
Empagliflozin
BI-10773
(2S,3R,4R,5S,6R)-2-[4-chloro-3-[[4-[(3S)-oxolan-3-yl]oxyphenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
M.Wt: 450.91
: C23H27ClO7
Sponsor/Developer: Eli Lilly and Boehringer Ingelheim
Mechanism of action: SGLT 2 inhibitor
Indication (Phase): Oral treatment of adults with type 2 diabetes (Phase III, expected to conclude by year’s end); Oral treatment of adults with type 2 diabetes plus high blood pressure (Phase IIb; trial results released Oct. 2)
NDA, MAA filings planned for 2013
Empagliflozin is a potent, selective sodium glucose co-transporter-2 inhibitor that is in development for the treatment of type 2 diabetes. Empagliflozin is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in the proximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra. The Empagliflozin phase III clinical trial program will include about 14,500 patients. The program consists of twelve ongoing international phase III clinical trials, including a large cardiovascular outcomes trial.
Empagliflozin is a novel SGLT2 inhibitor that is described for the treatment or improvement in glycemic control in patients with type 2 diabetes mellitus, for example in WO 05/092877, WO 06/117359, WO 06/120208, WO 2010/092126, WO 2010/092123, WO 2011/039107, WO 2011/039108. The use of a SGLT2 inhibitor in a method for treating obesity is described in WO 08/116,195
WO2005/092877
WO2006/117359 MP 149 DEG CENT
Empagliflozin is drug which is being investigated in clinical trials for the oral treatment oftype 2 diabetes by Boehringer Ingelheim and Eli Lilly and Company.[1][2] It is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in theproximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra.[3][4]
SGLT-2 inhibitors such as empagliflozin reduce blood glucose by blocking glucose reabsorption in the kidney and thereby excreting glucose (i.e., blood sugar) via the urine.[5]
As of December 2013, empagliflozin is in phase III clinical trials.[2]

When taken in dosages of 10 or 25 mg once a day, the incidence of adverse events was similar to placebo. However, there was a higher frequency of genital infections at both the 10 mg and the 25 mg dosages.
1-chloro-4-(β-D-glucopyranos-1-yl)-2-[4-((S)-tetrahydrofuran-3-yloxy)-benzyl]-benzene of the formula

as described for example in WO 2005/092877. Methods of synthesis are described in the literature, for example WO 06/120208 and WO 2011/039108. According to this invention, it is to be understood that the definition of empagliflozin also comprises its hydrates, solvates and polymorphic forms thereof, and prodrugs thereof. An advantageous crystalline form of empagliflozin is described in WO 2006/117359 and WO 2011/039107 which hereby are incorporated herein in their entirety. This crystalline form possesses good solubility properties which enables a good bioavailability of the SGLT2 inhibitor. Furthermore, the crystalline form is physico-chemically stable and thus provides a good shelf-life stability of the pharmaceutical composition. Preferred pharmaceutical compositions, such as solid formulations for oral administration, for example tablets, are described in WO 2010/092126,

http://www.google.com/patents/WO2011039108A2
Example 1 : Synthesis of the fluoride VIII.1
Oxalylchloride (176kg; 1386mol; 1 ,14eq) is added to a mixture of 2-chloro-5-iodo benzoic acid (343kg; 1214mol) (compound IX.1 ), fluorobenzene (858kg) and N,N-dimethylformamide (2kg) within 3 hours at a temperature in the range from about 25 to 30°C (gas formation). After completion of the addition, the reaction mixture is stirred for additional 2 hours at a temperature of about 25 to 30°C. The solvent (291 kg) is distilled off at a temperature between 40 and 45°C (p=200mbar). Then the reaction solution (91 1 kg) is added to aluminiumchloride AICI3 (181 kg) and fluorobenzene (192kg) at a temperature between about 25 and 30°C within 2 hours. The reaction solution is stirred at the same temperature for about an additional hour. Then the reaction mixture is added to an amount of 570 kg of water within about 2 hours at a temperature between about 20 and 30°C and stirred for an additional hour. After phase separation the organic phase (1200kg) is separated into two halves (600kg each). From the first half of the organic phase solvent (172kg) is distilled off at a temperature of about 40 to 50°C (p=200mbar). Then 2-propanole (640kg) is added. The solution is heated to about 50°C and then filtered through a charcoal cartouche (clear filtration). The cartouche may be exchanged during filtration and washed with a
fluorobenzene/2-propanole mixture (1 :4; 40kg) after filtration. Solvent (721 kg) is distilled off at a temperature of about 40 to 50°C and p=200mbar. Then 2-propanole (240kg) is added at a temperature in the range between about 40 to 50°C. If the content of fluorobenzene is greater than 1 % as determined via GC, another 140kg of solvent are distilled off and 2- propanole (140kg) is added. Then the solution is cooled from about 50°C to 40°C within one hour and seeding crystals (50g) are added. The solution is further cooled from about 40°C to 20°C within 2 hours. Water (450kg) is added at about 20°C within 1 hour and the suspension is stirred at about 20°C for an additional hour before the suspension is filtered. The filter cake is washed with 2-propanole/water (1 :1 ; 800kg). The product is dried until a water level of <0.06%w/w is obtained. The second half of the organic phase is processed identically. A total of 410kg (94%yield) of product which has a white to off-white crystalline appearance, is obtained. The identity of the product is determined via infrared spectrometry.
