert-butyl dicarbonate (1.4 eq, 1.9 wt) to a drum, followed by acetonitrile (10.0 vol, 7.8 wt) and agitate until fully dissolved. Charge this di-tert-butyl dicarbonate solution to vessel A, 2 to 6 h while distilling under vacuum at 35 to 45°C maintaining the volume of the reaction at 7.0 vol. Load acetonitrile (3.0 vol, 2.4 wt) over 20 to 40 min. as a line rinse while distilling under vacuum at 35 to 45°C, maintaining the volume of the reaction at 7.0 vol.
[00106] Charge di-tert-butyl dicarbonate, (0.14 eq, 0.19 wt) to a drum, followed by acetonitrile (1.0 vol, 0.74 wt) and agitate until fully dissolved. Charge this di-tert-butyl dicarbonate solution to vessel A over 20 to 40 min.. Charge acetonitrile (0.3 vol, 0.24 wt) over 10 to 20 min as a line rinse while distilling under vacuum at 35 to 45°C, maintaining the volume of the reaction at 7.0 vol.
[00107] Concentrate the reaction mixture to 5.0 vol distilling under vacuum at 35 to 45°C.
[00108] Charge n-heptane, (7.5 vol, 5.1 wt) to the reaction mixture, and concentrate the reaction mixture to 5.0 vol under reduced pressure at 40°C. This step is repeated once as described below.
[00109] Charge n-heptane, (7.5 vol, 5.1 wt) to the reaction mixture, and concentrate the reaction mixture to 5.0 vol under reduced pressure at 40°C.
[00110] Charge decolorizing, activated charcoal (0.2 wt) to the solution and stir for 1 to 2 h at 40°C. Filter the reaction mixture at 40°C. Charge n-heptane, (2.0 vol, 1.4 wt) to the reactor vessel and stir for 5 to 15 min. at 20°C before charging to the filter as a line rinse. Combine the filtrate and wash, and as required adjust to 20°C.
[00111] Take the resulting solution of Cl into next reaction without isolation.
Step 1C: Preparation of Bl
[00112] Charge 15% v/v acetic acid (2.0 vol) caution gas evolution, to vessel A containing solution of Cl from Step IB, maintaining the temperature at 20°C and stir for 10 min. at 20°C. Allow the phases to separate for 15 min. at 20°C. Discharge the aqueous phase to waste, retaining the organic phase in vessel A. This step is repeated once as described below.
[00113] Charge 15% v/v acetic acid (2.0 vol) maintaining 20°C and stir for 10 min. at 20°C. Allow the phases to separate for 15 min. at 20°C. Discharge the aqueous phase to waste, retaining the organic phase in vessel A.
[00114] Adjust the reaction to 30°C. Charge 4% w/w sodium chloride solution (2.1 vol) to the vessel maintaining the temperature at 30°C. Charge glacial acetic acid (4.1 vol, 4.3 wt) to the vessel maintaining 30°C. Stir the reaction mixture for 2 h maintaining the temperature at 30°C.
[00115] Charge purified water, (6.0 vol) at 30°C. Stir the contents for 5 to 10 min. at 30°C, and separate the phases, retaining the upper organic phase in vessel A. Charge the lower aqueous phase to vessel B.
[00116] Charge purified water (4.0 vol) at 30°C and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, retaining the upper organic phase in vessel A. Charge the lower aqueous phase to vessel B.
[00117] Adjust the temperature to 30°C of vessel B containing combined aqueous phases. Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min.. Charge the upper organic phase to vessel A and recharge the lower aqueous phase to vessel B. This step is repeated two additional times as described below.
[00118] Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min.. Charge the upper organic phase to vessel A and recharge the lower aqueous phase to vessel B.
[00119] Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min., discharge the lower aqueous phase to waste and charge the upper organic layer to vessel A.
[00120] Concentrate the combined organic phases in vessel A to 3.0 vol at 10 to 20°C under reduced pressure. Offload the solution to new HDPE drum(s) and line rinse with n-heptane (0.5
vol, 0.4 wt) at 20°C. Homogenize the drum and store as “Bl solution in n-heptane,” and take into next reaction without isolation.
Step ID: Preparation of F-2d
[00121] Calculate a new 1.00 wt based on the above assay.
[00122] Charge “Bl solution in n-heptane” from Step 1C (1.00 wt, 1.00 eq, corrected for w/w assay, ca. 3.0 vol), into an appropriate vessel. THF load (3.0 vol, 2.7 wt). Heat the reaction mixture to 40°C.
[00123] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C. This step was repeated four additional times to add the reagent in five portions total, as detailed below.
[00124] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00125] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00126] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00127] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 36 hours at 40°C.
[00128] Cool the reaction mixture to 20°C over 3 to 4 h at a target constant rate. Filter the reaction mixture at 20°C on a 1-2 pm cloth.
[00129] Wash the solid with a pre-mixed mixture of THF (0.5 vol, 0.5 wt) and n-heptane (0.5 vol, 0.3 wt) maintaining the temperature at 20°C. This step was repeated an additional three times, as detailed below.
[00130] Wash the solid with n-heptane, (2.0 vol, 1.4 wt) as a line rinse and apply to the filtercake at 20°C.
[00131] Wash the solid with n-heptane, (2.0 vol, 1.4 wt) as a line rinse and apply to the filtercake at 20°C.
[00132] Wash the solid with acetonitrile, (2.0 vol, 1.6 wt) as a line rinse and apply to the filtercake at 20°C.
[00133] Dry the solid at 38°C under a flow of nitrogen for 12 h.
[00137] Charge J, (1.00 wt, 1.00 eq) to vessel A. Charge purified water, (1.0 vol, 1.0 wt) to vessel A and as necessary adjust the temperature to 20°C. Charge concentrated hydrochloric acid, (4.0 eq, 3.0 vol, 3.6 wt) to vessel A maintaining the temperature at 20°C. Line rinse with purified water, (0.5 vol, 0.5 wt) maintaining the contents of vessel A at 15 to 25°C.
[00138] Charge chloroacetic acid, (1.3 wt, 1.5 eq) and purified water, (1.0 vol, 1.0 wt) to vessel B and as necessary, adjust the temperature to 20°C. Stir until fully dissolved, expected 10 to 20 min.
[00139] Charge the chloroacetic acid solution to vessel A maintaining the temperature of vessel A at 20°C. Line rinse vessel A with purified water, (0.5 vol, 0.5 wt) at 15 to 25°C and charge to vessel B at 20°C. Heat the reaction mixture to 80°C. Stir the reaction mixture at 80°C for 20 h.
[00140] Cool the reaction mixture to 10°C over 1.5 h. Load 47% w/w potassium phosphate solution (6.0 vol) over 60 min. targeting a constant rate maintaining 10°C. Adjust the pH of the reaction mixture by charging 47% w/w potassium phosphate solution to pH 7.0 maintaining the reaction temperature at 10°C. Expected charge is 2.0 to 3.5 vol 47% w/w potassium phosphate solution.
[00141] Stir the slurry for >30 min. maintaining 10°C and rechecking the pH, pass criterion pH 7.0. Filter the reaction mixture through 20 pm cloth at 10°C. Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C. This step is repeated additional three times as described below.
[00142] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00143] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00144] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00145] Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C. The filter-cake was washed with purified water additional five times as described below.
[00146] Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C.
[00147] Wash the filter-cake with acetonitrile, (2×1.3 vol, 2×1.0 wt) at 10°C.
[00148] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 20°C until the water content is <15.0% w/w by Karl-Fisher analysis.
[00149] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 30°C until the water content is <5.0% w/w by Karl-Fisher analysis.
[00150] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 50°C until the water content is <1.0% w/w by Karl-Fisher analysis.
[00151] Yield of compound Gl: about 75%.
Step 2B: Preparation of F-3a
Charge di-/c/7-butyl dicarbonate, (1.85 wt, 1.4 eq) to vessel A followed by N,N-dimethylformamide, (2.6 wt, 2.7 vol) and stir at 20°C for 20 min. until dissolution achieved. Add A,A-diisopropylethylamine, (0.08 wt, 0.11 vol, 0.1 eq) to contents of vessel A at 20°C. Heat the contents of vessel A to 40°C.
[00153] Charge Gl, (1.00 wt) to vessel B followed by YW-di methyl form am ide, (5.2 wt, 5.5 vol) and adjust to 14°C.
[00154] Charge the Gl/DMF solution from vessel B to vessel A over 5 h at 40°C, at an approximately constant rate. Line rinse with Y,Y-di methyl form am ide, (0.4 wt, 0.4 vol), maintaining vessel A at 40°C. Stir the resulting reaction mixture at 40°C for 16 h.
[00155] Charge decolorizing charcoal activated, (0.20 wt). Adjust the mixture to 40°C and stir at 40°C for 60 to 90 min..
[00156] Clarify (filter) the reaction mixture into vessel B at 40°C. Charge N,N-dimethylformamide, (0.9 wt, 1.0 vol) via vessel A and filter at 40°C. Charge purified water, (3.5 vol) to the combined filtrates, over 60 min., maintaining the temperature at 40°C. As required, cool the mixture to 35°C over 30 to 60 min..
[00157] Filler F-3a, (0.02 wt) as seed material at 35°C. Stir at 34°C for 1.5 h then check for crystallization. Cool slurry to 30°C over 40 min.
[00158] Filler F-3a, (0.02 wt) as seed material at 30°C. Stir at 30°C for 1.5 h then check for crystallization.
[00159] Cool slurry at 20°C over 3.5 h at a targeted constant rate. Stir at 20°C for 3 hours. Charge purified water, (1.0 vol), maintaining the temperature at 20°C over 60 min..
Stir at 20°C for 3 hours.
[00160] Cool slurry to 2°C over 2.5 h. Stir at 2°C for 2.5 hours. Filter through 20 pm cloth and pull dry until no further filtrate passes. Wash the solid with pre-mixed Y,Y-di methyl form am ide / purified water, (2.0 vol, 1:2 v:v) at 2°C. Wash the solid with purified water, (2 x 3.0 vol) at 2°C. Dry under vacuum at 28°C until KF <0.2% w/w, and Y,Y-di methyl form am ide <0.4% w/w.
[00161] Yield of compound F-3a: 62-70%.
Example 3: Synthesis of Mavorixafor:
Scheme VI:
nce
Step 3A: Preparation of imine Q-1
[00162] To vessel A charge purified water, (8.7 vol, 8.7 wt) followed by potassium phosphate, (5.52 eq, 5.3 wt) portion-wise and cool to 15°C. Charge tetrahydrofuran, (4.3 vol, 3.8 wt) and n-heptane, (2.2 vol, 1.5 wt) to vessel A and cool the biphasic mixture to 0°C. Charge Fl, (1.00 eq, 1.00 wt) to the vessel in 2 portions maintaining 0°C.
[00163] Charge F-2d, (1.10 eq, 1.95 wt) to the vessel in 4 portions maintaining 0°C, ensuring portions are spaced by 10 min.. Stir the resulting biphasic mixture for 1.5 h at 0°C. Allow the layers to separate for 45 min. at 0°C before separating the layers. Retain the upper organic phase within vessel A.
[00164] Take the resulting solution of Ql into next reaction without isolation.
Step 3B: Preparation of amine P-1
[00165] To vessel B, charge tetrahydrofuran, (6.0 vol, 5.3 wt) and adjust to 15°C. Charge zinc chloride, (1.5 eq, 0.92 wt) to vessel B in 4 portions, maintaining 10 to 30°C. Adjust the reaction mixture in vessel B to 15°C. Stir the mixture at 15°C for 1 hour. Charge sodium borohydride,(1.0 eq, 0.17 wt) to vessel B in 2 portions maintaining 15°C. Cool the reaction mixture in vessel B to 15°C. Stir the mixture for 1 hour maintaining 15°C. Cool the reaction mixture in vessel B to -5°C.
[00166] Cool the retained organic solution of Ql in vessel A, from Step 3A, to -5°C.
[00167] Charge the organic solution in vessel A into vessel B over 1 to 2 h maintaining -5°C. Charge tetrahydrofuran, (1.0 vol, 0.9 wt) to vessel A as a line rinse and adjust to -5°C. Transfer the contents of vessel A to vessel B maintaining -5°C.
[00168] Stir the resulting reaction mixture in vessel B for 1.5 h maintaining -5°C.
[00169] Charge purified water, (4.5 vol, 4.5 wt) and glacial acetic acid, (1.0 eq, 0.27 wt, 0.26 vol) to the cleaned vessel A and cool to 0°C. Charge the contents of vessel B to vessel A over 1 to 2 h maintaining 0°C. Charge tetrahydrofuran, (1.0 vol, 0.9 wt) to vessel B as a vessel rinse, cool to 0°C and transfer to vessel A maintaining 0°C.
[00170] Warm the resulting mixture in vessel A to 30°C. Stir the resulting mixture in vessel A at 30°C for 1 h. Allow the layers to settle for 15 min. at 30°C before separating the layers. Retain the upper organic phase.
[00171] Cool the retained organic phase to 15°C. Charge to the vessel 25% w/w ammonia solution (3.0 vol) at 10 to 30°C. Cool the reaction mixture to 20°C. Charge to the vessel 25% w/w ammonium chloride solution (3.0 vol) at 20°C and stir for 1 h. Separate the layers for 15 min. at 20°C, retain the upper organic phase. Wash the retained organic phase with 10% w/w sodium chloride solution (3.0 vol) at 20°C for 10 min.. Allow the layers to settle for 10 min. at 20°C before separating and retaining the upper organic phase within the vessel.
[00172] Charge tert-butyl methyl ether, (0.5 vol, 0.4 wt) to the organic phase. Cool the mixture to 5°C. Adjust the pH of the reaction mixture to pH 5 with hydrochloric acid aqueous solution (expected ca. 9.0 vol) over 1 h at a targeted constant rate at 5°C. Stir the mixture at 5°C for 45 min.. Measure the pH of the aqueous phase to confirm the value is pH 5.
[00173] Charge sodium chloride, (2.1 wt) to the reaction mixture at 5°C and stir the mixture until everything is dissolved. Adjust the temperature of the reaction mixture to 20°C. Separate the layers at 20°C and retain the organic phase within the vessel. Tetrahydrofuran charge, (1.5 vol, 1.3 wt) maintaining 20°C.
[00174] Charge to the vessel 24% w/w sodium chloride solution (7.5 vol) at 20°C and stir for 10 min.. Separate the layers at 20°C and retain the organic phase in the vessel. This step is repeated additional one more time as described below.
[00175] Charge to the vessel 24% w/w sodium chloride solution (7.5 vol) at 20°C and stir for 10 min.. Separate the layers at 20°C and retain the organic phase in the vessel.
[00176] Heat the retained organic phase to 35°C and concentrate the mixture to 6.0 vol under reduced pressure maintaining 35°C.
[00177] Tetrahydrofuran charge, (15.0 vol, 13.2 wt) maintaining 35°C. Concentrate the mixture to 6.0 vol under reduced pressure maintaining 35°C.
[00178] Tetrahydrofuran charge, (15.0 vol, 13.2 wt) maintaining 35°C. Concentrate the mixture to 11.0 vol under reduced pressure maintaining 35°C.
[00179] Cool the mixture to -5°C. Load tert-butyl methyl ether, (10.0 vol, 7.4 wt) over 1 h maintaining -5°C. Stir the mixture at -5°C for 1.5 hours. Filter the solid on 1 to 2 pm filter cloth at -5°C. Wash the solid with pre-mixed tetrahydrofuran, (1.9 vol, 1.7 wt) and tert-butyl methyl ether, (3.1 vol, 1.9 wt) at -5°C as a displacement wash.
[00180] Wash the solid with tert-butyl methyl ether, (5.0 vol, 3.7 wt) at -5°C.
[00181] Dry the solid on the filter under a flow of nitrogen at 23°C.
[00182] Yield of compound P-1: 76-87%.
