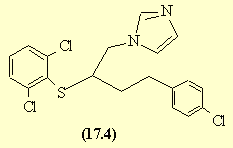
1-(4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl)-1H-imidazole
64872-77-1 NITRATE ,
64872-76-0 (free base)
Butoconazole nitrate, RS-35887-00-10-3, RS-35887, Gynomyk, Gynazole-1, Femstat

Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| - | G01AF15 | C 19 H 17 Cl 3 N 2 S | 411.78 g / mol | 64872-76-0 |
| mononitrate | G01AF15 | C 19 H 17 Cl 3 N 2 S ⋅ HNO 3 | 474.80 g / mol | 64872-77-1 |
No Exclusivity found
| Drug Name | Femstat 3 (from Drugs@FDA) |
|---|---|
| Active Ingredient | Butoconazole nitrate |
| Dosage Form | Cream |
| Route | Vaginal |
| Strength | 2% |
| Market Status | Over the Counter |
| Company | Bayer |
- 64872-77-1 , butoconazole nitrate
- CAS Number: 64872-77-1
- Molecular Weight: 474.79
- Molecular Formula: C19H17Cl3N2S ·HNO3
| Patent No | Patent Expiry | |
|---|---|---|
| 5993856 | Nov 17, 2017 |
Laszlo Czibula, Laszlo Dobay, Eva Werkne Papp, Judit Nagyne Bagdy, Ferenc Sebok, “High Purity Butoconazole Nitrate with Specified Particle Size and a Process for the Preparation Thereof.” U.S. Patent US20080221190, issued September 11, 2008.

 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1-[4-(4-Chlorophenyl)-2-(2,6-dichlorophenyl)sulfanylbutyl]imidazole | |
| Clinical data | |
| Trade names | Gynazole-1, Mycelex-3 |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a682012 |
| Pregnancy cat. |
|
| Legal status | |
| Routes | Vaginal cream |
| Identifiers | |
| CAS number | 67085-13-6  |
| ATC code | G01AF15 |
| PubChem | CID 47472 |
| DrugBank | DB00639 |
| ChemSpider | 43192 |
| UNII | 0Q771797PH |
| KEGG | D00880  |
| ChEBI | CHEBI:3240 |
| ChEMBL | CHEMBL1295 |
| Chemical data | |
| Formula | C19H17Cl3N2S |
| Mol. mass | 411.776 g/mol |
Use
-
an antifungal agent for topical use
Classes substance
-
Eter chlorothiophenol
-
Imidazoles
-
Synthesis pathway
| Synthesis of a) |
|---|
  |
Trade names
| Country | Trade name | Manufacturer |
|---|---|---|
| France | Ginomik | Cassenne |
| USA | Femstat | Syntex |
| Ukraine | Gіnofort | BAT “Gideon Rіhter” Ugorschina |
Formulations
-
2% vaginal cream
Reference for syn
-
Synthesis of a)
-
Walker, KAM et al .: J. Med. Chem. (JMCMAR) 21, 840 (1978).
-
US 4,078,071 (Syntex; USA-prior. 28.7.1975).
-
DOS 2,800,755
-
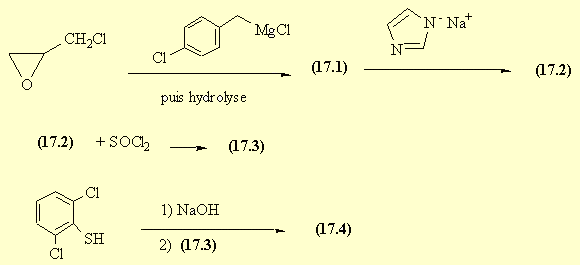
………………………
Patent
http://www.google.com/patents/EP1709005A1?cl=en
Butoconazole nitrate (chemical name: l-[4-(4-chlorophenyl)-2-(2,6-dichloro- -phenylthio)-n-butyl]-imidazol nitrate) is a compound of the formula (I),