Example 2: Synthesis of the ketone VII.1
To a solution of the fluoride VIII.1 (208kg), tetrahydrofuran (407kg) and (S)-3- hydroxytetrahydrofuran (56kg) is added potassium-ie f-butanolate solution (20%) in tetrahydrofuran (388kg) within 3 hrs at 16 to 25°C temperature. After completion of the addition, the mixture is stirred for 60min at 20°C temperature. Then the conversion is determined via HPLC analysis. Water (355kg) is added within 20 min at a temperature of 21 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature: 20°C). The stirrer is switched off and the mixture is left stand for 60 min (temperature: 20°C). The phases are separated and solvent is distilled off from the organic phase at 19 to 45°C temperature under reduced pressure. 2-Propanol (703kg) is added to the residue at 40 to 46°C temperature and solvent is distilled off at 41 to 50°C temperature under reduced pressure. 2-Propanol (162kg) is added to the residue at 47°C temperature and solvent is distilled off at 40 to 47°C temperature under reduced pressure. Then the mixture is cooled to 0°C within 1 hr 55 min. The product is collected on a centrifuge, washed with a mixture of 2- propanol (158kg) and subsequently with ie f.-butylmethylether (88kg) and dried at 19 to 43°C under reduced pressure. 227kg (91 ,8%) of product are obtained as colourless solid. The identity of the product is determined via infrared spectrometry.
Example 3: Synthesis of the iodide V.1
To a solution of ketone VII.1 (217,4kg) and aluminium chloride (AICI3; 81 ,5kg) in toluene (366,8kg) is added 1 ,1 ,3,3-tetramethyldisiloxane (TMDS, 82,5kg) within 1 hr 30 min
(temperature: 18-26°C). After completion of the addition, the mixture is stirred for additional 1 hr at a temperature of 24°C. Then the conversion is determined via HPLC analysis.
Subsequently the reaction mixture is treated with acetone (15,0kg), stirred for 1 hr 5 min at 27°C temperature and the residual TMDS content is analyzed via GC. Then a mixture of water (573kg) and concentrated HCI (34kg) is added to the reaction mixture at a temperature of 20 to 51 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature:
51 °C). The stirrer is switched off and the mixture is left stand for 20 min (temperature: 52°C). The phases are separated and solvent is distilled off from the organic phase at 53-73°C temperature under reduced pressure. Toluene (52,8kg) and ethanol (435,7kg) are added to the residue at 61 to 70°C temperature. The reaction mixture is cooled to 36°C temperature and seeding crystals (0,25kg) are added. Stirring is continued at this temperature for 35 min. Then the mixture is cooled to 0 to 5°C and stirred for additional 30 min. The product is collected on a centrifuge, washed with ethanol (157kg) and dried at 15 to 37°C under reduced pressure. 181 kg (82,6%) of product are obtained as colourless solid. The identity of the product is determined via the HPLC retention time.
Example 4: Synthesis of the lactone IV.1
A suspension of the D-(+)-gluconic acid-delta-lactone IVa.1 (42,0kg), tetrahydrofuran (277,2kg), 4-methylmorpholine (NMM; 152,4kg) and 4-dimethylaminopyridine (DMAP;
1 ,44kg) is treated with chlorotrimethylsilane (TMSCI; 130,8kg) within 50 min at 13 to 19°C. After completion of the addition stirring is continued for 1 hr 30 min at 20 to 22°C and the conversion is determined via HPLC analysis. Then n-heptane (216,4kg) is added and the mixture is cooled to 5°C. Water (143kg) is added at 3 to 5°C within 15 min. After completion of the addition the mixture is heated to 15°C and stirred for 15 min. The stirrer is switched off and the mixture is left stand for 15 min. Then the phases are separated and the organic layer is washed in succession two times with water (143kg each). Then solvent is distilled off at 38°C under reduced pressure and n-heptane (130kg) is added to the residue. The resulting solution is filtered and the filter is rinsed with n-heptane (63kg) (filter solution and product solution are combined). Then solvent is distilled off at 39 to 40°C under reduced pressure. The water content of the residue is determined via Karl-Fischer analysis (result: 0,0%).