Step 3C: Preparation of compound 0-1
[00183] Charge purified water, (2.0 vol, 2.0 wt) followed by potassium phosphate, (3.3 eq, 1.54 wt), carefully portion-wise, maintaining <15°C, to vessel A. Charge toluene, (4.5 vol, 3.9 wt) to the vessel maintaining <15°C. As necessary, adjust the temperature to 10°C.
[00184] Charge P-1, (1.00 eq, 1.00 wt) to the vessel in two portions maintaining 10°C. Stir the reaction mixture at 10°C for 15 min..
[00185] Load F-3a, (1.1 eq, 0.64 wt) in 4 equal portions ensuring portions are spaced by 10 min. at 10°C.
[00186] Tetrabutylammonium iodide (TBAI) filler (0.20 eq, 0.16 wt). Heat the reaction mixture to 40°C. Stir the reaction mixture at 40°C for 30 h.
[00187] Charge pre-mixed 2-mercaptoacetic acid, (0.40 eq, 0.08 wt, 0.06 vol), and toluene, (0.5 vol, 0.4 wt) over 20 min. to Vessel A at 40°C. Line rinse with toluene, (0.5 vol, 0.4 wt) at 40°C. Adjust the temperature of the reaction mixture to 50°C. Stir the mixture at 50°C for 2.5 hours.
[00188] Adjust the temperature of Vessel A to 20°C. Charge purified water, (3.0 vol, 3.0 wt) maintaining 20°C. Stir the reaction mixture at 20°C for 15 min. and transfer to a new, clean HDPE container. Line/vessel rinse with toluene, (0.5 vol, 0.4 wt) at 20°C. Clarify (filter) the reaction mixture via a 1 pm filter at 20°C into clean Vessel A. Wash the vessel and the filter with toluene, (0.5 vol, 0.4 wt) at 20°C. Allow the layers to separate for 15 min. at 20°C, retaining the upper organic layer (organic layer 1).
[00189] Wash the aqueous layer with toluene, (2.5 vol, 2.2 wt) at 20°C for 15 min.. Allow the layers to separate for 15 min. at 20°C. Retain the upper organic layer (organic layer 2).
[00190] Combine the organic layer 1 and organic layer 2 and adjust the temperature to 20°C. Wash the combined organic layers with 10% w/w sodium chloride solution (5.0 vol) at 20°C for 15 min.. Allow the layers to settle for 15 min. at 20°C. Retain the upper organic layer.
[00191] Take the resulting solution of Ol into next reaction without isolation.
Step 3D: Preparation of compound Kl
[00192] Charge n-butanol, (2.4 wt, 3.0 vol) to vessel B and adjust to 5°C. Charge concentrated sulfuric acid, (1.1 wt, 5.0 eq, 0.6 vol) slowly to Vessel B maintaining <15°C. Line rinse with toluene, (0.4 wt, 0.5 vol) maintaining <15°C. Adjust the temperature of Vessel B to 25°C.
[00193] Heat the n-butanol/ sulfuric acid solution in Vessel B to 55°C. Charge the organic layer from Vessel A (from Step 3C) to the butanol/ sulfuric acid solution in Vessel B over 60 to 90 min. maintaining 55°C. Charge toluene, (1.3 wt, 1.5 vol) to Vessel A as a line rinse and transfer to Vessel B maintaining 55°C. Stir the contents of Vessel B at 55°C for 1.5 h.
[00194] Stir the mixture in Vessel B for 4.5 h at 55°C. Cool the contents of Vessel B to 20°C over 10 h. Filter the slurry over 1-2 pm filter cloth under nitrogen at 20°C. Wash the filter cake with pre-mixed toluene, (3.5 wt, 4.0 vol) and n-butanol, (1.0 vol, 0.8 wt) at 20°C. Wash the filter cake with toluene, (4.3 wt, 5.0 vol) at 20°C. Dry the solid at 30°C under vacuum.
[00195] Correct the output weight for assay. Expected 50-55% w/w.
[00196] Yield of compound K1: 89-92%.
Step 3E: Preparation of Mavorixafor Drug Substance
[00197] Charge Kl, (1.00 eq, 1.00 wt, corrected for HPLC assay) in vessel A followed by nitrogen-purged purified water, (2.0 wt, 2.0 vol) and if necessary, adjust the temperature to 20°C. Charge nitrogen-purged toluene, (12.0 wt, 14.0 vol) to the solution maintaining 20°C. Charge nitrogen-purged n-butanol, (0.8 wt, 1.0 vol) to the solution maintaining 20°C. Heat the biphasic mixture to 30°C. Charge nitrogen-purged 3.0 M aqueous sodium hydroxide solution (6.2 eq, 5.9 vol) maintaining 30°C. Check the pH (expected 12 to 13). Adjust the pH of the aqueous layer to pH 10.0 with nitrogen-purged 0.3 M sulfuric acid solution (expected up to 2.5 vol) maintaining 30°C. Stir the mixture at 30°C for 45 min..
[00198] Measure the pH to confirm the value is pH 10.0.
[00199] Allow the layers to settle at 30°C for 30 min. and separate the layers retaining the organic phase in the vessel, and discharge the aqueous layer into a separate container (container C).
[00200] Charge pre-mixed toluene, (4.1 wt, 4.7 vol) and n-butanol, (0.24 wt, 0.3 vol) to a separate vessel; heat the contents to 30°C and charge the aqueous layer from container C. As required adjust the temperature to 30°C and stir for 5 to 10 min. at 30°C. Allow the phases to separate for 10 to 15 min. at 30°C. Discharge the aqueous phase to waste and combine the organic phase to the organic phase in vessel A.
[00201] Charge nitrogen-purged purified water, (2.0 wt, 2.0 vol) to the organic layer maintaining the temperature at 30°C and stir for 5 to 10 min. at 30°C. Allow the phases to separate for 10 to 15 min. at 30°C. Discharge the aqueous phase to waste retaining the organic phase in the vessel. Heat the retained organic solution to 40°C. Concentrate the resulting organic phase to 7.0 vol by vacuum distillation at 40°C.
[00202] Charge nitrogen -purged toluene, (13.0 wt, 15.0 vol) to the mixture and concentrate the solution 7.0 vol by vacuum distillation at 40°C. This step is repeated additional one time as described below.
[00203] Charge nitrogen -purged toluene, (13.0 wt, 15.0 vol) to the mixture and concentrate the solution 7.0 vol by vacuum distillation at 40°C.
[00204] Charge nitrogen-purged toluene, (7.0 wt, 8.0 vol) to the mixture at 40°C, heat to 55°C and clarify the hot reaction mixture under nitrogen via a 1 pm filter.
[00205] Charge clarified nitrogen-purged toluene, (1.7 wt, 2.0 vol) to the mixture as a line and vessel rinse at 40°C. Concentrate the solution to 7.0 vol by vacuum distillation at 40°C. At the end of the distillation the product is expected to have precipitated. Heat the mixture to 63°C.
[00206] Adjust the temperature to 60.5°C. This batch will be referred to as the main batch.
[00207] Load seed material, (0.02 wt) to a new clean container. Charge clarified nitrogen-purged toluene, (0.09 wt, 0.10 vol) to this seed material and gently shake.
[00208] Seed the main batch with the slurry maintaining the temperature at 60.5 ± 2°C. Stir the reaction at the 60.5± 2°C for 1 hour.
[00209] Cool to 40°C for 2.5 h. Stir the reaction at 40°C for 1 hour.
[00210] Cool to 30°C over 2 h.. Stir the reaction at 30°C for 1 h.
[00211] Cool to 25°C 50 min. Stir the reaction at 25°C over 2 hours.
[00212] Cool to 2°C over 4 h. Stir the mixture for 12 hours at 2°C.
[00213] Filter the mixture at 2°C over 1 to 2 pm cloth. Wash the filter cake with clarified nitrogen-purged toluene, (2.0 vol, 1.7 wt) at 2°C. Dry the filter cake under vacuum and a flow of nitrogen for 1.5 h.
[00214] Dry the solid at 40°C under vacuum and a flow of nitrogen until drying specification is achieved.
[00215] Yield of the final compound mavorixafor: 72%.
[00216] When toluene is used as the recrystallization solvent, optionally with a dissolution aid such butanol or methanol, for maxorixa for recrystallization, advantages were found compared to using dichloromethane and isopropyl acetate. We have found that these solvents do not react with the API, and accordingly we believe that this change has caused the significant reduction of impurities A (imine), B (N-formyl) and C (acetamide) that we have observed.
[00217] In some embodiments, the mavorixafor composition included 7000, 6000, 5000, 4500, 4450, 4000, 3500, 3000, 2500, 2000, 1750, 1700, 1650, 1600, 1550, 1500, 1450, 1400, 1400, 1400, 1400 gold 50 ppm of toluene or less. In some embodiments, the mavorixafor composition comprises a detectable amount of toluene. In some embodiments, the mavorixafor composition comprises from a detectable amount of toluene to 1350 ppm of toluene.
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Mavorixafor (AMD-070) is a potent, selective and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively.
125I-SDF-CXCR413 nM (IC50)HIV-1 (NL4.3 strain)1 nM (IC50, in MT-4 cells)HIV-1 (NL4.3 strain)9 nM (IC50, in PBMCs)HIV-1 (NL4.3 strain)3 nM (IC90, in MT-4 cells)HIV-1 (NL4.3 strain)26 nM (IC90, in PBMCs)
In Vitro
Mavorixafor (AMD-070) is a potent and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4 125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively. Mavorixafor (AMD-070) shows no effect on other chemokine receptors (CCR1, CCR2b, CCR4, CCR5, CXCR1, and CXCR2)[1]. Mavorixafor (AMD-070) (6.6 µM) significantly suppresses the anchorage-dependent growth, the migration and matrigel invasion of the B88-SDF-1 cells[2].MCE has not independently confirmed the accuracy of these methods. They are for reference only.
In Vivo
Mavorixafor (AMD-070) (2 mg/kg, p.o.) significantly reduces the number of metastatic lung nodules in mice, and lowers the expression of human Alu DNA in mice, without body weight loss[2].MCE has not independently confirmed the accuracy of these methods. They are for reference only.
ZAVZPRET is indicated for the acute treatment of migraine with or without aura in adults.
The recommended dose of ZAVZPRET is 10 mg given as a single spray in one nostril, as needed. The maximum dose that may be given in a 24-hour period is 10 mg (one spray). The safety of treating more than 8 migraines in a 30-day period has not been established, Nasal spray: 10 mg of zavegepant per device. Each unit-dose nasal spray device delivers a single spray containing 10 mg of zavegepant.
ZAVZPRET (zavegepant) nasal spray contains zavegepant hydrochloride, a calcitonin generelated peptide receptor antagonist. Zavegepant hydrochloride is described chemically as (R)-N- (3-(7-methyl-1H-indazol-5-yl)-1-(4-(1-methylpiperidin-4-yl) piperazin-1-yl)-1-oxopropan-2-yl)- 4-(2-oxo-1,2-dihydroquinolin-3-yl) piperidine-1-carboxamide hydrochloride and its structural formula is:
Its molecular formula is C36H46N8O3․HCl, representing a molecular weight of 675. 28 g/mol. Zavegepant free base has a molecular weight of 638.82 g/mol. Zavegepant hydrochloride is a white to off-white powder, freely soluble in water, and has pKa values of 4.8 and 8.8. Each unit-dose ZAVZPRET device for nasal administration delivers 10 mg of zavegepant (equivalent to 10.6 mg of zavegepant hydrochloride) in a buffered aqueous solution containing dextrose, hydrochloric acid, sodium hydroxide, and succinic acid in water for injection. The solution has a pH of 5.3 to 6.7.
Active ingredients in ZAVZPRET: zavegepant Inactive ingredients in ZAVZPRET: dextrose, hydrochloric acid, sodium hydroxide, and succinic acid in water for injection.
The most common adverse reactions include taste disorders, nausea, nasal discomfort, and vomiting.[1]
Zavegepant was approved for medical use in the United States in March 2023.[1][2][3]
Medical usesZavegepant is a Calcitonin Gene-related Peptide Receptor Antagonist. The mechanism of action of zavegepant is as a Calcitonin Gene-related Peptide Receptor Antagonist.
Zavegepant is indicated for the acute treatment of migraine with or without aura in adults.[1]
Zavegepant is an antagonist of the calcitonin gene-related peptide (CGRP) receptor currently in phase 3 trials in an intranasal formulation for the treatment of migraine. If FDA approved, it will join other previously-approved “-gepant” drugs [rimegepant] and [ubrogepant] as an additional treatment alternative for patients with migraine, particularly those for whom traditional triptan therapy has proven ineffective. On April 15th, 2020, a phase 2 clinical trial (NCT04346615: Safety and Efficacy Trial of Vazegepant Intranasal for Hospitalized Patients With COVID-19 Requiring Supplemental Oxygen) began to investigate the use of intranasally administered zavegepant to combat the acute respiratory distress syndrome (ARDS) sometimes seen in patients with COVID-19. Acute lung injury activates the release of CGRP, which plays a role in the development of ARDS – CGRP antagonists, then, may help to blunt the significant inflammation associated with COVID-19. The clinical trial is expected to complete in September 2020.
Zavegepant is a highly soluble small molecule calcitonin gene related peptide (CGRP) receptor antagonist, with potential analgesic and immunomodulating activities. Upon administration, zavegepant targets, binds to and inhibits the activity of CGRP receptors located on mast cells in the brain. This may inhibit neurogenic inflammation caused by trigeminal nerve release of CGRP. In addition, by blocking the CGRP receptors located in smooth muscle cells within vessel walls, zavegepant inhibits the pathologic dilation of intracranial arteries. Zavegepant, by blocking the CGRP receptors, also suppresses the transmission of pain by inhibiting the central relay of pain signals from the trigeminal nerve to the caudal trigeminal nucleus. Altogether, this may relieve migraine. As CGRP receptors induce the release of pro-inflammatory mediators, such as interleukin-6 (IL-6), from inflammatory cells, zavegepant may prevent an IL-6-mediated inflammatory response. Zavegepant may also inhibit the CGRP-mediated induction of eosinophil migration and the stimulation of beta-integrin-mediated T cell adhesion to fibronectin at the site of inflammation, and may abrogate the CGRP-mediated polarization of the T cell response towards the pro-inflammatory state characterized by Th17 and IL-17. This may improve lung inflammation and oxygenation, prevent edema, and further lung injury. CGRP, a 37 amino-acid peptide expressed in and released from a subset of polymodal primary sensory neurons of the trigeminal ganglion and nerve fibers projecting to the airways and by pulmonary neuroendocrine cells, plays an important role in pain transmission, inflammation, and neurogenic vasodilatation. It is released upon acute lung injury and upregulation of transient receptor potential (TRP) channels.
Azepino-indazoles as calcitonin gene-related peptide (CGRP) receptor antagonists
PMID: 33096162Publication Date: 2021-01-01Journal: Bioorganic & medicinal chemistry lettersDiscovery of (R)-N-(3-(7-methyl-1H-indazol-5-yl)-1-(4-(1-methylpiperidin-4-yl)-1-oxopropan-2-yl)-4-(2-oxo-1,2-dihydroquinolin-3-yl)piperidine-1-carboxamide (BMS-742413): a potent human CGRP antagonist with superior safety profile for the treatment of migraine through intranasal delivery PMID: 23632269Publication Date: 2013-06-01Journal: Bioorganic & medicinal chemistry letters
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Migraine is a chronic and debilitating disorder characterized by recurrent attacks lasting four to 72 hours with multiple symptoms, including typically one-sided, pulsating headaches of moderate to severe pain intensity that are associated with nausea or vomiting, and/or sensitivity to sound (phonophobia) and sensitivity to light (photophobia). Migraines are often preceded by transient neurological warning symptoms, known as auras, which typically involve visual disturbances such as flashing lights, but may also involve numbness or tingling in parts of the body. Migraine is both widespread and disabling. The Migraine Research Foundation ranks migraine as the world’s third most prevalent illness, and the Global Burden of Disease Study 2015 rates migraine as the seventh highest specific cause of disability worldwide. According to the Migraine Research Foundation, in the United States, approximately 36 million individuals suffer from migraine attacks. While most sufferers experience migraine attacks once or twice per month, more than 4 million people have chronic migraine, defined as experiencing at least 15 headache days per month, of which at least eight are migraine, for more than three months. Others have episodic migraine, which is characterized by experiencing less than 15 migraine days per month. People with episodic migraine may progress to chronic migraine over time. Migraine attacks can last four hours or up to three days. More than 90% of individuals suffering from migraine attacks are unable to work or function normally during a migraine attack, with many experiencing comorbid conditions such as depression, anxiety and insomnia. Also, those suffering from migraine often have accompanying nausea and have an aversion to consuming food or liquids during an attack.