(I)
it belongs among the aryl-ethylimidazole compounds, has fungicidal activity and may be used for the treatment of vaginal infections caused primarily by Candida albicans. Azoles exert their antifungal effect via modifying the ergosterol synthesis of fungus cells; more particularly, imidazoles inhibit the 14α-demethylase enzyme, thereby bringing about an increased level of 14α-methyl sterols which, in turn, causes an alteration of cell membrane permeability leading to the destruction of the fungus cells (Tetrahedron: Asymmetry Vol 4, No. 7, pp. 1521-1526, 1993). The first process for the preparation of the butoconazole nitrate is a multistep synthesis disclosed in the US 4,078,071 patent specification. Here two reaction routes are given for the preparation of the key intermediate of the formula (TV) (l-[4-(4-chlorophenyl)-2-hydroxy-n- -butyl] -imidazole) .

(IN)
According to one of them first an epoxy compound is prepared from an aromatic aldehyde or from an olefinic compound having a terminal double bond; then the epoxy compound is reacted with imidazole to yield the key intermediate. The aromatic aldehyde (VIII)

(VIII)
is treated with expensive and hazardous reagents (trimethylsulfoxonium iodide and sodium hydride) in dry dimethyl sulfoxide and the epoxide formed in the reaction is isolated after a complicated work-up. The epoxide so obtained is converted to the imidazole derivate in a time consuming reaction in the presence of dimethylformamide, then the key intermediate of the formula (IN) (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) is isolated and purified in an additional step. From the compounds having terminal double bond (Nil)

(Nil) the epoxide is obtained via a highly explosive peracidic oxidation step and the epoxide is then converted into (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IV) in a manner described above. In the other reaction route a poisoning aromatic α-halo-keto compound is used as starting material which is reacted with imidazole to give the corresponding keto-imidazole which, in turn, is reduced with a complex metal hydride – a reagent with potential hazards – to yield the key intermediate (IN). The reaction mixture is worked up in an involved manner. The synthesis way described in J. Med. Chem., 1978, Vol. 21, No. 8, pp 840-843 is as follows: l-chloro-4-chlorophenyl-2-butanol (II)

(II) is treated with the imidazole (III)

(HI)
in the presence of sodium hydride reagent in dimethylformamide solvent. This substitution reaction takes a long time and gives the (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]- imidazole) (IN) with a poor yield (51.7 %). In the next step of the butoconazole nitrate synthesis
(l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) is treated with thionyl chloride (which is at once a reagent and a solvent) at 65-70 °C to yield l-[4-(4-chlorophenyl)-2-chloro- -n-butyl] -imidazole of the formula (N).

(V)
The reaction mixture is then evaporated to dryness. The removal of the excess thionyl chloride, a highly corrosive substance, requires special equipment; the same applies to waste treatment, an operation which also involves an environmental risk. The residue is dissolved in dichloromethane, the solution is made alkaline by adding aqueous potassium carbonate solution. Phases are separated, the organic layer is washed with water, dried on magnesium sulphate and evaporated to give l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole (N), as a gum. Said gum is dissolved in acetone and reacted with 2,6-dichlorothiophenol in the presence of potassium carbonate with a long reaction time. After the reaction has been finished, the inorganic salts are removed by filtration, the solvent is evaporated, and the residue is partitioned between water and ether. Butoconazole nitrate is precipitated with nitric acid from the ethereal layer. The end-product crystals in white plates from a mixture of acetone and ethyl acetate (yield: 84 %). Our aim was to provide a process by which the active agent can be prepared in high purity via reaction steps producing good yields and besides that said steps require neither solvents that are highly flammable and explosive (ether), carcinogenic (dimethylformamide) or corrosive (thionylchloride), nor reagents (e. g. sodium hydride) that are highly flammable or explosive. We have surprisingly found that when the starting material l-chloro-4-chlorophenyl-2-
-butanol (II) is reacted with the imidazole (III) in a mixture of toluene and aqueous sodium hydroxide solution in the presence of a phase transfer catalyst, the
(l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) key intermediate is obtained with short reaction time and excellent yield (95 %). Next we studied alternative solvents to replace the thionyl chloride in solvent function in the reaction step converting (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) into (l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole) (N). In the inert solvents which could be taken into account such as dichloromethane, toluene, chlorobenzene and dimethylformamide, the chlorinating reaction yielded a sticky reaction mixture which couldn’t be processed. We have surprisingly found, however that when (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) is dissolved in 1 ,2-dichloroethane and reacted with approximately equimolar amount of thionyl chloride reagent in the presence of catalytic amount of dimethylformamide at 30-35 °C temperature, a crystal suspension is obtained which is easy-to-stir during the whole reaction time, resulting in that chlorination proceeds completely giving l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole (N) in quantitative yield. Being the compound sufficiently pure, it is not isolated, but separated by extraction and reacted directly with 2,6-dichlorothiophenol in methyl isobutyl ketone to give 1 -[4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl]-imidazole (VI) (butoconazole).