1 12,4kg of the product is obtained as an oil (containing residual n-heptane, which explains the yield of >100%). The identity of the product is determined via infrared spectrometry.
Example 5a: Synthesis of the glucoside 11.1
To a solution of the iodide V.1 (267kg) in tetrahydrofuran (429kg) is added Turbogrignard solution (isopropylmagnesium chloride/lithium chloride solution, 14 weight-% iPrMgCI in THF, molar ratio LiCI : iPrMgCI = 0,9 – 1 .1 mol/mol) (472kg) at -21 to -15°C temperature within 1 hr 50 min. On completion of the addition the conversion is determined via HPLC analysis. The reaction is regarded as completed when the area of the peak corresponding to the iodide V.1 is smaller than 5,0% of the total area of both peaks, iodide V.1 and the corresponding desiodo compound of iodide V.1 . If the reaction is not completed, additional Turbogrignard solution is added until the criterion is met. In this particular case the result is 3,45%. Then the lactone IV.1 (320kg) is added at -25 to -18°C temperature within 1 hr 25 min. The resulting mixture is stirred for further 1 hr 30 min at -13 to -18°C. On completion the conversion is determined via HPLC analysis (for information). On completion, a solution of citric acid in water (938L; concentration: 10 %-weight) is added to the reaction mixture of a volume of about 2500L at -13 to 19°C within 1 hr 25 min.
The solvent is partially distilled off from the reaction mixture (residual volume: 1816-1905L) at 20 to 30°C under reduced pressure and 2-methyltetrahydrofuran (532kg) is added. Then the stirrer is switched off and the phases are separated at 29°C. After phase separation the pH value of the organic phase is measured with a pH electrode (Mettler Toledo MT HA 405 DPA SC) or alternatively with pH indicator paper (such as pH-Fix 0-14, Macherey and Nagel). The measured pH value is 2 to 3. Then solvent is distilled off from the organic phase at 30 to 33°C under reduced pressure and methanol (1202kg) is added followed by the addition of a solution of 1 ,25N HCI in methanol (75kg) at 20°C (pH = 0). Full conversion to the acetale 111.1 is achieved by subsequent distillation at 20 to 32°C under reduced pressure and addition of methanol (409kg).
Completion of the reaction is obtained when two criteria are fulfilled:
1 ) The ratio of the sum of the HPLC-area of the alpha-form + beta-form of intermediate 111.1 relative to the area of intermediate llla.1 is greater or equal to 96,0% : 4,0%. 2) The ratio of the HPLC-area of the alpha-form of intermediate 111.1 to the beta-form of 111.1 is greater or equal to 97,0% to 3,0%.
In this particular case both criteria are met. Triethylamin (14kg) is added (pH = 7,4) and solvent is distilled off under reduced pressure, acetonitrile (835kg) is added and further distilled under reduced pressure. This procedure is repeated (addition of acetonitrile: 694kg) and methylene chloride (640kg) is added to the resulting mixture to yield a mixture of the acetale 111.1 in acetonitrile and methylene chloride. The water content of the mixture is determined via Karl Fischer titration (result: 0,27%).
The reaction mixture is then added within 1 hr 40 min at 10 to 19°C to a preformed mixture of AICI3 (176kg), methylene chloride (474kg), acetonitrile (340kg), and triethylsilane (205kg). The resulting mixture is stirred at 18 to 20°C for 70 min. After completion of the reaction, water (1263L) is added at 20 to 30°C within 1 hr 30 min and the mixture is partially distilled at 30 to 53°C under atmospheric pressure and the phases are separated. Toluene (698kg) is added to the organic phase and solvent is distilled off under reduced pressure at 22 to 33°C. The product is then crystallized by addition of seeding crystals (0,5kg) at 31 °C and water (267kg) added after cooling to 20°C. The reaction mixture is cooled to 5°C within 55 min and stirred at 3 to 5°C for 12 hrs. Finally the product is collected on a centrifuge as colourless, crystalline solid, washed with toluene (348kg) and dried at 22 to 58°C. 21 1 kg (73%) of product are obtained. The identity of the product is determined via the HPLC retention time.