CGRP (calcitonin gene-related peptide) is a 37 amino acid neuropeptide, which belongs to a family of peptides that includes calcitonin, adrenomedullin and amylin. In humans, two forms of CGRP (a-CGRP and 0-CGRP) exist and have similar activities. They vary by three amino acids and exhibit differential distribution. At least two CGRP receptor subtypes may also account for differential activities. The CGRP receptor is located within pain-signaling pathways, intracranial arteries and mast cells and its activation is thought to play a causal role in migraine pathophysiology. For example, research and clinical studies have shown: serum levels of CGRP are elevated during migraine attacks, infusion of intravenous CGRP produces persistent pain in migraine sufferers and non-migraine sufferers, and treatment with anti-migraine drugs normalizes CGRP activity.
Currently, clinicians use a number of pharmacologic agents for the acute treatment of migraine. A study published by the American Headache Society in 2015 concluded that the medications deemed effective for the acute treatment of migraine fell into the following classes: triptans, ergotamine derivatives, non-steroidal anti-inflammatory drugs (“NSAIDs”), opioids and combination medications. The current standard of care for the acute treatment of migraine is prescription of triptans, which are serotonin 5-HT IB/ID receptor agonists. Triptans have been developed and approved for the acute treatment of migraine over the past two decades. The initial introduction of triptans represented a shift toward drugs more selectively targeting the suspected pathophysiology of migraine. While triptans account for almost 80% of anti-migraine therapies prescribed at office visits by healthcare providers, issues such as an incomplete effect or headache recurrence remain important clinical limitations. In fact, only about 30% of patients from clinical trials are pain free at two hours after taking triptans. In addition, triptans are contraindicated in patients with cardiovascular disease, cerebrovascular disease, or significant risk factors for either because of potential systemic and cerebrovascular vasoconstriction from the 5-HT IB -mediated effects. Also, according to a January 2017 study published in the journal Headache, an estimated 2.6 million migraine sufferers in the United States have a cardiovascular event, condition or procedure that limits the potential of triptans as a treatment option.
Accordingly, there remains a significant unmet medical need for a novel migraine-specific medication that provides enhanced patient benefits compared to existing therapies.
Possible CGRP involvement in migraine has been the basis for the development and clinical testing of a number of compounds, including for example, advanced clinical candidates rimegepant (BHV-3000) and zavegepant (BHV-3500), which are developed by Biohaven Pharmaceutical Holding Company Ltd., New Haven, CT.
Zavegepant (also known as vazegepant) is a third generation, high affinity, selective and structurally unique small molecule CGRP receptor antagonist having the following formula I:
I
Zavegepant is described, for example, in WO 03/104236 published December 18, 2003 and US 8,481,546 issued July 9, 2013, which are incorporated herein in their entireties by reference.
While zavegepant is a highly soluble molecule, its bioavailability characteristics may render it challenging to prepare the drug in an oral dosage form. Enhancing the bioavailability of zavegepant and other CGRP inhibitors by different administration routes would therefore be desirable.
Calcitonin gene-related peptide (CGRP) is widely distributed in nociceptive pathways in human peripheral and central nervous system and its receptors are also expressed in pain pathways. While CGRP is involved in migraine pathophysiology, its role in non-headache pain has not been quite clear. There remains a need for new medicines to treat various pain disorders in patients in need thereof.
Scheme 1
Scheme 3
Scheme 4
tert-butyl 4-(2-methoxy-2-oxoethylidene)piperidine-l -carboxylate. Sodium hydride in mineral oil (60%, 7.92 g, 198.02 mmoles) was washed with hexanes then suspended in dimethylformamide (220 mL). The mixture was cooled to 0°C. Trimethyl phosphonoacetate (29.0 mL, 189.82 mmoles) was added dropwise to the stirred reaction mixture. After 20 min at 0°C, a solution of A-/c/7-butoxycarbonyl-4-pi peri done (30.41 g, 152.62 mmoles) in dimethylformamide (80 mL) was added to the mixture dropwise. The reaction was stirred at room temperature for 3 h and then diluted with diethyl ether (650 mL). The mixture was washed once with water and the aqueous layer was extracted once with diethyl ether. The combined organic layers were washed 4 times with water and the aqueous phase was discarded. The organic phase was washed with brine and dried over magnesium sulfate, filtered, and concentrated to dryness. The title compound was obtained as a white solid in 92% yield. 1 H- NMR (300 MHz, CDCh): 5 = 5.68 (s, 1 H), 3.66 (s, 3 H), 3.40-3.51 (m, 4 H), 2.90 (t, J= 5.49, 2 H), 2.25 (t, J= 5.49, 2 H), 1.44 (s, 9 H).
ed-butyl 4-(2-methoxy-2-oxoethyl)piperidine-l -carboxylate. A solution of tert-butyl 4- (2-methoxy-2-oxoethylidene)piperidine-l -carboxylate (35.71 g, 140 mmoles) in a mixture of 1 : 1 ethyl acetate/methanol (220 mL) was carefully treated with 50% wet 10% palladium on carbon (3.3 g). The reaction vessel was charged with 55 psi of hydrogen gas and the mixture was shaken on a Parr apparatus at room temperature for 16 h. The reaction mixture was then filtered to remove the catalyst and the filtrate concentrated in vacuo. The title compound was obtained as a clear colorless oil in 97% yield. ‘H-NMR (300 MHz, CDCh): 5 = 4.04 (d, J= 10.25, 2 H), 3.64 (s, 3 H), 2.68 (t, J= 12.44, 2 H), 2.21 (d, J= 6.95, 2 H), 1.98-1.77 (m, 1 H), 1.64 (d, J= 13.54, 2 H), 1.41 (s, 9 H), 1.25-0.99 (m, 2 H).
4-[2-Hydroxy-l-methoxycarbonyl-2-(2-nitro-phenyl)-ethyl]-piperidine-l-carboxylic acid tert-butyl ester. A A-diisopropylamine (4.40 mL, 31.3 mmoles) was dissolved in tetrahydrofuran (50 mL). The mixture was cooled to -78°C. Butyllithium (2.5 M in hexanes, 12.4 mL, 31 mmoles) was added dropwise to the stirred solution. After stirring at -78°C for 30 min, a solution of tert-butyl 4-(2-methoxy-2-oxoethyl)piperidine-l -carboxylate (6.65 g, 25.8 mmoles) in tetrahydrofuran (15 mL) was added dropwise to the mixture. Stirring was continued at -78°C for 1 h. A solution of 2-nitrobenzaldehyde (3.90 g, 25.8 mmoles) in tetrahydrofuran (20 mL) was then added to the mixture dropwise, and then stirring was continued at -78°C for a further 2.5 h. The reaction was quenched with cold aqueous ammonium chloride and then diluted with water. The mixture was extracted twice with ethyl acetate and the aqueous phase was discarded. The material was dried (magnesium sulfate) filtered, and concentrated to dryness. Silica gel chromatography afforded the desired product in 94% yield as light yellow foam. MS m/e (M- C4H8+H)+= 353.1.
4-(4-Hydroxy-2-oxo-l , 2, 3, 4-tetrahydro-quinolin-3-yl)-piperidine-l -carboxylic acid tertbutyl ester. In a 3 neck flask fitted with a nitrogen inlet, thermometer, and a mechanical stirrer, 4-[2-hydroxy-l -methoxy carbonyl-2-(2-nitro-phenyl)-ethyl]-piperidine-l -carboxylic acid tertbutyl ester (9.93 g, 24.3 mmoles) was dissolved in acetic acid (1.75 moles, 100 mL). Iron powder (8.90 g, 159 mmoles) was added to the vessel with stirring. The stirred mixture was slowly heated to 80°C for 30 min and then cooled to room temperature. It was then diluted with ethyl acetate and filtered through a pad of celite. Solids were washed with 20% methanol/ethyl acetate, and then with methanol. The filtrate was concentrated and the residue partitioned between ethyl acetate and aqueous sodium bicarbonate. The layers were separated. The resulting aqueous phase was extracted twice with ethyl acetate. The organic layers were combined. The mixture was washed twice with water and the aqueous phase was discarded. The material was dried (magnesium sulfate) filtered, and concentrated to dryness. Silica gel chromatography afforded the title compound as light yellow foam in 77% yield. MS m/e (M-H)’ = 345.1.
3-(Piperidin-4-yl)quinolin-2(lH) hydrochloride . A stirred solution of 4-(4-hydroxy-2- oxo-l,2,3,4-tetrahydro-quinolin-3-yl)-piperidine-l-carboxylic acid tert-butyl ester (5.60 g, 16.2 mmoles) in ethyl acetate (70 mL) was treated with HC1 in dioxane (4N, 40 mmoles, 10 mL). The mixture was stirred at room temperature for 45 min. More HC1 in dioxane (4N, 120 mmoles, 30 mL) was then added and stirring was continued at room temperature for 16 h. The resulting solid was collected by filtration and washed with ethyl acetate. It was then suspended in 5% water-isopropanol (100 mL) and the mixture was warmed to reflux and stirred for 20 min. The mixture was cooled to room temperature and stirred at room temperature for 16 h. The solid was collected by filtration, washed with isopropanol, and dried under high vacuum. The title compound was obtained as white solid in 75% yield. ‘H-NMR (DMSO-de) 5 11.85 (s, 1 H), 9.02 (bs, 1 H), 8.88 (bs, 1 H), 7.70 (t, J= 3.81 Hz, 2 H), 7.53 – 7.30 (d, J= 8.24 Hz, 1 H), 7.17 (t, J= 7.48 Hz, 2 H), 3.36 (d, J= 12.51 Hz, 2 H), 3.10 – 2.94 (m, 3 H), 2.01 (d, J= 13.43 Hz, 2 H), 1.87 – 1.73 (m, 2 H); MS m/e (M+H)+ = 229.0.
4-Iodo-2,6-dimethylbenzenamine hydrochloride . To a suspension of sodium bicarbonate (126 g, 1.5 moles) and 2,6-dimethylaniline (61.5 mL, 500 mmoles) in methanol (700 mL) was added iodine monochloride (1.0 M in dichloromethane, 550 mL, 550 mmoles) at room temperature over 1 h. After addition was complete, stirring was continued for 3 h. The reaction was filtered to remove excess sodium bicarbonate and the solvent removed in vacuo. The residue was re-dissolved in diethyl ether (1.5 L) and treated with hydrochloric acid (2M in ether, 375 mL, 750 mmoles). The resulting suspension was stored in the freezer (-15°C) overnight. The solid was filtered and washed with diethyl ether until it became colorless, to give 126.5 g (89%) as a grey-green powder. ‘H-NMR (DMSO-de) 5 2.33 (s, 6 H), 7.48 (s, 2 H), 9.05 (bs, 3 H); 13C-NMR (DMSO-de) 5 17.4, 91.5, 133.1, 131.2, 136.9.
Methyl 2 -(benzyloxy carbonyl) acrylate . To a flame dried three-neck round bottom flask, fitted with a mechanical stirrer, was added (S)-methyl 2-(benzyloxycarbonyl)-3- hydroxypropanoate (129 g, 509 mmoles), anhydrous dichloromethane (2 L), and methanesulfonyl chloride (49.3 mL, 636 mmoles). The mixture was cooled to -15°C, and treated with tri ethylamine (213 mL, 1527 mmoles), dropwise, to ensure the temperature of the reaction mixture did not exceed 0°C. The addition of the first equivalent of triethylamine was exothermic. After addition of tri ethylamine, the mixture was stirred at 0°C for 30 min. The cooling bath was removed and the mixture stirred at room temperature for 1.5 h. The reaction was quenched by addition of methanol (21 mL). The mixture was washed with 0.5% aqueous potassium bisulfate until the washings were pH 5, then saturated sodium bicarbonate, and brine, dried over sodium sulfate, and concentrated. Flash chromatography (silica gel, 1 :9 ethyl acetate/hexanes) gave I l l g (92%) as a viscous colorless oil, which crystallized upon standing. ’H-NMR (DMSO-de) 5 3.71 (s, 3 H), 5.10 (s, 2 H), 5.60 (s, 1 H), 5.76 (s, 1 H), 7.39-7.35 (m, 5 H), 8.96 (s, 1 H); 13C-NMR (DMSO-de) 5 52.3, 65.9, 127.8, 128.1, 128.3, 128.8, 133.3, 136.3, 153.5, 163.7.
(Z)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl) acrylate. A 2 L round bottom flask was charged 4-iodo-2,6-dimethylbenzenamine hydrochloride salt (55 g, 194 mmoles), methyl 2-(benzyloxycarbonyl)acrylate (59.2 g, 252 mmoles), tetrabutylammonium chloride (59.2 g, 213 mmoles), palladium (II) acetate (4.34 g, 19.4 mmoles), and tetrahydrofuran (1.2 L, degassed by a flow of nitrogen for 30 min). The mixture was stirred so that a suspension was formed and then degassed by a flow of nitrogen for 30 min. Triethylamine (110 mL, 789 mmoles) was added and the resulting mixture was heated at reflux for 3 h. After cooling to room temperature, the reaction mixture was filtered through a pad of celite, washed with tetrahydrofuran (2 x 100 mL), and concentrated. The residue was dissolved in di chloromethane, washed with water (3X) and brine (2X), dried over sodium sulfate, and concentrated. Flash chromatography (silica gel, using 1 :9 ethyl acetate/dichloromethane) gave a tan solid. The solid was recrystallized from warm methanol (210 mL) and water (100 mL). The mixture was held at room temperature overnight, then at 0°C for 2 h, and finally at -15°C for 2 h. The resulting solid was filtered, washed with ice cold 1 : 1 methanol/water, and dried under high vacuum overnight to give 44.7 g (65%) as a light tan solid which was a mixture of ZZE isomers (73 :27). ’H-NMR (DMSO-de) 5, 2.05 (s, 6 H), 3.61 (s, 0.8 H), 3.68 (s, 2.2 H), 5.00 (s, 0.54 H), 5.13 (s, 1.46 H), 5.24 (s, 2 H), 7.40-7.21 (m, 8 H), 8.51 (s, 0.27 H), 8.79 (s, 0.73 H); 13C-NMR (DMSO-de) 5 17.8, 51.7, 65.3, 119.4, 120.0, 120.3, 127.3, 127.7, 128.3, 130.9, 135.8, 137.2, 146.9, 154.7, 166.0.
(R)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl)propanoate. A flame- dried 2 L Parr hydrogenation bottle was charged with (Z)-methyl 3-(4-amino-3,5- dimethylphenyl)-2-(benzyloxycarbonyl)acrylate (84.5 g, 239 mmoles), di chloromethane (300 mL), and methanol (300 mL). The bottle was swirled so that a light brown suspension was formed. The mixture was degassed using a flow of nitrogen for 30 min. To this was quickly added (-)-l,2-bis((2A,5A)-2,5-diethylphospholano)-bezene(cyclooctadiene) rhodium (I) tetrafluoroborate ([(2A,5A)-Et-DuPhosRh]BF4) (2.11 g, 3.20 mmoles). The bottle was immediately attached to a Parr Hydrogenator. After 5 cycles of hydrogen (60 psi) and vacuum, the bottle was pressurized to 65 psi and the suspension was agitated at room temperature for 16 h. The reaction had become homogeneous. The reaction mixture was concentrated, and the resulting residue purified by flash chromatography (silica gel, 1 :9 ethyl acetate/dichloromethane) to give 82.9 g (98%). ‘H-NMR (DMSO-de) 5 2.04 (s, 6 H), 2.65 (dd, J= 13.4, 9.8 Hz, 1H), 2.82 (dd, J= 13.7, 5.2 Hz, 1 H), 3.62 (s, 3 H), 4.15-4.10 (m, 1H), 4.41 (s, 2 H), 5.00 (s, 2 H), 6.68 (s, 2 H), 7.37-7.28 (m, 5 H), 7.70 (d, J= 7.9 Hz, 1 H); 13C-NMR (DMSO-de) 5 17.7, 35.9, 51.7, 56.1, 65.3, 120.4, 124.0, 127.5, 127.7, 128.2, 128.3, 136.9, 142.6, 155.9, 172.5.