(NI)
Example 1. Preparation of (1 4-(4-chlorophenyl)-2-hvdroxy-n-butyll-imidazole) (IV) To a solution of 56.7 g (0.26 mol) of l-chloro-4-chloroρhenyl-2-butanol (J. of Medicinal Chemistry, 1978. Nol. 21. No. 8. p. 842) in 200 ml of toluene 36.2 g (0.9 mol) of sodium hydroxide dissolved in 100 ml of water, 6.4 g (0.028 mol) of benzyltriethyammom‘um chloride and 35.2 g (0.51 mol) of imidazole (III) are added. The reaction mixture is heated at 93-95 °C for one hour then the temperature is returned to about 60 °C, the phases are separated and to the organic layer water (100 ml) is added. The mixture is first stirred at 22-25 °C for 1 hour then at 0-5 °C for two hours. The crystals are separated by filtration, washed with water (2 x 35 ml) of 0-5 °C to yield 74 g of wet (l-[4-(4-chloroρhenyl)-2-hydroxy-n-butyl]-imidazole) which is dried at maximum 50 °C in vacuo to give 61.6 g (95 %) of the product. Recrystallization from ethyl acetate gives 52.4 g (85 %) of dry product melting at 104-106 °C.
Example 2. Preparation of l-[4-(4-chlorophenvπ-2-(2,6-(McMorophenyl o)-n-butyl1-ϊmidazole nitrate (I) 25 g (0.1 mol) of l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole (IN) is suspended in 1,2-dichloroethane (125 ml), to this suspension dimethylformamide (1 ml) and thionyl chloride (13.6 g; 0.11 mol) are added at 30-32 °C and the reaction mixture is kept at 35-38 °C for 1.5 hour under stirring. After the chlorination has been finished the homogenous solution is cooled to 15-18 °C, the excess of thionyl choride is decomposed with water (10 ml) then again water (80 ml) is added to the solution. After stirring at 20-22 °C for 0.5 hour the phases are separated and the organic layer is extracted with water (30 ml). To the aqueous solution methyl isobutyl ketone (250 ml) is added and the pH of the mixture is adjusted to 8.5 – 9 with 15 g (0.14 mol) of sodium carbonate dissolved in water (70 ml). The mixture is stirred at 22-25 °C for 0.5 hour, phases are separated, from the organic layer an 50 ml portion is distilled off to remove water and to the remaining solution 26.8 g (0.15 mol) of 2,6-dichloro-thiophenol and 40 g (0.29 mol) of dry potassium carbonate are added. The suspension is stirred at 105 – 108 °C under nitrogen for 3-4 hours. After the reaction has been finished the inorganic salts are removed by filtration at 22-25 °C, the filtrate is washed and clarified with activated carbon and the pH of the clear solution is adjusted to 3 – 3.5 by adding about 8 – 9 ml of 65 % nitric acid. The solution is stirred at the same temperature for 1 hour then the temperature is lowered to 8 – 12 °C. The crystals obtained are filtered and washed to give 48 g of wet l-[4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl]- -imidazole nitrate corresponding to 42.6 g (90 %) of dry product.
HPLC
Details of the HPLC method: Type of the apparatus: Spectra System/TSP (manufacturer: Thermo Separation Products, USA) Column: LiChrospher RP-18, 250×4.0 mm ID., 5 μm (Merck, Germany, Cat. No. : 1.50983) Mobile phase: methanol : buffer = 8:2 Bujfer: 2.18 g KH2PO4 + 4.18 g K2HPO4-3H2O dissolved in 1000 ml of distilled water; MeOH (HPLC Gradient grade, Merck, Germany, Cat. No.: 1.06007.2500) KH2PO4 (p.a., Merck, Germany, Cat. No.: 1.04877.1000) K2HPO4-3H2O (p.a., Merck, Germany, Cat. No.: 1.05099.1000) Flow rate: 1.0 ml/min Temperature: 40 °C Detection: UN 229 nm Solvent for sampling: eluent Sample concentration: 1.0 mg/ml Injected volume: 10 μl Duration of analysis: 40 min Evaluation: area normalization method. Approximative retention time: 11.9 min B. Particle size: Particle size was determined by sieve analysis using an Alpine sieve operated by a jet of air.
……………………..
WALKER K A M ET AL: “1-[4-(4-Chlorophenyl)-2-(2,6-dichloro phenylthio)-n-butyl]-1H-imidazole nitrate, a new potent antifungal agent” JOURNAL OF MEDICINAL CHEMISTRY, vol. 21, no. 8, August 1978 (1978-08), pages 840-843,
http://pubs.acs.org/doi/pdf/10.1021/jm00206a028
1- [4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-b~-
tyll-lH-imidazole nitrate (I).
I as colorless blades
(9.6 g, 84%): mp 162-163 “C (foaming). Anal. (C19H18C13N303S)
C, H, N. The free base prepared by neutralization of a suspension
of 1 in ether with aqueous potassium carbonate and recrystallization
from cyclohexane had mp 68-70.5 “C (foaming).
……………….
FULL SYNTHESIS
SEE
http://www.chemdrug.com/databases/8_0_yyfgohllmfsvfvsx.html