Example 5b: Synthesis of the glucoside 11.1
To a solution of the iodide V.1 (30g) in tetrahydrofuran (55ml_) is added Turbogrignard solution (isopropylmagnesium chloride/lithium chloride solution, 14 weight-% iPrMgCI in THF, molar ratio LiCI : iPrMgCI = 0,9 – 1 .1 mol/mol) (53g) at -14 to -13°C temperature within 35 min. On completion of the addition the conversion is determined via HPLC analysis. The reaction is regarded as completed when the area of the peak corresponding to the iodide V.1 is smaller than 5,0% of the total area of both peaks, iodide V.1 and the corresponding desiodo compound of iodide V.1 . If the reaction is not completed, additional Turbogrignard solution is added until the criterion is met. In this particular case the result is 0,35%. Then the lactone IV.1 (36g) is added at -15 to -6°C temperature within 15 min. The resulting mixture is stirred for further 1 hr at -6 to -7°C. On completion, the conversion is determined via HPLC analysis (for information). On completion, a solution of citric acid in water (105mL;
concentration: 10 %-weight) is added to the reaction mixture at -15 to 10°C within 30 min. The solvent is partially distilled off from the reaction mixture (residual volume: 200mL) at 20 to 35°C under reduced pressure and 2-methyltetrahydrofuran (71 mL) is added. Then the mixture is stirred for 25min at 30°C. Then the stirrer is switched off and the phases are separated at 30°C. After phase separation the pH value of the organic phase is measured with a pH electrode (Mettler Toledo MT HA 405 DPA SC) or alternatively with pH indicator paper (such as pH-Fix 0-14, Macherey and Nagel). The measured pH value is 3. Then solvent is distilled off from the organic phase at 35°C under reduced pressure and methanol (126ml_) is added followed by the addition of a solution of 1 ,25N HCI in methanol (10,1 ml_) at 25°C (pH = 1 -2). Full conversion to the acetale 111.1 is achieved by subsequent distillation at 35°C under reduced pressure and addition of methanol (47ml_).
Completion of the reaction is obtained when two criteria are fulfilled:
1 ) The ratio of the sum of the HPLC-area of the alpha-form + beta-form of intermediate 111.1 relative to the area of intermediate llla.1 is greater or equal to 96,0% : 4,0%. In this particular case the ratio is 99,6% : 0,43%.
2) The ratio of the HPLC-area of the alpha-form of intermediate 111.1 to the beta-form of III.1 is greater or equal to 97,0% to 3,0%. In this particular case the ratio is 98,7% : 1 ,3%.
Triethylamin (2,1 mL) is added (pH = 9) and solvent is distilled off at 35°C under reduced pressure, acetonitrile (120ml_) is added and further distilled under reduced pressure at 30 to 35°C. This procedure is repeated (addition of acetonitrile: 102ml_) and methylene chloride (55ml_) is added to the resulting mixture to yield a mixture of the acetale 111.1 in acetonitrile and methylene chloride. The water content of the mixture is determined via Karl Fischer titration (result: 0,04%).
The reaction mixture is then added within 1 hr 5 min at 20°C to a preformed mixture of AICI3 (19,8g), methylene chloride (49ml_), acetonitrile (51 mL), and triethylsilane (23g). The resulting mixture is stirred at 20 to 30°C for 60 min. After completion of the reaction, water (156mL) is added at 20°C within 25 min and the mixture is partially distilled at 55°C under atmospheric pressure and the phases are separated at 33°C. The mixture is heated to 43°C and toluene (90mL) is added and solvent is distilled off under reduced pressure at 41 to 43°C. Then acetonitrile (1 OmL) is added at 41 °C and the percentage of acetonitrile is determined via GC measurement. In this particular case, the acetonitrile percentage is 27%- weight. The product is then crystallized by addition of seeding crystals (0,1 g) at 44°C and the mixture is further stirred at 44°C for 15min. The mixture is then cooled to 20°C within 60min and water (142mL) is added at 20°C within 30min. The reaction mixture is cooled to 0 to 5°C within 60 min and stirred at 3°C for 16 hrs. Finally the product is collected on a filter as colourless, crystalline solid, washed with toluene (80mL) and dried at 20 to 70°C. 20, 4g (62,6%) of product are obtained. The identity of the product is determined via the HPLC retention time.
- Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P (January 2012). “Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors”. Diabetes Obes Metab 14 (1): 83–90. doi:10.1111/j.1463-1326.2011.01517.x. PMID 21985634.
- “Empagliflozin”. clinicaltrials.gov. U.S. National Institutes of Health. Retrieved 22 September 2012.
- Nair S, Wilding JP (January 2010). “Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus”. J. Clin. Endocrinol. Metab. 95 (1): 34–42. doi:10.1210/jc.2009-0473. PMID 19892839.
- Bays H (March 2009). “From victim to ally: the kidney as an emerging target for the treatment of diabetes mellitus”. Curr Med Res Opin25 (3): 671–81. doi:10.1185/03007990802710422. PMID 19232040.