(R)-Methyl 2-(benzyloxycarbonyl)-3-(7-methyl-lH-indazol-5-yl)propanoate. (R)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl)propanoate (50.0 g, 140 mmoles) was weighed into a flame-dried 5 L three neck round bottom flask, followed by the addition of toluene (2.4 L) and glacial acetic acid (120 mL, 2.1 moles). The mixture was mechanically stirred to form a clear solution, and then potassium acetate (103 g, 1.05 moles) was added. To the resulting white suspension, z.w-amyl nitrite (20.7 mL, 154 mmoles) was added dropwise at room temperature, and the resulting mixture was stirred at room temperature for 16 h. Saturated sodium bicarbonate (I L) was added, followed by the careful addition of solid sodium bicarbonate to neutralize the acetic acid. The mixture was extracted with a mixture of di chloromethane (2 L) and brine (1.5 L). After separation, the aqueous layer was extracted with di chloromethane (500 mL). The combined organic layers were dried over anhydrous sodium sulfate and filtered. Solvents were removed to afford a tan solid, which was washed with hexanes (2 L) and toluene (150 mL). The solid was recrystallized from hot acetone (260 mL) and hexanes (700 mL). The slightly cloudy mixture was allowed to cool to room temperature slowly, then to 0°C for 1.5 h, and finally to -15°C for 1.5 h. The resulting solid was filtered and washed with ice-cold acetone/hexanes (1 : 1, 200 mL) to afford 39.1 g (76% yield). Analytical HPLC showed >98% UV purity. The enantiomeric excess (ee) was determined to be 99.8% (conditions: Chiralpak AD column, 4.6 x 250 mm, 10 pm; A = ethanol, B = 0.05% diethylamine/heptane; 85%B @1.0 mL/min. for 55 min. The retention times for R was 44.6 min and for S was 28.8 min). ‘H-NMR (DMSO-de) 5 2.48 (s, 3 H), 2.93 (dd, J= 13.4, 10.7 Hz, 1H), 3.10 (dd, J= 13.7, 4.9 Hz, 1H), 3.63 (s, 3H), 4.32-4.27 (m, 1 H), 4.97 (s, 2 H), 7.03 (s, 1 H), 7.24-7.22 (m, 2 H), 7.29 -7.27 (m, 3 H), 7.41 (s, 1 H), 7.83 (d, J= 8.2 Hz, 1H), 7.99 (s, 1H), 13.1 (s, 1 H); 13C-NMR (DMSO-de) 5 16.7, 36.5, 51.8, 56.0, 65.3, 117.6, 119.6, 122.7, 127.2, 127.4, 127.6, 128.2, 129.3, 133.4, 136.8, 139.2, 155.9, 172.4. Mass spec.: 368.16 (MH)+.
(R)-Methyl 2-amino-3-(7-methyl-lH-indazol-5-yl)propanoate. A Parr hydrogenation bottle was charged with (R)-methyl 2-(benzyloxycarbonyl)-3-(7-methyl-lH-indazol-5- yl)propanoate (11.0 g, 29.9 mmoles) and methanol (75 mL). The suspension was purged with nitrogen and treated with palladium (10% on charcoal, 700 mg). The bottle was shaken under hydrogen (15 psi) overnight. The mixture was filtered through a pad of celite to remove the catalyst. Concentration of the eluent gave 7.7 g (quant.) as an oil which was used without further purification. XH-NMR (CD3OD) 5 2.54 (s, 3 H), 2.98 (dd, J= 13.5, 7.0 Hz, 1 H), 3.09 (dd, J= 13.5, 5.9 Hz, 1 H), 3.68 (s, 3 H), 3.75 (dd, J= 7.0, 6.2 Hz, 1 H), 7.01 (s, 1 H), 7.39 (s, 1 H), 7.98 (s, 1 H). Mass spec.: 232.34 (M-H)’.
(R)-methyl 3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3- yl)piperidine-l-carboxamido)propanoate. To a solution of (R)-methyl 2-amino-3-(7-methyl-lH- indazol-5-yl)propanoate hydrochloride (7.26 g, 27.0 mmoles) in dimethylformamide (50 mL) at room temperature was added N, A’-disuccinimidyl carbonate (7.60 g, 29.7 mmoles) followed by triethylamine (11.29 mL, 81 mmoles). The resulting mixture was stirred for 30 min and treated with 3-(piperidin-4-yl)quinolin-2(lH)-one (6.77 g, 29.9 mmoles) in portions. The reaction was allowed to stir for 24 h. The mixture was concentrated, dissolved in ethyl acetate, and washed sequentially with water, brine, and 0.5 N HC1 (2X). The organic phase was dried over magnesium sulfate, filtered, and concentrated. The resulting residue was purified by flash chromatography (silica gel, 20: 1 ethyl acetate/methanol) to give 11.9 g (78%). 1 H-NMR (CD3OD) 5 13.0 (s, 1 H), 11.8 (s, 1 H), 7.98 (s, 1 H), 7.63 (d, J= 7.6 Hz, 1 H), 7.57 (s, 1 H), 7.45 – 7.41 (m, 2 H), 7.27 (d, J= 8.2Hz, 1 H), 7.16 (t, J= 7.9 Hz, 1 H), 7.03 (s, 1 H), 6.85 (d, J= 7.9 Hz, 1 H), 4.31 – 4.26 (m, 1 H), 4.10 – 4.08 (m, 2 H), 3.60 (s, 3 H), 3.07 – 3.01 (m, 2 H), 2.93 – 2.88 (m, 1 H), 2.77 – 2.67 (m, 2 H), 2.48 (s, 3 H), 1.78 – 1.72 (m, 2 H), 1.34 – 1.26 (m, 2 H). Mass spec.: 488.52 (MH)+.
(R)-3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3-yl)piperidine-l- carboxamido)propanoic acid. A solution of (R)-methyl 3-(7-methyl-lH-indazol-5-yl)-2-(4-(2- oxo-1, 2-dihydroquinolin-3-yl)piperidine-l-carboxamido)propanoate_(5.50 g, 11.3 mmoles) in tetrahydrofuran (50 mL) and methanol (10 mL) was cooled to 0°C. To this was added a cold (0°C) solution of lithium hydroxide monohydrate (0.95 g, 22.6 mmoles) in water (20 mL), dropwise over 15 min. The reaction was stirred at room temperature for additional 3 h. The mixture was concentrated to remove the organic solvents. The resulting residue was dissolved in a minimum amount of water, cooled to 0°C, and treated with cold (0°C) IN HC1 until pH 2 was attained. The resulting solid was collected by filtration, washed with cold water and ether, and then dried overnight under high vacuum to give 5.0 g (94%) as a white solid. ’H-NMR (DMSO- d6) 5 13.05 (bs, 1 H), 11.77 (s, 1 H), 7.98 (s, 1 H), 7.62 (d, J= 8.0 Hz, 1 H), 7.55 (s, 1 H), 7.44 (d, J= 8.2Hz, 1 H), 7.42 (s, 1 H), 7.27 (d, J= 8.2 Hz, 1 H), 7.16 (t, J= 7.6 Hz, 1 H), 7.05 (s, 1 H), 6.65 (d, J= 7.9 Hz, 1 H), 4.27 – 4.22 (m, 1 H), 4.10 – 4.07 (m, 2 H), 3.12 – 3.07 (m, 1 H), 3.03 – 2.99 (m, 1 H), 2.93 – 2.88 (m, 1 H), 2.77 – 2.66 (m, 2 H), 2.47 (s, 3 H), 1.77 – 1.74 (m, 2 H), 1.34 – 1.27 (m, 2 H). Mass spec.: 474.30 (MH)+.
(R)-N-(3-(7-methyl-lH-indazol-5-yl)-l-(4-(l-methylpiperidin-4-yl)piperazin-l-yl)-l- oxopropan-2-yl)-4-(2-oxo-l,2-dihydroquinolin-3-yl)piperidine-l-carboxamide (I). A flask was charged with (R)-3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3- yl)piperidine-l-carboxamido)propanoic acid (2.9 g, 6.11 mmoles), triethylamine (3.00 mL, 21.5 mmoles), l-(l-methylpiperidin-4-yl)piperazine (1.23 g, 6.72 mmoles), and dimethylformamide (10 mL). The resulting solution was treated with 2-(lH-benzotriazole-l-yl)-l, 1,3,3- tetramethyluronium tetrafluoroborate (2.26 g, 7.03 mmoles) in portions. The reaction was allowed to stir at room temperature overnight. The mixture was concentrated under vacuum to remove dimethylformamide. The crude product was dissolved in 7% methanol in di chloromethane and purified by flash chromatography using 7% methanol in di chloromethane containing 2% of aqueous ammonium hydroxide as eluent. The pure fractions were collected and solvent was removed under vacuum. The desired product was crystallized from hot acetone to give the compound having Formula I in 77% yield. Analytical HPLC showed 99.0 % UV purity at 230 nm. The enantiomeric excess (ee) was determined to be >99.9% (conditions: Chiralpak AD column, 4.6 x 250 mm, 10 pm; eluent: 70% (0.05% diethylamine)/heptane/30%ethanol; @1.0 mL/min. for 45 min. The retention times were 18.7 min for R and 28.1 min for S). ‘H-NMR (500 MHz, DMSO-de) 5 ppm 13.01 (s, 1 H), 11.76 (s, 1 H), 7.96 (s, 1 H), 7.62 (d, J= 7.10 Hz, 1 H), 7.60 (s, 1 H), 7.42 (m, 1 H), 7.36 (s, 1 H), 7.26 (d, J = 8.25 Hz, 1 H), 7.14 (m, 1 H), 7.00 (s, 1 H), 6.69 (d, J= 8.25 Hz, 1 H), 4.78 (q, J= 7.79 Hz, 1 H), 4.14 (d, J= 12.37 Hz, 2 H), 3.54 (dd, J= 9.16, 4.58 Hz, 1 H), 3.24 (m, 1 H), 3.11 (m, 1 H), 2.97 (m, 1 H), 2.89 (m, 2 H), 2.69 (m, 4 H), 2.32 (m, 1 H), 2.21 (m, 1 H), 2.07 (m, 4 H), 1.95 (t, J= 8.25 Hz, 1 H), 1.87 (m, J= 11.28, 11.28, 3.55, 3.44 Hz, 1 H), 1.76 (t, J= 12.03 Hz, 2 H), 1.68 (t, J= 11.11 Hz, 2 H), 1.53 (t, J= 8.25 Hz, 1 H), 1.32 (m, 4 H), 1.16 (m, 2 H); 13C-NMR (DMSO-de) 5 16.80, 27.30, 30.51, 30.51, 30.67, 35.50, 38.04, 41.74, 44.00, 44.16, 45.35, 45.78, 48.14, 48.39, 51.45, 54.76, 54.76, 60.61, 114.53, 117.79, 119.29, 119.34, 121.57, 122.78, 127.46, 127.79, 129.29, 129.79, 133.31, 133.72, 136.98, 137.41, 139.12, 156.50, 161.50, 170.42.
Accurate mass analysis: m/z 639.3770, [MH]+, A = -0.2 ppm. Optical rotation: -27.36° @ 589 nm, concentration = 4.71 mg/mL in methanol. DESCRIPTION AND DOSAGE FORM
The physical and chemical properties of zavegepant (BHV-3500) drug substance mono-hydrochloride salt form are provided in Table 1.
To reduce the volume of new heterotopic ossification in adults and pediatric patients (aged 8 years and older for females and 10 years and older for males) with fibrodysplasia ossificans progressiva
It was approved for medical use in Canada in June 2022,[4] and in the United States in August 2023.[5]
Medical uses
Palovarotene is indicated for the treatment of heterotopic ossification and fibrodysplasia ossificans progressiva.[4][5]
History
Palovarotene is a retinoic acid receptor gamma (RARγ) agonist licensed to Clementia Pharmaceuticals from Roche Pharmaceuticals. At Roche, palovarotene was evaluated in more than 800 individuals including healthy volunteers and patients with chronic obstructive pulmonary disease (COPD).[7] A one-year trial did not demonstrate a significant benefit on lung density in moderate-to-severe emphysema secondary to severe α(1)-antitrypsin deficiency.[8]
In 2011, animal studies demonstrated that RARγ agonists, including palovarotene, blocked new bone formation in both an injury-induced mouse model of heterotopic ossification (HO) and a genetically modified biological mouse model of fibrodysplasia ossificans progressiva containing a continuously active ACVR1/ALK2 receptor in a dose-dependent manner.[9][10] A 2016 study demonstrated that palovarotene also inhibited spontaneous heterotopic ossification, maintained limb mobility and functioning, and restored skeletal growth in fibrodysplasia ossificans progressiva mouse models.[11]
Society and culture
Legal status
Palovarotene is being developed by Ipsen Biopharmaceuticals and was granted priority review and orphan drug designations by the United States Food and Drug Administration (FDA) for the treatment of fibrodysplasia ossificans progressiva[12][13] and orphan medicinal product designation by the European Medicines Agency (EMA) in 2014.[14][15][16][17]Phase II clinical studies failed to show a significant change in heterotopic bone volume, the main outcome measure, but prompted further investigation in a phase III clinical trial.[18] In December 2022, the FDA declined to approve palovarotene for the fibrodysplasia ossificans progressive without additional clinical trial data.[19] In January 2023, the European Medicines Agency (EMA) recommended the refusal of the marketing authorization for palovarotene for the treatment of fibrodysplasia ossificans progressiva.[20]
Research
Phase II
Clementia submitted a new drug application for palovarotene for the treatment of fibrodysplasia ossificans progressiva after observing positive phase II results.[21]
Phase III
In December 2019, Ipsen issued a partial clinical hold for people under the age of 14, due to reports of early fusion of growth plates.[22] Ipsen acquired Clementia in 2019.[23]
SYN
Desjardins, C., Grogan, D. R., Packman, J. N., & Harnett, M. (2017). Methods for treating heterotopic ossification (WO2017210792A1). World Intellectual Property Organization. https://patents.google.com/patent/WO2017210792A1
Chemical Communications (Cambridge, United Kingdom) (2019), 55(38), 5420-5422
XAMPLE 12: PREPARATION OF 4-r(E)-2-(5,5.8.8-TETRAMETHYL-3-PYRAZOL-l-YLMETHYL -5.6.7.8-TETRAHYDRO-NAPHTHALEN-2-YL VINYLl BENZOIC ACID (6)
A mixture of 2.0 g (4.5 mmol) of (E)- methyl-4-[2-(3-bromomethyl-5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoate and 0.65 g (9.5 mmol) of pyrazole in 15 mL of N-methyl pyrrolidine was heated at 100°. After 2 hours, the reaction mixture was cooled to room temperature, poured into brine and extracted with ethyl acetate. The organic extracts were washed with brine, dried over sodium sulfate and concentrated under reduced pressure. The residue was stirred with hexane and the product was filtered off, washed with hexane and dried to give 1.6 g (83%) of methyl-4-[2-(5,5,8,8-Tetramethyl-3-pyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoate (M+ = 429). A mixture of 27.6 g (64.4 mmol) of methyl-4-[2-(5,5,8,8-tetramethyl-3-pyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoate and 97 mL (193 mmol) of 2 N sodium hydroxide in 300 mL of ethyl alcohol was heated at reflux. After 1 hour, the reaction mixture was cooled to room temperature and diluted with 900 mL of water. The reaction mixture was acidified with 2 N HCl and the product was isolated by filtration, washed with water and pentane and dried to give 25.9 g (97%) of 4-[(E)-2-(5,5,8,8-tetramethyl-3-pyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid (m.p. = 246.5-248 °C) 6. Proceeding as described in the example above but substituting pyrazole with pyrrole, 4-methylpyrazole, 1,2,4-triazole, moφholine, 2-pyrrohdone, 3,5-dimethylpyrzole, δ – valerolactone, 2-methyhmidazole and 4-methylimidzole gave 4-[(E)-2-(5,5,8,8-tetramethyl-3-pyrrol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 7, 4-{(E)-2-[5,5,8,8-Tetramemyl-3-(4-methylpyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 20, 4-[(E)-2-(5,5,8,8-Tetxamethyl-3-[l,2,4]triazol-l-ylmethyl-5,6,7,8Jetrahydro-naphthalen-2-yl]vinyl}benzoic acid 39, 4-[(E)-2-(5,5,8,8-tetramethyl-3-moφhohn-4-ylmethyl- 5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 138, 4-[(E)-2-(5,5,8,8-tetramethyl-3- (2-oxo-pyrrohdin-l-yl-methyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 139, 4-{(E)-2-[5,5,8,8-Tetramet yl-3-(3,5-mmemylpyτazol-l-yhnethyl-5,6,7,8-tetrahydro-napn^ 2-yl)vinyl]benzoic acid 143, 4-[(E)-2-(5,5,8,8-tetramethyl-3-(2-oxo-piperidin-l-yl-methyl-5,6,7,8-tetrahydro-naρhthalen-2-yl)vinyl]benzoic acid 146 4-{(E)-2-[5,5,8,8-Tetramethyl-3-(2-methyhmidazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 149and 4-{(E)-2-[5,5,8,8-Tetramethyl-3-(4-methyhmidazol-l-ylmethyl-5,6,7,8-tettahydro-naphthalen-2-yl)vinyl]benzoic acid 150 respectively.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
^ Hind M, Stinchcombe S (November 2009). “Palovarotene, a novel retinoic acid receptor gamma agonist for the treatment of emphysema”. Current Opinion in Investigational Drugs. 10 (11): 1243–50. PMID19876792.