The chlorohydrin (II) is obtained by the reaction of p-chlorobenzylmagnesium chloride (I) with epichlorohydrin (A) in ether. This is then converted to the crystalline alcohol (III) by reaction with sodium imidazole (B) in DMF. On treatment with thionyl chloride is converted to the corresponding chloro compound (IV). When (IV) is reacted with 2,6-dichloro thiophenol (C) in the presence of anhydrous potassium carbonate in acetone, the free base of butoconazole is formed. Neutralization of the free base (V) with nitric acid gives butoconazole.
References
- Seidman, L. S.; Skokos, C. K. (2005). “An evaluation of butoconazole nitrate 2% site release vaginal cream (Gynazole-1) compared to fluconazole 150 mg tablets (Diflucan) in the time to relief of symptoms in patients with vulvovaginal candidiasis”. Infectious diseases in obstetrics and gynecology 13 (4): 197–206. doi:10.1080/10647440500240615. PMC 1784583. PMID 16338779.
- Butoconazole monograph
Literature References:
Imidazole derivative with antifungal properties. Prepn: K. A. M. Walker, US 4078071 (1978 to Syntex).
Prepn, toxicity, activity vs Candida albicans in mice: K. A. M. Walker et al., J. Med. Chem. 21, 840 (1978).
In vitro comparison with other antifungal agents: F. C. Odds et al., J. Antimicrob. Chemother. 14, 105 (1984).
Clinical trials in treatment of vulvovaginal candidiasis: W. Droegemueller et al., Obstet. Gynecol. 64, 530 (1984); J. B. Jacobson et al., Acta Obstet. Gynecol. Scand. 64, 241 (1985).
Comparison with miconazole, q.v.: C. S. Bradbeer et al., Genitourin. Med. 61, 270 (1985).
Filed under: Uncategorized Tagged: Butoconazole, Butoconazole Nitrate