- Abdul-Ghani MA, DeFronzo RA (September 2008). “Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus”. Endocr Pract 14 (6): 782–90. PMID 18996802.
[1]. Grempler R, Thomas L, Eckhardt M et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012 Jan;14(1):83-90.
Abstract
AIMS: Empagliflozin is a selective sodium glucose cotransporter-2 (SGLT-2) inhibitor in clinical development for the treatment of type 2 diabetes mellitus. This study assessed pharmacological properties of empagliflozin in vitro and pharmacokinetic properties in vivo and compared its potency and selectivity with other SGLT-2 inhibitors. METHODS: [(14)C]-alpha-methyl glucopyranoside (AMG) uptake experiments were performed with stable cell lines over-expressing human (h) SGLT-1, 2 and 4. Two new cell lines over-expressing hSGLT-5 and hSGLT-6 were established and [(14)C]-mannose and [(14)C]-myo-inositol uptake assays developed. Binding kinetics were analysed using a radioligand binding assay with [(3)H]-labelled empagliflozin and HEK293-hSGLT-2 cell membranes. Acute in vivo assessment of pharmacokinetics was performed with normoglycaemic beagle dogs and Zucker diabetic fatty (ZDF) rats. RESULTS: Empagliflozin has an IC(50) of 3.1 nM for hSGLT-2. Its binding to SGLT-2 is competitive with glucose (half-life approximately 1 h). Compared with other SGLT-2 inhibitors, empagliflozin has a high degree of selectivity over SGLT-1, 4, 5 and 6. Species differences in SGLT-1 selectivity were identified. Empagliflozin pharmacokinetics in ZDF rats were characterised by moderate total plasma clearance (CL) and bioavailability (BA), while in beagle dogs CL was low and BA was high. CONCLUSIONS: Empagliflozin is a potent and competitive SGLT-2 inhibitor with an excellent selectivity profile and the highest selectivity window of the tested SGLT-2 inhibitors over hSGLT-1. Empagliflozin represents an innovative therapeutic approach to treat diabetes.
[2]. Thomas L, Grempler R, Eckhardt M et al. Long-term treatment with empagliflozin, a novel, potent and selective SGLT-2 inhibitor, improves glycaemic control and features of metabolic syndrome in diabetic rats. Diabetes Obes Metab. 2012 Jan;14(1):94-6.
Abstract
Empagliflozin is a potent, selective sodium glucose co-transporter-2 inhibitor that is in development for the treatment of type 2 diabetes. This series of studies was conducted to assess the in vivo pharmacological effects of single or multiple doses of empagliflozin in Zucker diabetic fatty rats. Single doses of empagliflozin resulted in dose-dependent increases in urinary glucose excretion and reductions in blood glucose levels. After multiple doses (5 weeks), fasting blood glucose levels were reduced by 26 and 39% with 1 and 3 mg/kg empagliflozin, respectively, relative to vehicle. After 5 weeks, HbA1c levels were reduced (from a baseline of 7.9%) by 0.3 and 1.1% with 1 and 3 mg/kg empagliflozin, respectively, versus an increase of 1.1% with vehicle. Hyperinsulinaemic-euglycaemic clamp indicated improved insulin sensitivity with empagliflozin after multiple doses versus vehicle. These findings support the development of empagliflozin for the treatment of type 2 diabetes.
[3]. Luippold G, Klein T, Mark M, Grempler R. Empagliflozin, a novel potent and selective SGLT-2 inhibitor, improves glycaemic control alone and in combination with insulin in streptozotocin-induced diabetic rats, a model of type 1 diabetes mellitus. Diabetes Obes Metab. 2012 Jul;14(7):601-7.
Abstract
AIM: Sodium glucose cotransporter-2 (SGLT-2) is key to reabsorption of glucose in the kidney. SGLT-2 inhibitors are in clinical development for treatment of type 2 diabetes mellitus (T2DM). The mechanism may be of value also in the treatment of type 1 diabetes mellitus (T1DM). This study investigated effects of the SGLT-2 inhibitor, empagliflozin, alone and in combination with insulin, on glucose homeostasis in an animal model of T1DM. METHODS: Sprague-Dawley rats were administered a single intraperitoneal injection of streptozotocin (STZ; 60 mg/kg). Acutely, STZ rats received two doses of insulin glargine with or without empagliflozin, and blood glucose was measured. In a subchronic study, STZ rats received empagliflozin alone, one or two insulin-releasing implants or a combination of one implant and empagliflozin over 28 days; blood glucose and HbA(1c) were measured. RESULTS: In the acute setting, empagliflozin in combination with 1.5 IU insulin induced a similar glucose-lowering effect as 6 IU insulin. Both interventions were more efficacious than monotherapy with 1.5 IU insulin. In the subchronic study, 12-h blood glucose profile on day 28 in the combination group was lower than with one implant, and similar to two implants. Plasma HbA(1c) was improved in the combination group and in animals with two implants. CONCLUSIONS: Empagliflozin reduced blood glucose levels in a T1DM animal model. Empagliflozin combined with low-dose insulin showed comparable glucose-lowering efficacy to treatment with high-dose insulin. Our data suggest that empagliflozin is an efficacious adjunctive-to-insulin therapy with the clinical potential for the treatment of T1DM.