Clinical trial number NCT03312634 for “An Efficacy and Safety Study of Palovarotene for the Treatment of Fibrodysplasia Ossificans Progressiva. (MOVE)” at ClinicalTrials.gov
Hepatitis B virus (HBV) is an infectious disease that targets the liver resulting in either an acute infection, with symptoms arising in 45 to 160 days, or a chronic infection, which 350 million people worldwide are affected by. Estimates indicate that 600,000 deaths occur each year as a result of consequences related to HBV infection. HBV possesses a 3.2- kb relaxed circular DNA (rcDNA) genome that is used to form covalently closed circular DNA (cccDNA) in a host cell. The cccDNA is then transcribed by RNA polymerase II, a host DNA-dependent RNA polymerase, to produce pregenomic RNA (pgRNA). The pgRNA is then used by the virally encoded reverse transcriptase to form rcDNA. The goals of current treatments for chronic HBV infections are to reduce HBV replication and reduce liver damage.
Current treatments for chronic HBV infections include pegylated alpha interferon and nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs). NRTIs are converted to their corresponding 5 ‘-triphosphate, or diphosphate in the case of phosphonate containing NRTIs, and reduce viral replication by inhibiting the HBV encoded polymerase. Clevudine is an NRTI that is no longer being developed for the treatment of chronic HBV because of drug-related skeletal myopathy that was a result of mitochondrial dysfunction in patients.
Interestingly, clevudine triphosphate has been shown to be a competitive nonsubstrate inhibitor of the HBV encoded polymerase, and due to its long intracellular half-life, is able to suppress HBV replication for an extended period of time after drug withdrawal.
The discovery and synthesis of the (S,S) and (S,R) diastereomers of clevudine phosphoramidate has been previously reported. These studies were undertaken to address the myopathy concerns associated with clevudine. The phosphoramidate moiety was utilized to deliver clevudine, as its 5 ‘-monophosphate, to the liver reducing 1) systemic exposure to clevudine and 2) the possibility of skeletal myopathy. Both phosphoramidates showed anti-HBV activity similar to clevudine with the (S,S) diastereomer being slightly more potent.
[0374] Preparation of intermediate ATI-2173 from Compound-10.
[0375] Experimental Procedure
[0376] (H-l) Crude Compound-10 in THF (see Example 10) was divided into three aliquots and stirred with 2% HC1 aq. solution at pH 5-6, 4-5, and 3-4; for 16 h at 20-25 °C; the aliquots were combined and stirred at 15-20 °C for 72 h, with no degradation of ATI-2173 observed over the second time period;
[0377] (H-2) the pH of the mixture was adjusted to 7 with 7% NaHCCh;
[0378] (H-3) phase separation was carried out using 2-MeTHF, the organic phase was washed with NA2SO4 aqueous soltion, then concentrated to 1-3 V, and MTBE (5 V) was added; this operation was repeated twice;
[0379] (H-5) ATI-2173 was precipitated gradually upon addition of seed crystal and addition of n-heptane (5 V);
[0380] (H-6) the product was filtered; and
[0381] (H-7) the wetcake was dried, resulting in ATI-2173 in 99.73a% purity and
84.4% yield.
[0382] EXAMPLE 12:
[0383] Crystallization of ATI-2173
[0384] Initial studies examined the use of single or mixed solvent systems to crystalize the amorphous product, ATI-2173. Several solvent conditions were screened, including single solvent and mixed solvent systems, in order to determine the potential for obtaining a crystalline material from the amorphous material. None of the solvents tested worked and all conditions produced an oil product. The results are shown below in Tables 8 and 9.
[0385] Table 8: Single Solvent Systems
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Zuranolone was approved for medical use in the United States for the treatment of postpartum depression in August 2023.[2] It was developed by Sage Therapeutics and Biogen.[9]
The most common side effects include drowsiness, dizziness, diarrhea, fatigue, nasopharyngitis (cold-like symptoms), and urinary tract infection.[2]
The US FDA label contains a boxed warning noting that zuranolone can impact a person’s ability to drive and perform other potentially hazardous activities.[2] Use of zuranolone may cause suicidal thoughts and behavior.[2] Zuranolone may cause fetal harm.[2]
History
Zuranolone was developed as an improvement on the intravenously administered neurosteroid brexanolone, with high oral bioavailability and a biological half-life suitable for once-daily administration.[7][10] Its half-life is around 16 to 23hours, compared to approximately 9hours for brexanolone.[4][5]
The efficacy of zuranolone for the treatment of postpartum depression in adults was demonstrated in two randomized, double-blind, placebo-controlled, multicenter studies.[2] The trial participants were women with postpartum depression who met the Diagnostic and Statistical Manual of Mental Disorders criteria for a major depressive episode and whose symptoms began in the third trimester or within four weeks of delivery.[2] In study 1, participants received 50 mg of zuranolone or placebo once daily in the evening for 14 days.[2] In study 2, participants received another zuranolone product that was approximately equal to 40 mg of zuranolone or placebo, also for 14 days.[2] Participants in both studies were monitored for at least four weeks after the 14-day treatment.[2] The primary endpoint of both studies was the change in depressive symptoms using the total score from the 17-item Hamilton depression rating scale (HAMD-17), measured at day 15.[2] Participants in the zuranolone groups showed significantly more improvement in their symptoms compared to those in the placebo groups.[2] The treatment effect was maintained at day 42—four weeks after the last dose of zuranolone.[2]
Zuranolone was approved by the US Food and Drug Administration (FDA) for the treatment of postpartum depression in August 2023.[2][12] The FDA granted the application for zuranolone priority review and fast track designations.[2] Approval of Zurzuvae was granted to Sage Therapeutics, Inc.[2]
Zuranolone has also been under development for the treatment of major depressive disorder, but the application for this use was given a Complete Response Letter (CRL) by the FDA due to insufficient evidence of effectiveness.[13]
Research
In a randomized, placebo-controlled phase III trial to assess its efficacy and safety for the treatment of major depressive disorder, subjects in the zuranolone group (50 mg oral zuranolone once daily for 14 days) experienced statistically significant and sustained improvements in depressive symptoms (as measured by HAM-D score) throughout the treatment and follow-up periods of the study.[14]
Example 1. Synthesis of 1-(2-((3R,5R,8R,9R,10S,13S,14S,17S)-3-hydroxy-3,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethyl)-1H-pyrazole-4-carbonitrile (Compound 1).
[00488] To a suspension of K2CO3 (50 mg, 0.36 mmol) in THF (5 mL) was added 1H-pyrazole-4-carbonitrile (100 mg, 0.97 mmol) and 2-bromo-1-((3R,5R,8R,9R,10S,13S,14S,17S)-3-hydroxy-3,13-dimethylhexadecahydro-1H-cyclopenta[ ^]phenanthren-17-yl)ethan-1-one (50 mg, 0.12 mmol). The mixture was stirred at room temperature for 15 hours. The reaction mixture was poured into 5 mL H2O and extracted with ethyl acetate (2×10 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue mixture was purified by reverse-phase preparative HPLC to afford Compound 1 as a white solid (9 mg, 17.4% yield).1H NMR (500 MHZ, CDCl3) δ (ppm) 7.87 (1H, s), 7.82 (1H, s), 5.02 (1H, AB), 4.2 (1H, AB), 2.61 (1H, t), 2.16-2.24 (1H, m), 2.05 (1H, dxt), 1.70-1.88 (6H, m), 1.61-1.69 (2H, m), 1.38-1.52 (6H, m), 1.23-1.38 (5H, m), 1.28 (3H, s), 1.06-1.17 (3H, m), 0.67 (3H, s). LCMS: rt=2.24 min, m/z=410.1 [M+H]+.
PAPER
Journal of Medicinal Chemistry (2017), 60(18), 7810-7819
Certain classes of neuroactive steroids (NASs) are positive allosteric modulators (PAM) of synaptic and extrasynaptic GABAA receptors. Herein, we report new SAR insights in a series of 5β-nor-19-pregnan-20-one analogues bearing substituted pyrazoles and triazoles at C-21, culminating in the discovery of 3α-hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5β-pregnan-20-one (SAGE-217, 3), a potent GABAA receptor modulator at both synaptic and extrasynaptic receptor subtypes, with excellent oral DMPK properties. Compound 3 has completed a phase 1 single ascending dose (SAD) and multiple ascending dose (MAD) clinical trial and is currently being studied in parallel phase 2 clinical trials for the treatment of postpartum depression (PPD), major depressive disorder (MDD), and essential tremor (ET).
The compounds of the invention can be prepared in accordance with methods described in the art (Upasmi et al., J. Med. Chem. 1997, 40:73-84; and Hogenkamp et al., J. Med. Chem. 1997, 40:61- 72) and using the appropriate reagents, starting materials, and purification methods known to those skilled in the art. In some embodiments, compounds described herein can be prepared using methods shown in general Schemes 1-4, comprising a nucleophilic substitution of 19-nor pregnane bromide with a neucleophile. In certain embodiments, the nucleophile reacts with the 19-nor pregnane bromide in the presence of K2CO3 in THF.
Synthesis of compound SA-B. Compound SA (50 g, 184 mmol) and palladium black (2.5 g) in tetrahydrofuran (300 mL) and concentrated hydrobromic acid (1.0 mL) was hydrogenated with 10 atm hydrogen. After stirring at room temperature for 24h, the mixture was filtered through a pad of celite and the filtrate was concentrated in vacuo to afford the crude compound. Recrystallization from acetone gave compound SA-B (42.0 g, yield: 83.4%) as white powder.
Synthesis of compound SA-C. A solution of SA-B (42.0 g, 153.06 mmol) in 600 mL anhydrous toluene was added dropwise to the methyl aluminum bis(2,6-di-tert-butyl-4-methylphenoxide (MAD) (459.19 mmol, 3.0 eq, freshly prepared) solution under N2 at -78°C. After the addition was completed, the reaction mixture was stirred for 1 hr at -78°C. Then 3.0 M MeMgBr (153.06 mL, 459.19 mmol) was slowly added dropwise to the above mixture under N2 at -78°C. Then the reaction mixture was stirred for 3 hr at this temperature. TLC (Petroleum ether/ethyl acetate = 3:1) showed the reaction was completed. Then saturated aqueous NH4Cl was slowly added dropwise
to the above mixture at -78°C. After the addition was completed, the mixture was filtered, the filter cake was washed with EtOAc, the organic layer was washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated, purified by flash Chromatography on silica gel (Petroleum ether/ ethyl acetate20:1 to 3:1) to afford compound SA-C (40.2 g, yield: 90.4%) as white powder. 1H NMR: (400 MHz, CDCl3) δ 2.47-2.41 (m, 1H), 2.13-2.03 (m, 1H), 1.96-1.74 (m, 6H), 1.70-1.62 (m, 1H), 1.54-1.47 (m, 3H), 1.45-1.37 (m, 4H), 1.35-1.23 (m, 8H), 1.22-1.10 (m, 2H), 1.10-1.01 (m, 1H), 0.87 (s, 3H).
Synthesis of compound SA-D. To a solution of PPh3EtBr (204.52 g, 550.89 mmol) in THF (500 mL) was added a solution of t-BuOK (61.82 g, 550.89 mmol) in THF (300 mL) at 0°C. After the addition was completed, the reaction mixture was stirred for 1 h 60 °C, then SA-C (40.0 g, 137.72 mmol) dissolved in THF (300 mL) was added dropwise at 60°C. The reaction mixture was heated to 60 °C for 18 h. The reaction mixture was cooled to room temperature and quenched with Sat. NH4Cl, extracted with EtOAc (3*500 mL). The combined organic layers were washed with brine, dried and concentrated to give the crude product, which was purified by a flash column chromatography (Petroleum ether/ ethyl acetate50:1 to 10:1) to afford compound SA-D (38.4 g, yield:92%) as a white powder. 1H NMR: (400 MHz, CDCl3) δ 5.17-5.06 (m, 1H), 2.42-2.30 (m, 1H), 2.27-2.13 (m, 2H), 1.89-1.80 (m, 3H), 1.76-1.61 (m, 6H), 1.55-1.43 (m, 4H), 1.42-1.34 (m, 3H), 1.33-1.26 (m, 6H), 1.22-1.05 (m, 5H), 0.87 (s, 3H).
Synthesis of compound SA-E. To a solution of SA-D (38.0 g, 125.62 mmol) in dry THF (800 mL) was added dropwise a solution of BH3.Me2S (126 mL, 1.26 mol) under ice-bath. After the addition was completed, the reaction mixture was stirred for 3 h at room temperature (14-20 °C). TLC (Petroleum ether/ ethyl acetate3:1) showed the reaction was completed. The mixture was cooled to 0 °C and 3.0 M aqueous NaOH solution (400 mL) followed by 30% aqueous H2O2 (30%, 300 mL) was added. The mixture was stirred for 2 h at room temperature (14-20 °C), and then filtered, extracted with EtOAc (3*500 mL). The combined organic layers were washed with saturated aqueous Na2S2O3, brine, dried over Na2SO4 and concentrated in vacuum to give the crude product (43 g , crude) as colorless oil. The crude product was used in the next step without further purification.
Synthesis of compound SA-F. To a solution of SA-E (43.0 g, 134.16 mmol) in dichloromethane (800 mL) at 0 °C and PCC (53.8 g, 268.32 mmol) was added portion wise. Then the reaction mixture was stirred at room temperature (16-22 °C) for 3 h. TLC (Petroleum ether/ ethyl acetate3:1) showed the reaction was completed, then the reaction mixture was filtered, washed with DCM. The organic phase was washed with saturated aqueous Na2S2O3, brine, dried over Na2SO4 and concentrated in vacuum to give the crude product. The crude product was purified by a flash column chromatography (Petroleum ether/ ethyl acetate50:1 to 8:1) to afford compound SA-F (25.0 g, yield:62.5%, over two steps) as a white powder. 1H NMR (SA-F): (400 MHz, CDCl3) δ 2.57-2.50 (m, 1H), 2.19-2.11 (m, 4H), 2.03-1.97 (m, 1H), 1.89-1.80 (m, 3H), 1.76-1.58 (m, 5H), 1.47-1.42 (m, 3H), 1.35-1.19 (m, 10H), 1.13-1.04 (m, 3H), 0.88-0.84 (m, 1H), 0.61 (s, 3H).
Synthesis of compound SA. To a solution of SA-F (10 g, 31.4 mmol) and aq. HBr (5 drops, 48% in water) in 200 mL of MeOH was added dropwise bromine (5.52 g, 34.54 mmol). The reaction mixture was stirred at 17 °C for 1.5 h. The resulting solution was quenched with saturated aqueous NaHCO3 at 0°C and extracted with EtOAc (150 mLx2). The combined organic layers were dried and concentrated. The residue was purified by column chromatography on silica gel eluted with (PE: EA=15:1 to 6:1) to afford compound SA (9.5 g, yield: 76.14%) as a white solid. LC/MS: rt 5.4 mm ; m/z 379.0, 381.1, 396.1.
To a suspension of K2CO3 (50 mg, 0.36mmol) in THF (5 mL) was added ethyl 1H-pyrazole-4-carbonitrile (100 mg, 0.97 mmol ) and SA (50 mg,0.12 mmol). The mixture was stirred at rt for 15h. The reaction mixture was poured in to 5 mL H2O and extracted with EtOAc (2 x 10 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue mixture was purified with by reverse-phase prep-HPLC to afford the title compound as a white solid (9mg, 17.4%). 1H NMR (500 MHz, CDCl3), δ (ppm) 7.87 (1H, s),
7.82 (1H, s), 5.02 (1H, AB), 4.92 (1H, AB), 2.61 (1H, t), 2.16-2.24 (1H, m), 2.05 (1H, dXt), 1.70-1.88 (6H, m), 1.61-1.69 (2H, m), 1.38-1.52 (6H, m), 1.23-1.38 (5H, m), 1.28 (3H, s), 1.06-1.17 (3H, m), 0.67 (3H, s). LCMS: rt = 2.24 mm, m/z = 410.1 [M+H]+.
PATENT
WO2020150210
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
^ Jump up to:abBlanco MJ, La D, Coughlin Q, Newman CA, Griffin AM, Harrison BL, et al. (2018). “Breakthroughs in neuroactive steroid drug discovery”. Bioorganic & Medicinal Chemistry Letters. 28 (2): 61–70. doi:10.1016/j.bmcl.2017.11.043. PMID29223589.
^Martinez Botella G, Salituro FG, Harrison BL, Beresis RT, Bai Z, Blanco MJ, et al. (2017). “Neuroactive Steroids. 2. 3α-Hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5β-pregnan-20-one (SAGE-217): A Clinical Next Generation Neuroactive Steroid Positive Allosteric Modulator of the (γ-Aminobutyric Acid)A Receptor”. Journal of Medicinal Chemistry. 60 (18): 7810–7819. doi:10.1021/acs.jmedchem.7b00846. PMID28753313.
^World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
^Clayton AH, Lasser R, Parikh SV, Iosifescu DV, Jung J, Kotecha M, et al. (May 2023). “Zuranolone for the Treatment of Adults With Major Depressive Disorder: A Randomized, Placebo-Controlled Phase 3 Trial”. The American Journal of Psychiatry: appiajp20220459. doi:10.1176/appi.ajp.20220459. PMID37132201. S2CID258461851.
Clinical trial number NCT04442503 for “A Study to Evaluate the Efficacy and Safety of SAGE-217 in Participants With Severe Postpartum Depression (PPD)” at ClinicalTrials.gov
Clinical trial number NCT02978326 for “A Study to Evaluate SAGE-217 in Participants With Severe Postpartum Depression” at ClinicalTrials.gov
Evobrutinib is under investigation in clinical trial NCT03934502 (Effect of Meal Composition and Timing on Evobrutinib Bioavailability).
Evobrutinib is an inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, evobrutinib inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways, which leads to the inhibition of the growth of malignant B-cells that overexpress BTK. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival.
Evobrutinib is in clinical development to investigate its potential as a treatment for multiple sclerosis (MS). It is an oral, highly selective inhibitor of Bruton’s tyrosine kinase (BTK) which is important in the development and functioning of various immune cells including B lymphocytes and macrophages.
Evobrutinib is designed to inhibit primary B cell responses such as proliferation and antibody and cytokine release, without directly affecting T cells. BTK inhibition is thought to suppress autoantibody-producing cells, which preclinical research suggests may be therapeutically useful in certain autoimmune diseases.
U.S. Patent No. 9073947 discloses a pyrimidine derivative of Evobrutinib which chemically named as l-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)
piperidin-l-yl)prop-2-en-l-one and pharmaceutically acceptable salts, solvates and pharmaceutical compositions thereof.
U.S. Patent No. 9073947 and ‘Journal of Medicinal Chemistry 2019, 62(17), 7643-7655’ discloses process for the preparation of Evobrutinib which involves column purifications and lyophilisation methods to provide Evobrutinib with low yield, which is not viable at large scale production.
Radiosynthesis of [11C]Evobrutinib. [11C]Evobrutinib was synthesized similarly to the Tolebrutinib example above with the following exceptions. First, the precursor 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine (4) (1 mg, 2.7 μmol) was used and the crude reaction mixture after the carbonylation reaction was purified by semi-preparative HPLC (column: Luna C18(2), 5 μ (250 x 9.6 mm); mobile phase: 44% MeCN in 200 mM ammonium formate; flow rate: 5 ml/min; UV: 254 nm). The [11C]1-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)piperidin-1-yl)prop-2-en-1-one ([11C]evobrutinib) was isolated between the 15.5 and 18 min mark of the chromatogram and this sample was collected into a dilution flask that contained 50 ml of a 2 mg/ml sodium ascorbate aqueous solution. This solution was transferred to an HLB light (30 mg) SPE cartridge. After transfer, the cartridge was eluted with 1 ml of ethanol into the sterile product vial that contained 4 ml of sterile saline. Using this method, 2.2 ± 0.6 GBq (81.4 ± 22.2 mCi) [11C]evobrutinib was isolated (n = 3), and the product was analyzed via reverse phase HPLC using the following methods. Method A described above and Method B (Isocratic and molar activity): column: Luna C18(2) 3-μm (250×4.6 mm); mobile phase Isocratic: 36% acetonitrile in aqueous 0.1% TFA; flow rate: 1.3 ml/min; UV: 254 nm. Method A was used to confirm chemical identity using a co-injection of non-radioactive standard. Radiochemical purity and molar activity were determined by Method B. [11C]Evobrutinib was confirmed by co-injection with a verified non-radioactive reference standard. Am was determined using a 4-point standard curve (analytical HPLC peak area) (Y) vs. standard concentration (X: in nmol) by comparison with an evobrutinib reference standard of known concentration (2.3 mg in 1 ml). The isolated [11C] evobrutinib was co-eluted with a non-radioactive reference standard. The sample was >99% radiochemically pure, >95% chemically pure (HPLC, UV: 254 nm), with a molar activity of 496.5 ± 74 GBq/μmol (13.4 Ci/μmol) The overall synthesis time from the end of cyclotron bombardment was 37–46 min.
Patent
U.S. Patent No. 9073947
PAPER
Journal of Medicinal Chemistry 2019, 62(17), 7643-7655
To a 20 mL vial was added 5-(4-phenoxyphenyl)-N-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine (210.00 mg, 0.56 mmol, 1.00 equiv), sodium bicarbonate (70.48 mg, 0.84 mmol, 1.50 equiv), THF (8.00 mL, 98.74 mmol, 176.55 equiv), and water (0.80 mL, 44.41 mmol, 79.40 equiv). The mixture was cooled to 0 °C on an ice bath. Acryloyl chloride (0.15 mL, 1.83 mmol) was then added dropwise. The ice bath was removed, and the reaction was stirred at room temperature for 12 h before it was purified by silica gel chromatography (25 g KPNH silica, 0–100% methanol/ethyl acetate) to afford the title compound (A18) (21 mg, 8.7% yield) was synthesized with a similar protocol to prepared as described in the main body of the article. 1H NMR (DMSO-d6) δ 7.93 (s, 1 H), 7.40–7.08 (m, 9H), 6.76 (dd, J = 4 Hz, 1 H), 6.04 (d, J = 4 Hz, 1 H), 5.61 (d, J = 4 Hz, 1 H), 5.43 (s, 2H), 4.34 (d, J = 12 Hz, 1 H), 3.98 (d, J = 8 Hz, 1 H), 3.12 (m, 2H), 2.95 (m, 1 H), 2.56 (m, 1 H), 1.81 (m, 1 H), 1.59 (m, 2H), 0.92 (m, 2H). [ES-MS] (ESI+): m/z calcd for C25H28N5O2 [M + H]+ 430, found 430.
Example-1: Preparation of tert-butyl 4-(((6-amino-5-chloropyrimidin-4-yl)amino) met hy 1 jpiperid ine- 1 -carboxylate
Tert-butyl-4-(aminomethyl)piperidine-l -carboxylate (81 ml) and 1,8-diazabicyclo [5.4.0]undec-7-ene (60.34 g) were added to a mixture of 5,6-dichloropyrimidin-4-amine (50 g) in N,N-dimethylformamide (500 ml) at 25-35°C. Heated the mixture to 90-95°C and stirred for 22 hrs. Cooled the mixture to 25-30°C. Water was added to the mixture at 25-35°C and stirred for 5 hrs. Filtered the precipitated solid, washed with water and n-heptane and dried to get the title compound. Yield: 73.0 gms; Purity by HPLC: 98.7%
Example-2: Preparation of tert-butyl 4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl) amino)methyl)piperidine-l-carboxylate
(4-Phenoxyphenyl)boronic acid (75.12 g) was added to a mixture of tert-butyl 4-(((6-amino-5-chloropyrimidin-4-yl)amino)methyl)piperidine-l-carboxylate(100 g), 2-di cyclo hexylphosphino-2′,6′-dimethoxybiphenyl (12 g) and potassium carbonate (121.28 g) in 1,4-di oxane (1000 ml) at 25-30°C and stirred for 30 minutes under nitrogen atmosphere. Palladium acetate (1.96 g) was added to the mixture at 25-30°C. Heated the mixture to 100-105°C and stirred for 3 hrs. Cooled the mixture to 25-30°C. Water and ethyl acetate were added to the mixture at 25-35°C and stirred for 30 minutes. Filtered the mixture by using hyflow bed. Organic layer was separated from the filtrate. Organic layer was treated with carbon powder and distilled-off the solvent under reduced pressure, n-heptane (800 ml) was added to the obtained compound. Heated the mixture to 60-65°C and stirred for 90 minutes. Cooled the mixture to 25-30°C and stirred for 2 hrs. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound. Yield: 120 gms, Purity by HPEC: 97.6% Example-3: Preparation of 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine
Tert-butyl-4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl) piperidine- 1 -carboxylate (200 g) in methanol (600 ml) was cooled to 0-5°C. Hydrochloric acid in ethyl acetate (500 ml) was slowly added to the mixture at 0-5°C. Mixture allowed to warm to 25-30°C and stirred for 20 hours. Water was added to the mixture and treated the mixture with aqueous ammonia solution. Dichloromethane was added to the mixture at 25-30°C and stirred for 10 minutes. Layers were separated and distilled-off the organic layer under reduce pressure. Obtained compound was treated with isopropyl ether and dried to get the title compound. Yield: 150 gms, Purity by HPLC: 76.4%
Example-4: Preparation of Evobrutinib
Sodium bicarbonate (23.86 g) and water (301 ml) were added to the mixture of 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine (70 g) in tetrahydrofuran (2800 ml). Cooled the mixture to 0-5°C. Acryloyl chloride (23.62 g) was slowly added to the mixture. Mixture allowed to warm to 25-30°C and stirred for 20 hrs. Distilled-off the solvent from the mixture under reduced pressure. Ethyl acetate and water were added to the mixture and stirred for 10 minutes. Both the layers were separated. Organic layer was treated with aqueous hydrochloric acid solution and carbon powder. Distilled-off the organic layer under reduced pressure. Isopropyl ether was added to the mixture at 25-30°C and stirred for 14 hrs. Filtered the mixture and washed with isopropyl ether. Dried to get the title compound.
Yield: 41.8 gms, Purity by HPLC: 97.6%
AS ON AUG2023 4,071,221 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Motixafortide is a peptide inhibitor of CXCR4 used to mobilize hematopoietic stem cells prior to collection and autologous transplantation in multiple myeloma patients.
To use with filgrastim (G-CSF) to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation in patients with multiple myeloma
Motixafortide was approved for medical use in the United States in September 2023.[2][3]
Motixafortide is a cyclic peptide hematopoietic stem cell mobilizer used to improve stem cell collection prior to autologous transplantation.3 Hematopoietic stem cell transplantation (HSCT) is commonly employed in the context of hematologic cancers – high-dose chemotherapy regimens destroy cancerous blood cells, which are then replaced via infusion of the patient’s own stem cells (i.e. an autologous transplant).4 Similar in mechanism to the previously approved plerixafor, motixafortide is an inhibitor of C-X-C Motif Chemokine Receptor 4 (CXCR4), a protein that helps to anchor stem cells to bone marrow matrix.3 When administered alongside filgrastim, another agent used to aid in stem cell collection, motixafortide enabled the collection of an adequate number of stem cells in ~92% of patients within two apheresis procedures, compared to ~26% of patients receiving only filgrastim.1
Motixafortide was approved by the FDA in September 2023, in combination with filgrastim, for use in stem cell mobilization prior to autologous stem cell transplant in patients with multiple myeloma.5 It has also been investigated alongside pembrolizumab for the treatment of pancreatic cancer.2
^World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
Clinical trial number NCT03246529 for “A Phase III, Safety, Tolerability and Efficacy of Combination Treatment of BL-8040 and G-GSF as Compared to Placebo and G-CSF for thE MobilizatioN of HematopoiEtic Stem Cells for Autologous TransplantatIon in SubjectS With MM (GENESIS)” at ClinicalTrials.gov
Etrasimod is a synthetic next-generation selective Sphingosine 1-phosphate (S1P) receptor modulator that targets the S1P1,4,5 with no detectable activity on S1P2 and S1P3 receptors. S1P receptors are membrane-derived lysophospholipid signaling molecules that are involved in the sequestration of circulating peripheral lymphocytes in lymph nodes.1 Therefore, S1P receptor modulators like etrasimod were investigated in treating immune-mediated diseases like ulcerative colitis where a high level of inflammatory T cells is present in the gastrointestinal tract, thus causing diffuse mucosal inflammation.1 In fact, it has been observed that antigen-activated T cells within peripheral lymphoid organs can transiently downregulate S1P receptor levels to facilitate immune cells trafficking into the intestinal mucosa.2
Etrasimod was approved on October 13, 2023, by the FDA under the brand name VELSIPITY for the treatment of adults with moderately to severely active ulcerative colitis. This approval was based on favorable results obtained from Pfizer’s Elevate UC Phase III registrational program, consisting of the Elevate UC 52 and Elevate UC 12 clinical trials, that investigates the efficacy of a 2-mg daily dose regimen of etrasimod, with a 32% and 26% remission rate observed in UC 52 and UC 12 trials respectively.4
Medical uses
Etrasimod is used for the treatment of moderate to severe ulcerative colitis.[1]
Mechanism of action
It works by causing T cells to become trapped in the lymph nodes, preventing them from entering the bloodstream, from where they would travel to other tissues in the body and mediate inflammation.[3][4][5][6][7][8]
APD334 was discovered as part of our internal effort to identify potent, centrally available, functional antagonists of the S1P1 receptor for use as next generation therapeutics for treating multiple sclerosis (MS) and other autoimmune diseases. APD334 is a potent functional antagonist of S1P1 and has a favorable PK/PD profile, producing robust lymphocyte lowering at relatively low plasma concentrations in several preclinical species. This new agent was efficacious in a mouse experimental autoimmune encephalomyelitis (EAE) model of MS and a rat collagen induced arthritis (CIA) model and was found to have appreciable central exposure.