[4]. Macha S, Rose P, Mattheus M et al. Lack of drug-drug interaction between empagliflozin, a sodium glucose cotransporter-2 inhibitor, and warfarin in healthy volunteers. Diabetes Obes Metab. 2012 Oct 24. doi: 10.1111/dom.12028. [Epub ahead of print]
Abstract
AIM: To investigate potential drug-drug interactions between empagliflozin and warfarin. MATERIALS AND METHODS: Healthy subjects (n=18) received empagliflozin 25 mg qd for 5 days (treatment A), followed by empagliflozin 25 mg qd for 7 days (days 6-12) with a single 25 mg dose of warfarin on day 6 (B), and a single 25 mg dose of warfarin alone (C), in an open-label, crossover study. Subjects received treatments in sequence AB_C or C_AB with a washout period of ≥14 days between AB and C or C and AB. RESULTS: Warfarin had no effect on empagliflozin area under concentration-time curve or maximum plasma concentration at steady-state (AUC(τ) (,ss) or C(max,ss) ): geometric mean ratios (GMRs) (90% confidence intervals [CI]) were 100.89% (96.86, 105.10) and 100.64% (89.79, 112.80), respectively. Empagliflozin had no effect on AUC from 0 hours to infinity (AUC(0) (-∞) ) or C(max) for R-warfarin or S-warfarin (GMRs [90% CI] for AUC(0) (-∞) : 98.49% [95.29, 101.80] and 95.88% [93.40, 98.43], respectively; C(max) : 97.89% [91.12, 105.15] and 98.88% [91.84, 106.47], respectively). Empagliflozin had no clinically relevant effects on warfarin’s anticoagulant activity (international normalised ratio [INR]) (GMR [95% CI] for peak INR: 0.87 [0.73, 1.04]; area under the effect-time curve from 0 to 168 hours: 0.88 [0.79, 0.98]. No drug-related adverse events were reported for empagliflozin after monotherapy or combined administration. The combination of empagliflozin and warfarin was well tolerated. CONCLUSIONS: No drug-drug interactions were observed between empagliflozin and warfarin, indicating that empagliflozin and warfarin can be co-administered without dosage adjustments of either drug.
[5]. Sarashina A, Koiwai K, Seman LJ et al. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single Doses of Empagliflozin, a Sodium Glucose Cotransporter-2 (SGLT-2) Inhibitor, in Healthy Japanese Subjects. Drug Metab Pharmacokinet. 2012 Nov 13. [Epub ahead of print]
Abstract
This randomized, placebo-controlled within dose groups, double-blind, single rising dose study investigated the safety, tolerability, pharmacokinetics and pharmacodynamics of 1 mg to 100 mg doses of empagliflozin in 48 healthy Japanese male subjects. Empagliflozin was rapidly absorbed, reaching peak levels in 1.25 to 2.50 hours; thereafter, plasma concentrations declined in a biphasic fashion, with mean terminal elimination half-life ranging from 7.76 to 11.7 hours. Increase in empagliflozin exposure was proportional to dose. Oral clearance was dose independent and ranged from 140 to 172 mL/min. In the 24 hours following 100 mg empagliflozin administration, the mean (%CV) amount of glucose excreted in urine was 74.3 (17.1) g. The amount and the maximum rate of glucose excreted via urine increased with dose of empagliflozin. Nine adverse events, all of mild intensity, were reported by 8 subjects (7 with empagliflozin and 1 with placebo). No hypoglycemia was reported. In conclusion, 1 mg to 100 mg doses of empagliflozin had a good safety and tolerability profile in healthy Japanese male subjects. Exposure to empagliflozin was dose-proportional. The amount and rate of urinary glucose excretion were higher with empagliflozin than with placebo, and increased with empagliflozin dose.
…………………………….
formula A

Example I

(5-bromo-2-chloro-phenyl)-(4-methoxy-phenyl)-methanone
38.3 ml oxalyl chloride and 0.8 ml of dimethylformamide are added to a mixture of
100 g of 5-bromo-2-chloro-benzoic acid in 500 ml dichloromethane. The reaction mixture is stirred for 14 h, then filtered and separated from all volatile constituents in the rotary evaporator. The residue is dissolved in 150 ml dichloromethane, the solution is cooled to -5 0C, and 46.5 g of anisole are added. Then 51.5 g of aluminum trichloride are added batchwise so that the temperature does not exceed 5 0C. The solution is stirred for another 1 h at 1 to 5 0C and then poured onto crushed ice. The organic phase is separated, and the aqueous phase is extracted another three times with dichloromethane. The combined organic phases are washed with aqueous 1 M hydrochloric acid, twice with aqueous 1 M sodium hydroxide solution and with brine. Then the organic phase is dried, the solvent is removed and the residue is recrystallised in ethanol. Yield: 86.3 g (64% of theory)
Mass spectrum (ESI+): m/z = 325/327/329 (Br+CI) [M+H]+
Example Il
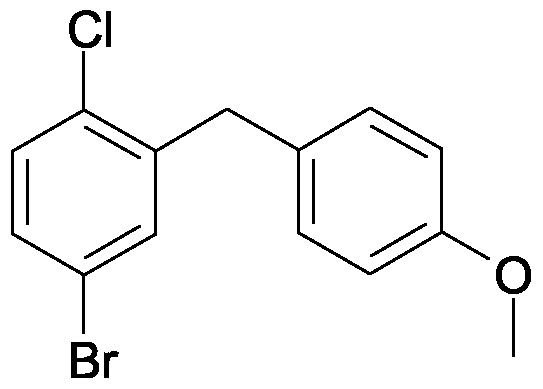
4-bromo-1-chloro-2-(4-methoxy-benzyl)-benzene
A solution of 86.2 g (5-bromo-2-chloro-phenyl)-(4-methoxy-phenyl)-methanone and 101.5 ml triethylsilane in 75 ml dichloromethane and 150 ml acetonitrile is cooled to 1O0C. Then with stirring 50.8 ml of boron trifluoride etherate are added so that the temperature does not exceed 2O0C. The solution is stirred for 14 h at ambient temperature, before another 9 ml triethylsilane and 4.4 ml boron trifluoride etherate are added. The solution is stirred for a further 3 h at 45 to 5O0C and then cooled to ambient temperature. A solution of 28 g potassium hydroxide in 70 ml of water is added, and the resulting mixture is stirred for 2 h. Then the organic phase is separated off and the aqueous phase is extracted another three times with diisopropylether. The combined organic phases are washed twice with aqueous 2 M potassium hydroxide solution and once with brine and then dried over sodium sulfate. After the solvent has been removed the residue is washed in ethanol, separated again and dried at 6O0C. Yield: 50.0 g (61 % of theory)
Mass spectrum (ESI+): m/z = 310/312/314 (Br+CI) [M+H]+
Example III

4-(5-bromo-2-chloro-benzyl)-phenol
A solution of 14.8 g 4-bromo-1-chloro-2-(4-methoxy-benzyl)-benzene in 150 ml dichloromethane is cooled in an ice bath. Then 50 ml of a 1 M solution of boron tribromide in dichloromethane are added, and the solution is stirred for 2 h at ambient temperature. The solution is then cooled in an ice bath again, and saturated aqueous potassium carbonate solution is added dropwise. At ambient temperature the mixture is adjusted with aqueous 1 M hydrochloric acid to a pH of 1 , the organic phase is separated, and the aqueous phase is extracted another three times with ethyl acetate. The combined organic phases are dried over sodium sulphate, and the solvent is removed completely. Yield: 13.9 g (98% of theory) Mass spectrum (ESI ): m/z = 295/297/299 (Br+CI) [M-HV
Example IV

r4-(5-bromo-2-chloro-benzyl)-phenoxyl-tert-butyl-dimethyl-silane
A solution of 13.9 g 4-(5-bromo-2-chloro-benzyl)-phenol in 140 ml dichloromethane is cooled in an ice bath. Then 7.54 g tert-butyldimethylsilylchlorid in 20 ml dichloromethane are added followed by 9.8 ml triethylamine and 0.5 g 4- dimethylaminopyridine. The solution is stirred for 16 h at ambient temperature and then diluted with 100 ml dichloromethane. The organic phase is washed twice with aqueous 1 M hydrochloric acid and once with aqueous sodium hydrogen carbonate solution and then dried over sodium sulfate. After the solvent has been removed the residue is filtered through silica gel (cyclohexane/ethyl acetate 100:1 ). Yield: 16.8 g (87% of theory) Mass spectrum (El): m/z = 410/412/414 (Br+CI) [M]+
Example V

2.3.4.6-tetrakis-O-(trimethylsilyl)-D-glucopyranone