^World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 78”. WHO Drug Information. 31 (3). hdl:10665/330961.
/////////Etrasimod, APD334, Velsipity, FDA 2023, APPROVALS 2023
Vodobatinib (K0706) is a potent, third generation and orally active Bcr-Abl1 tyrosine kinase inhibitor with an IC50 of 7 nM. Vodobatinib exhibits activity against most BCR-ABL1 point mutants, and has no activity against BCR-ABL1T315I. Vodobatinib can be used for chronic myeloid leukemia (CML) research. Vodobatinib is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Vodobatinib (K0706) is a potent, third generation and orally active Bcr-Abl1 tyrosine kinase inhibitor with an IC50 of 7 nM. Vodobatinib exhibits activity against most BCR-ABL1 point mutants, and has no activity against BCR-ABL1T315I. Vodobatinib can be used for chronic myeloid leukemia (CML) research[1][2]. Vodobatinib is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
A mixture of methyl 3-iodo-4-methylbenzoate (2.0g, 7mmol), trimethylsilylacetylene (1.2ml, 8mmol), Pd(PPh3)4 (0.42g, 0.3mmol), CuI (0.137g, 0.7mmol) and diisopropylethylamine (2.5ml, 11.4mmol) in THF (20ml) was heated at 50°C for 12hrs under nitrogen atmosphere. The reaction mixture was cooled to ambient temperature and filtered through a Celite® bed. The clear filtrate was concentrated and the residue purified by flash chromatography on silica gel (elution with 2% ethyl acetate in n-hexane) to provide methyl 4-methyl-3-[(trimethylsilyl)ethynyl]benzoate.
To the solution of methyl 4-methyl-3-[(trimethylsilyl)ethynyl]benzoate (2.3g) in THF (40ml) was added tetrabutylammonium fluoride (1.0M in THF, 3.2ml, 1 1mmol) at ambient
temperature and stirred for 15 minutes, concentrated and the residue purified by flash chromatography on silica gel (elution with 2% ethyl acetate in n-hexane) to provide methyl 3 – ethynyl- 4-methylbenzo at e .
Similarly were prepared the following ester compounds from their corresponding iodo esters:
Methyl 3-ethynyl-4-fluorobenzoate
Methyl 3-ethynyl-4-methoxybenzoate
Reƒerence Example 2
4-Methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid
A mixture of methyl 3-ethynyl-4-methylbenzoate (0.341 g, 2mmol), 3-iodoquinoline (0.5g, 2mmol), Pd(PPh3)4 (0.1 1g, 0.01mmol), CuI (0.179g, 0.1mmol) and diisopropylethylamine (0.5ml, 3mmol) in DMF (15ml) was stirred at ambient temperature for 12hrs under an atmosphere of nitrogen. The reaction mixture was concentrated and the crude product was purified by flash chromatography on silica gel (elution with 10% ethyl acetate in n-hexane) to provide methyl 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoate.
Sodium hydroxide (0.15g, 3.71mmol) was added to a solution of the above methyl ester in methanol (20ml) and water (3ml) and stirred at 50°C for 3hrs and then concentrated in vacuo. Water (10ml) was added to the residue, adjusted pH to 4.0-4.5 with citric acid. The solid obtained was filtered, washed successively with water and diethyl ether and dried at ambient temperature to obtain 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid. 1H NMR (500 MHz in DMSO-d6), δ 2.66 (s, 3H), 7.56 (d, J = 8.0 Hz, 1H), 7.75 (t, J; = 15.1 Hz, J2 = 8.2 Hz, 1H), 7.89 (t, J} = 13.7 Hz, J2 = 8.5 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 8.2 Hz, 1H), 8.12 (d, J = 8.1 Hz, 1H), 8.17 (s, 1H), 8.75 (s, 1H), 9.1 1 (s, 1H), 12.84 (s, 1H).
Reƒerence Example 3
4-Methyl-3-[2-(3-quinolyl)ethynyl]benzohydrazide
A mixture of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid (0.15g, 0.5mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (0.15g, 0.7mmol) and 1-hydroxybenzotriazole (0.1g, 0.7mmol) in N,N-dimethylformamide (15ml) was stirred at room temperature for 1hr. Hydrazine hydrate (1.52ml, 0.5mmol) was then added and the mixture stirred for another 3hrs. Concentration and trituration of the residue with water produced a solid which was filtered, washed successively with water and diethyl ether, and finally dried in vacuo to get the hydrazide as a pale yellow solid.
N’-(3-iodo-4-methylbenzoyl)-2,4,6-trichlorobenzohydrazide was prepared by the reaction of 3-iodo-4-methylbenzoic acid with 2,4,6-trichlorobenzohydrazide. The coupling was performed in a manner similar to that described in Reference Example 3.
A mixture of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid (0.15g, 0.5mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (0.15g, 0.7mmol) and 1-hydroxybenzotriazole (0.1g, 0.7mmol) in N,N-dimethylformamide (15ml) was stirred at ambient temperature for 1hr. 2,4,6-Trichlorobenzohydrazide (0.125g, 0.5mmol) was added and the mixture stirred for 12hrs at ambient temperature. Concentration and trituration of the residue with water produced a solid which was filtered, washed with water and the crude product was purified by flash chromatography on silica gel (elution with 10% methanol in dichloromethane) to get 2,4,6-trichloro-N-[4-methyl-3-[2-(3-quinolyl)ethynyl]benzoyl] benzohydrazide as a white solid.
Method B:
2,4,6-Trichloro-N’-[4-methyl-3-[2-(3-quinolyl)ethynyl]benzoyl] benzohydrazide was also prepared by the reaction of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid with 2,4,6-trichlorobenzohydrazide in diethyl cyanophosphonate. The condensation reaction was performed in a manner similar to that described in Method A.
Method C:
2,4,6-Trichloro-N-[4-methyl-3-[2-(3-quinolyl)ethynyl]benzoyl]benzohydrazide was also prepared by the reaction of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzohydrazide with 2,4,6- trichlorobenzoyl chloride. The condensation reaction was performed in a manner similar to that described in Method A.
The compounds 1.2 to 1.14, 1.21 to 1.34, 1.36 to 1.40, and 1.43 to 1.59 were prepared in a manner similar to Example I.1, by following either of the methods A, B or C, using the appropriate substrates.
Vodobatinib (N’-(2-chloro-6-methylbenzoyl)-4-methyl-3-[2-(3-quinolyl) ethynyl]-benzohydrazide), a c-Abl inhibitor is represented by Formula I (referred hereinafter interchangeably as vodobatinib or compound of Formula
International Publication Nos. WO 2017/208267A1, WO 2020/250133 Al and WO 2022/024072A1, which are hereby incorporated by reference, disclose methods of use of the compound of Formula I for the treatment of Parkinson’s disease, synucleinopathies and Alzheimer’s disease (AD) respectively.
There is a continuing need for effective and safe methods for the treatment of, and delaying the progression of, neurodegenerative diseases, including in the early-stage of the diseases.
Carebastine is the active metabolite of Ebastine. Carebastine is a histamine H1 receptor antagonist. Carebastine inhibits VEGF-induced HUVEC and HPAEC proliferation, migration and angiogenesis in a dose-dependent manner. Carebastine suppresses the expression of macrophage migration inhibitory factor.
Carebastine is the active metabolite of Ebastine. Carebastine is a histamine H1 receptor antagonist. Carebastine inhibits VEGF-induced HUVEC and HPAEC proliferation, migration and angiogenesis in a dose-dependent manner[1]. Carebastine suppresses the expression of macrophage migration inhibitory factor[2].
Literature References: Nonsedating type histamine H1-receptor antagonist. Prepn: J. M. P. Soto et al., EP 134124; eidem, US 4550116 (both 1985 to Fordonal). Metabolized in vivo to carebastine, its active carboxylic acid metabolite.
These schemes also illustrate the interrelatedness of the processes and intermediates.
EXAMPLE 1
One gram of 9 was dissolved in 20 mL of DMF and 18 mg of P(tBu)3, 41 mg of Pd(dba)2, 230 mg of ZnF2 and 1.2 g of 5 were added. A mixture was stirred at 80° for 18 hours, cooled to room temperature, diluted with ether and washed with water. The organic layer was dried over sodium sulfate, filtered and stripped in vacuo. The resulting product was flash chromatographed on silica gel using 4:1 hexane ethyl acetate to yield 1.0 g (91%) of 10. A repeat of the reaction on larger scale using 15 g of 9 provided 15.2 g (93%) of 10.
EXAMPLE 2
Five grams of 9 was dissolved in 50 mL of methylene chloride and cooled to 0° C. To the solution was added 5.78 g of trimethylsilyl iodide. The mixture was stirred for 30 minutes and excess sodium bisulfite solution was added with vigorous stirring at room temperature. The layers were separated and the aqueous layer extracted twice with methylene chloride. Combined organic layers were dried, filtered and stripped in vacuo to provide 7.7 g (98%) of 1. The reaction was repeated on a larger scale using 15 g of 9 to produce 22.5 g of 1 (96%) yield.
EXAMPLE 3
Six grams of potassium carbonate, 5.8 g of piperidine 2 and 7.6 g of 1 are combined in 100 mL of DMF. The suspension is stirred at room temperature until TLC in 4:1 hexane-ethyl acetate indicates a complete reaction. The reaction mixture is poured into 400 mL of water and extracted three times with methylene chloride. The combined organic extracts are dried, filtered and reduced in vacuo. The resulting product is flash chromatographed on silica gel using ethyl acetate containing 10% triethylamine to yield 3.
EXAMPLE 4
Seven grams of 3 is dissolved in 100 mL of methanol, cooled to 0° C. and 1.1 g of sodium borohydride is added. The mixture is stirred 1 hour, concentrated and partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The bicarbonate layer is extracted twice with ethyl acetate, the combined organic layers are dried over sodium sulfate and the solution is reduced in vacuo to provide 4.
EXAMPLE 5
Two grams of 4 is dissolved in 30 mL of DMF. To this are added 16.2 mg of P(tBu)3, 36.6 mg of Pd(dba)2, 209 mg of ZnF2 and 1.056 g of 5. The mixture is heated at 80° C., cooled, diluted with ether and worked up as in example 1. The resulting product is flash chromatographed on silica gel using 9:1 ethyl acetate-triethylamine to provide 7.
EXAMPLE 6
One hundred fifty milligrams of 6 is slurried in 5 mL of water and 10 mL of methanol. To the slurry is added 175 mg of sodium hydroxide. The slurry is refluxed for one hour, cooled to room temperature and the methanol removed in vacuo. The resulting aqueous solution is distributed between water and chloroform, the chloroform layer is discarded, the aqueous layer is adjusted to pH 2.3 and extracted with chloroform. The organic layer is dried, filtered and reduced in vacuo to provide carebastine.
EXAMPLE 7
Five grams of 1 was combined with 2.64 g of 2 and 2.0 g of potassium carbonate and 80 mL of DMF. The mixture was stirred at room temperature for two hours, poured into 400 mL of water and extracted three times into methylene chloride. The combined organic layers were dried, filtered and reduced in vacuo. The resulting product was flash chromatographed on silica gel using 9:1 ethyl acetate-triethylamine to provide 2.0 g (54%) of 3.
EXAMPLE 8
One and seven-tenths grams of 3, 90 mg of P(tBu)3, 300 mg of Pd(dba)2, 250 mg of ZnF2 and 1.1 g of 5 were dissolved in 330 mL of DMF under argon. The mixture was heated to 80° for two hours, cooled to room temperature, diluted with ether and worked up as described in example 1. The resulting product was filtered through silica to provide 1.2 g (67.8%) of 6.
EXAMPLE 9
Two grams of 20, 170 mg of P(tBu)3, 560 mg of Pd(acac)2, 474 mg of ZnF2 and 2.0 g of 5 were combined in 50 mL of DMF under argon. The mixture was heated to 80° C. and monitored by HPLC. When reaction was complete, the mixture was cooled to room temperature and 250 mL of water was added. The mixture was extracted three times with ether, dried, filtered and reduced in vacuo. The resulting product was flash chromatographed in 4:1 hexane-ethyl acetate to provide 1.89 g (85%) of 8.
EXAMPLE 10
Two grams of the triflate analog of 20 were reacted as in the foregoing example with 134 mg P(tBu)3, 433 mg of Pd(acac)2, 375 mg of ZnF2 and 1.58 g of 5 to provide 1.56 g (90% yield) of 8.
Example 11
Piperidinol 25 is reacted with chlorodiphenylmethane as described in Fujii et al. Arzneim.-Forsch. 44, 527-538 (1994) to provide 6.
Example 1: Potassium 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate (carristin potassium salt ) preparation
[0060]
[0061]
Step 1: Preparation of methyl 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate
[0062]
[0063]
Add 4-(diphenylmethoxy)piperidine hydrochloride (473mg, 1.77mmol), DMAC (4.5ml), K 3 PO 4 (1.13g, 5.3mmol), KI (29mg, 0.177mmol) to a 25ml single-neck bottle. , stir and heat to 100°C. Weigh 2-[4-(4-chloro-1-butyryl)phenyl]-2-methylpropionate methyl ester (600mg, 2.12mmol) and dissolve it in 1ml of DMAC. Add the reaction solution slowly and dropwise, and keep the reaction for 4~ 6h, TLC detects that the raw material reaction is complete. Cool to room temperature, add isopropyl acetate and water, and stir to separate layers. The aqueous phase was then extracted with isopropyl acetate, the organic phases were combined, washed twice with water, dried over anhydrous sodium sulfate, filtered, concentrated, and passed through a silica gel column to obtain 500 mg of the title product, yield 45%, purity: 97.3%.
Step 2: Preparation of 2-(4-(4-(4-(Diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (carristin)
[0067]
[0068]
Add (5-methyl-2-oxo-1,3-dioxo-4-yl)methyl-2-(4-(4-(4-(diphenylmethoxy))piperidine-1 to a 25ml three-necked flask) -Methyl)-butyryl)phenyl)-2-methylpropionate (320 mg, 0.62 mmol), 1.5 ml of methanol, 2 ml of 10% NaOH, heated to 60°C for 2 hours, and the TLC raw material reaction was completed. After the reaction is completed, cool to room temperature, concentrate to dryness, add EA, add hydrochloric acid to adjust the pH to 2~3, layer the layers, wash once with water, dry the organic phase, and concentrate to dryness to obtain 300 mg of the title product. Yield: 95%, purity 95.0%.
Add 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (499mg, 1mmol) and acetonitrile 3.5 to a 25ml three-necked flask. ml, heated to 60°C, added potassium hydroxide (56 mg, 1 mmol), stirred, cooled down, a white solid precipitated, filtered, and dried to obtain 500 mg of carristine potassium salt, with a yield of 90% and a purity of 98.67%.
Example 2: Sodium 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate (carristine sodium salt ) preparation
[0077]
[0078]
In this example, the preparation method of 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid is the same as in Example 1.