A solution of 20 g D-glucono-1 ,5-lactone and 98.5 ml Λ/-methylmorpholine in 200 ml of tetrahydrofuran is cooled to -5 0C. Then 85 ml trimethylsilylchloride are added dropwise so that the temperature does not exceed 5 0C. The solution is then stirred for 1 h at ambient temperature, 5 h at 35 0C and again for 14 h at ambient temperature. After the addition of 300 ml of toluene the solution is cooled in an ice bath, and 500 ml of water are added so that the temperature does not exceed 100C. The organic phase is then separated and washed in each case once with aqueous sodium dihydrogen phosphate solution, water and brine. The solvent is removed, the residue is taken up in 250 ml of toluene, and the solvent is again removed completely. Yield: 52.5 g (approx. 90% pure)
Mass spectrum (ESI+): m/z = 467 [M+H]+
Example Vl

1-chloro-4-(β-D-qlucopyranos-1-yl)-2-(4-hvdroxybenzyl)-benzene
A solution of 4.0 g [4-(5-bromo-2-chloro-benzyl)-phenoxy]-te/Tf-butyl-dimethyl-silane in 42 ml dry diethyl ether is cooled to -800C under argon. 11.6 ml of a 1.7 M solution of te/if-butyllithium in pentane are slowly added dropwise to the cooled solution, and then the solution is stirred for 30 min at -80 0C. This solution is then added dropwise through a transfer needle, which is cooled with dry ice, to a solution of 4.78 g
2,3,4,6-tetrakis-O-(trimethylsilyl)-D-glucopyranone in 38 ml diethyl ether chilled to – 80 0C. The resulting solution is stirred for 3 h at -78 0C. Then a solution of 1.1 ml methanesulphonic acid in 35 ml of methanol is added and the solution is stirred for 16 h at ambient temperature. The solution is then neutralised with solid sodium hydrogen carbonate, ethyl acetate is added and the methanol is removed together with the ether. Aqueous sodium hydrogen carbonate solution is added to the remaining solution, and the resulting mixture is extracted four times with ethyl acetate. The organic phases are dried over sodium sulphate and evaporated down. The residue is dissolved in 30 ml acetonitrile and 30 ml dichloromethane and the solution is cooled to -10 0C. After the addition of 4.4 ml triethylsilane 2.6 ml boron trifluoride etherate are added dropwise so that the temperature does not exceed -5 0C. After the addition is complete the solution is stirred for another 5 h at -5 to -10 0C and then quenched by the addition of aqueous sodium hydrogen carbonate solution. The organic phase is separated, and the aqueous phase is extracted four times with ethyl acetate. The combined organic phases are dried over sodium sulfate, the solvent is removed, and the residue is purified by chromatography on silica gel (dichoromethane/methanol 1 :0->3:1 ). The product then obtained is an approx. 6:1 mixture of β/α which can be converted into the pure β-anomer by global acetylation of the hydroxy groups with acetic anhydride and pyridine in dichloromethane and recrystallization of the product from ethanol. The product thus obtained is converted into the title compound by deacetylation in methanol with aqueous 4 M potassium hydroxide solution. Yield: 1.6 g (46% of theory)
Mass spectrum (ESI+): m/z = 398/400 (Cl) [M+H]+
Preparation of the compound A:

1-chloro-4-(β-D-qlucopyranos-1-yl)-2-r4-(<fS)-tetrahvdrofuran-3-yloxy)-benzyll- benzene
0.19 g (f?)-3-(4-methylphenylsulfonyloxy)-tetrahydrofuran are added to a mixture of 0.20 g 1-chloro-4-(β-D-glucopyranos-1-yl)-2-(4-hydroxybenzyl)-benzene and 0.29 g cesium carbonate in 2.5 ml dimethylformamide. The mixture is stirred at 75 0C for 4 h, before another 0.29 g caesium carbonate and 0.19 g (f?)-3-(4-methylphenyl- sulfonyloxy)-tetrahydrofuran are added. After an additional 14 h stirring at 75 0C the mixture is cooled to ambient temperature and brine is added. The resulting mixture is extracted with ethyl acetate, the combined organic extracts are dried over sodium sulfate, and the solvent is removed. The residue is purified by chromatography on silica gel (dichloromethane/methanol 1 :0 -> 5:1 ). Yield: 0.12 g (49% of theory) Mass spectrum (ESI+): m/z = 451/453 (Cl) [M+H] +
Filed under: phase3 drugs Tagged: empagliflozin, PHASE 3