[0079]
Add 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (499mg, 1mmol) and acetonitrile 3.5 to a 25ml three-necked flask. ml, heated to 60°C, added sodium hydroxide (40 mg, 1 mmol) and stirred for 1 hour, concentrated to dryness, added methyl tert-butyl ether and stirred, filtered, and dried to obtain 458 mg of carristin sodium salt, yield 85%, purity 96.98 %.
fda approved, To treat paroxysmal nocturnal hemoglobinuria, 12/5/2023, Fabhalta ‘
Iptacopan is a small-molecule factor B inhibitor previously investigated as a potential treatment for the rare blood disease paroxysmal nocturnal hemoglobinuria (PNH) by inhibiting the complement factor B.1 Factor B is a positive regulator of the alternative complement pathway, where it activates C3 convertase and subsequently C5 convertase.2 This is of particular importance to PNH, where one of the disease hallmarks is the mutation of the PIGA gene. Due to this mutation, all progeny erythrocytes will lack the glycosyl phosphatidylinositol–anchored proteins that normally anchor 2 membrane proteins, CD55 and CD59, that protect blood cells against the alternative complement pathway.3 Additionally, iptacopan has the benefit of targeting factor B, which only affect the alternative complement pathway, leaving the classic and lectin pathway untouched for the body to still mount adequate immune responses against pathogens.2
On December 6th, 2023, Iptacopan under the brand name Fabhalta was approved by the FDA for the treatment of adults with PNH. This approval was based on favorable results obtained from the phase III APPL-PNH and APPOINT-PNH studies, where 82.3% and 77.5% of patients experienced a sustained hemoglobin improvement without transfusions respectively.5
Iptacopan was approved by the US Food and Drug Administration (FDA) for the treatment of adults with paroxysmal nocturnal hemoglobinuria in December 2023.[2][3]
Medical uses
Iptacopan is indicated for the treatment of adults with paroxysmal nocturnal hemoglobinuria.[1][4]
In a clinical study with twelve participants, iptacopan as a single drug led to the normalization of hemolytic markers in most patients, and no serious adverse events occurred during the 12-week study.[5][6]
Iptacopan is also investigated as a drug in other complement-mediated diseases, like age-related macular degeneration and some types of glomerulopathies.[7]
The alternative pathway (AP) of the complement system is a key contributor to the pathogenesis of several human diseases including age-related macular degeneration, paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and various glomerular diseases. The serine protease factor B (FB) is a key node in the AP and is integral to the formation of C3 and C5 convertase. Despite the prominent role of FB in the AP, selective orally bioavailable inhibitors, beyond our own efforts, have not been reported previously. Herein we describe in more detail our efforts to identify FB inhibitors by high-throughput screening (HTS) and leveraging insights from several X-ray cocrystal structures during optimization efforts. This work culminated in the discovery of LNP023 (41), which is currently being evaluated clinically in several diverse AP mediated indications.
a Reagents and conditions: (a) i PrMgCl·LiCl, Cbz-Cl, THF; (b) Zn, AcOH; (c) LiBH4, THF; (d) TBDPS-Cl, imidazole, DMF; (e) separation of diastereomers by flash chromatography; (f) TBAF, THF; (g) NaH, EtI, DMF; (h) Ba(OH)2, i PrOH, H2O; (i) K2CO3, MeI, DMF; (j) H2, Pd/C, MeOH; (k) (±)-50, DIPEA, DMA; (l) K2CO3, MeOH; then TMS-diazomethane, toluene, MeOH; (m) chiral SFC; (n) LiOH, H2O, MeOH, THF; (o) (2S,4S)-50, NaBH(OAc)3, DCE.
4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). Step 1: tert-Butyl 4-(((2S,4S)-4-Ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58). To a solution of tert-butyl 4-formyl-5-methoxy-7-methyl1H-indole-1-carboxylate (57) (1.5 g, 5.18 mmol) and methyl 4- ((2S,4S)-4-ethoxypiperidin-2-yl)benzoate ((2S,4S)-50) (1.185 g, 4.50 mmol) in DCE (20 mL) was added NaBH(OAc)3 (3 g, 14.1 mmol), and this was stirred at rt for 21.5h. Additional tert-butyl 4-formyl-5- methoxy-7-methyl-1H-indole-1-carboxylate (57) (500 mg, 1.90 mmol) was added, and this was stirred for 20 h. The reaction was diluted with EtOAc, washed successively with 5% aqueous NaHCO3, H2O, and brine, dried over Na2SO4, filtered, and concentrated to provide the title compound (2.415 g, quant) which was used without further purification. MS (ESI+) m/z 537.4 (M + H). The absolutestereochemistry was ultimately determined via cocrystallization of 41 with the catalytic domain of FB. Step 2: 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). To a solution of tert-butyl 4-(((2S,4S)-4-ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58) (2.415 g, 4.50 mmol) in THF (10 mL) and MeOH (20 mL) was added 1 M LiOH in H2O (15 mL, 15 mmol), and this was stirred at 70 °C for 8 h. The reaction was cooled to rt, diluted with H2O, half saturated aqueous KHSO4 and citric acid, saturated with sodium chloride, then extracted with 9:1 DCM/TFE, dried with Na2SO4, filtered, and concentrated. RP-HPLC-B purification provided the title compound (730 mg, 38% for 2 steps). 1 H NMR (400 MHz, D2O) δ 7.96 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 3.2 Hz, 1H), 6.66 (s, 1H), 6.20 (s, 1H), 4.62−4.47 (m, 1H), 4.06 (d, J = 13.2 Hz, 1H), 3.97−3.76 (m, 2H), 3.66−3.48 (m, 5H), 3.43−3.29 (m, 1H), 3.26−3.15 (m, 1H), 2.35 (s, 3H), 2.31−2.11 (m, 2H), 2.00 (d, J = 15.4 Hz, 1H), 1.93−1.74 (m, 1H), 1.25−1.07 (m, 3H). HRMS calcd for C25H31N2O4 (M + H)+ 423.2284, found 423.2263. 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid Hydrochloride (41· HCl). To a solution of 41 (620 mg, 1.47 mmol) in H2O (10 mL) and acetonitrile (3 mL) was added 5 M aqueous HCl (0.5 mL, 2.5 mmol). The mixture was then lyophilized, and the resulting solid was suspended in i PrOH and heated to 70 °C. The mixture turned into a solution after 1.5 h and was then cooled to rt with stirring. After about 5 h, the mixture turned into a suspension and the solid was collected by filtration and dried under high vacuum at 50 °C to provide the title compound as the hydrochloride salt (450 mg, 65%). 1 H NMR (400 MHz, methanol-d4) δ 10.73 (s, 1H), 8.23 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.3 Hz, 2H), 7.36−7.31 (m, 1H), 6.77 (s, 1H), 6.42−6.31 (m, 1H), 4.40−4.19 (m, 2H), 3.87−3.80 (m, 1H), 3.76 (s, 3H), 3.68− 3.50 (m, 4H), 3.45−3.38 (m, 1H), 2.51 (s, 3H), 2.30−2.18 (m, 2H), 2.13−1.89 (m, 2H), 1.31 (t, J = 7.0 Hz, 3H). MS (ESI+) m/z 423.3 (M + H).
//////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Clinical trial number NCT04558918 for “Study of Efficacy and Safety of Twice Daily Oral LNP023 in Adult PNH Patients With Residual Anemia Despite Anti-C5 Antibody Treatment (APPLY-PNH)” at ClinicalTrials.gov
Clinical trial number NCT04820530 for “Study of Efficacy and Safety of Twice Daily Oral Iptacopan (LNP023) in Adult PNH Patients Who Are Naive to Complement Inhibitor Therapy (APPOINT-PNH)” at ClinicalTrials.gov
Eplontersen, FDA APP, 12/21/2023, To treat polyneuropathy of hereditary transthyretin-mediated amyloidosis, Wainua
AKCEA-TTR-LRx is under investigation in clinical trial NCT04136184 (Neuro-ttransform: A Study to Evaluate the Efficacy and Safety of Akcea-ttr-lrx in Participants With Hereditary Transthyretin-mediated Amyloid Polyneuropathy).
^ Coelho, Teresa; Marques, Wilson; Dasgupta, Noel R.; Chao, Chi-Chao; Parman, Yeşim; França, Marcondes Cavalcante; et al. (October 2023). “Eplontersen for Hereditary Transthyretin Amyloidosis With Polyneuropathy”. The Journal of the American Medical Association. 330 (15): 1448–1458. doi:10.1001/jama.2023.18688. PMC 10540057. PMID37768671.
^ Diep, John K.; Yu, Rosie Z.; Viney, Nicholas J.; Schneider, Eugene; Guo, Shuling; Henry, Scott; et al. (December 2022). “Population pharmacokinetic/pharmacodynamic modelling of eplontersen, an antisense oligonucleotide in development for transthyretin amyloidosis”. British Journal of Clinical Pharmacology. 88 (12): 5389–5398. doi:10.1111/bcp.15468. PMID35869634. S2CID250989659.
^World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85”. WHO Drug Information. 35 (1). hdl:10665/340684.
External links
Clinical trial number NCT04136184 for “NEURO-TTRansform: A Study to Evaluate the Efficacy and Safety of Eplontersen (Formerly Known as ION-682884, IONIS-TTR-LRx and AKCEA-TTR-LRx) in Participants With Hereditary Transthyretin-Mediated Amyloid Polyneuropathy” at ClinicalTrials.gov
Clinical trial number NCT01737398 for “Efficacy and Safety of Inotersen in Familial Amyloid Polyneuropathy” at ClinicalTrials.gov
Capivasertib is a novel pyrrolopyrimidine derivative, and an orally available inhibitor of the serine/threonine protein kinase AKT (protein kinase B) with potential antineoplastic activity. Capivasertib binds to and inhibits all AKT isoforms. Inhibition of AKT prevents the phosphorylation of AKT substrates that mediate cellular processes, such as cell division, apoptosis, and glucose and fatty acid metabolism. A wide range of solid and hematological malignancies show dysregulated PI3K/AKT/mTOR signaling due to mutations in multiple signaling components. By targeting AKT, the key node in the PIK3/AKT signaling network, this agent may be used as monotherapy or combination therapy for a variety of human cancers.
Medical uses
Capivasertib, used in combination with fulvestrant (Faslodex), is indicated for adults with hormone receptor-positive, human epidermal growth factor receptor 2-negative locally advanced or metastatic breast cancer with one or more PIK3CA/AKT1/PTEN-alterations, as detected by an FDA-approved test, following progression on at least one endocrine-based regimen in the metastatic setting or recurrence on or within twelve months of completing adjuvant therapy.[1][3]
History
Efficacy was evaluated in CAPItello-291 (NCT04305496), a randomized, double-blind, placebo-controlled, multicenter trial in 708 participants with locally advanced or metastatic HR-positive, HER2-negative breast cancer, of which 289 participants had tumors with PIK3CA/AKT1/PTEN-alterations.[3] All participants were required to have progression on aromatase inhibitor-based treatment.[3] Participants could have received up to two prior lines of endocrine therapy and up to one line of chemotherapy for locally advanced or metastatic disease.[3]
HCl (4M in Dioxane) (3.00 mL, 12.00 mmol) was added to (S)-tert-butyl 4-(1-(4-chlorophenyl)-3-hydroxypropylcarbamoyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-ylcarbamate (Intermediate 22) (1.27 g, 2.40 mmol) in dichloromethane (20 mL). The resulting suspension was stirred at 20° C. for 16 hours. The reaction mixture was filtered through a PTFE filter cup and the crude solid was purified by preparative HPLC (Waters XTerra C18 column, 5 μm silica, 19 mm diameter, 100 mm length), using decreasingly polar mixtures of water (containing 1% TFA) and MeCN as eluents. Fractions containing the desired compound were purified by ion exchange chromatography, using an SCX column. The desired product was eluted from the column using 7M NH 3/MeOH and pure fractions were evaporated to dryness to afford (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (0.200 g, 19.4%) as a white solid. 1H NMR (399.9 MHz, DMSO-d6) δ 1.45 (2H, d), 1.86 (1H, d), 1.90-1.93 (1H, m), 2.19 (2H, s), 3.38 (2H, q), 3.51-3.58 (2H, m), 4.35-4.38 (2H, m), 4.53 (1H, t), 4.88 (1H, d), 6.58 (1H, t), 7.16 (1H, t), 7.32-7.38 (4H, m), 8.12 (1H, s), 8.43 (1H, d), 11.63 (1H, s), m/z (ESI+) (M+H)+=429; HPLC tR=1.46 min.
EXAMPLE 9 ALTERNATIVE ROUTE 1: (S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE
N-Ethyldiisopropylamine (1.676 ml, 9.62 mmol) was added to (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)piperidine-4-carboxamide (Intermediate 49) (1 g, 3.21 mmol) and 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (0.493 g, 3.21 mmol) in butan-1-ol (15 ml). The resulting solution was stirred at 60° C. for 18 hours. The reaction mixture was diluted with EtOAc (50 mL), and washed sequentially with water (25 mL) and saturated brine (25 mL). The organic layer was dried over MgSO 4, filtered and evaporated to afford crude product. The crude product was purified by flash silica chromatography, elution gradient 0 to 6% MeOH with ammonia in DCM. Pure fractions were evaporated to dryness to afford (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (842 mg) as a white foam. (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide was stirred in ethyl acetate (7 mL) for 18 hours. The solid was collected by filtration, washed with a small amount of ethyl acetate and vacuum oven dried at 55° C. for 18 hours to afford (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (0.585 g, 42.5%) as a white solid.
m/z (ES+) (M+H)+=429; HPLC tR=1.60 min.
1H NMR (400.13 MHz, DMSO-d 6) δ 1.39-1.47 (2H, m), 1.80-2.02 (4H, m), 2.17 (2H, s), 3.35-3.40 (2H, m), 3.50-3.59 (2H, m), 4.34-4.41 (2H, m), 4.53 (1H, t), 4.88 (1H, d), 6.57 (1H, m), 7.14-7.16 (1H, m), 7.31-7.37 (4H, m), 8.12 (1H, s), 8.42 (1H, d), 11.62 (1H, s)
EXAMPLE 9 ALTERNATIVE ROUTE 2: (S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE
(S)-3-Amino-3-(4-chlorophenyl)propan-1-ol (Intermediate 47) (2.055 g, 11.07 mmol) was added in one portion to 4-(tert-butoxycarbonylamino)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxylic acid (Intermediate 1) (4 g, 11.07 mmol) and DIPEA (5.80 ml, 33.20 mmol) in DMA (40 ml). HATU (4.63 g, 12.18 mmol) was added and the resulting solution was stirred at 20° C. for 24 hours. The reaction mixture was evaporated to dryness then diluted with EtOAc (300 mL), and washed sequentially with water (50 mL) and saturated brine (50 mL). The organic layer was dried over MgSO 4, filtered and evaporated to afford crude product. The crude product was purified by flash silica chromatography, elution gradient 2 to 6% MeOH with ammonia in DCM. Pure fractions were evaporated to dryness and triturated with dioxane (40 ml) to afford (S)-tert-butyl 4-(1-(4-chlorophenyl)-3-hydroxypropylcarbamoyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-ylcarbamate (Intermediate 22) (4.82 g, 82%) as a white solid. (S)-tert-butyl 4-(1-(4-chlorophenyl)-3-hydroxypropylcarbamoyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-ylcarbamate (Intermediate 22) (4.82 g, 82%) was suspended in dioxane (40.0 ml) and 4M hydrogen chloride in dioxane (7.69 ml, 221.36 mmol) added. The reaction was stirred at ambient temperature for 2 hours. The crude product was purified by ion exchange chromatography, using an SCX column. The desired product was eluted from the column using 3.5M NH 3/MeOH and pure fractions were evaporated to dryness. The crude product was purified by preparative HPLC (Waters XBridge Prep C18 OBD column, 5 μm silica, 19 mm diameter, 100 mm length), using decreasingly polar mixtures of water (containing 1% NH 3) and MeCN as eluents. Fractions containing the desired compound were evaporated to dryness to afford (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (1.200 g, 25.3%) as a white solid.
m/z (ES+) (M+H)+=429; HPLC tR=1.67 min.
1H NMR matches previous.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Clinical trial number NCT04305496 for “Capivasertib+Fulvestrant vs Placebo+Fulvestrant as Treatment for Locally Advanced (Inoperable) or Metastatic HR+/HER2- Breast Cancer (CAPItello-291)” at ClinicalTrials.gov
///////Capivasertib, Truqap, FDA 2023, APPROVALS 2023, AZD 5363