
Lascufloxacin
CAS 848416-07-9
Kyorin Pharmaceutical Co., Ltd., 杏林製薬株式会社
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-
7-((3S,4S)-3-((Cyclopropylamino)methyl)-4-fluoropyrrolidin-1-yl)-6-fluoro-1-(2-fluoroethyl)-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
{(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
(KRP-AM1977X)
-
C21-H24-F3-N3-O4
- 439.4316
- SMILES……COc1c2c(cc(c1N3C[C@H](C(C3)CNC4CC4)F)F)c(=O)c(cn2CCF)C(=O)O
![]()
…………………………
Lascufloxacin hydrochloride
-
C21-H24-F3-N3-O4.Cl-H
- 475.8925
- CAS 1433857-09-0
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, hydrochloride (1:1)
……………….
Lascufloxacin mesylate
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, methanesulfonate (1:1)
-
C21-H24-F3-N3-O4.C-H4-O3-S
- 535.5372
- CAS 1433857-41-0
The other non-fluorinated quinolone under clinical development is KRP-AM1977, by Kyorin, which is in Phase I of clinical trials. The oral formulation of the compound (KRP-AM1977X) is being tested for treatment of respiratory infections and the I.V. formulation is under development for treatment of MRSA infections [1,2].
………………………………..
PATENT
WO 2013069297
http://www.google.co.in/patents/WO2013069297A1?cl=en
The present invention is represented by Formula (1) – {(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (hereinafter, compound (1) crystals of a salt also referred to), and a method for their preparation.
Typically, the pharmaceutical, in addition to the therapeutic effects on diseases, such as safety and quality are required. Therefore, the compound is the active ingredient of drugs, a variety of conditions and that is excellent in storage stability in the (light, temperature, humidity etc. influence the compound) are determined. Also, if the medicament is a dosage form such as oral preparations and injections, it is preferred that higher solubility in active ingredients of the water contained.
Compound (1) is safe, not only exhibit a strong antimicrobial action, conventional hard Gram-positive bacteria antimicrobial agents shown efficacy, particularly MRSA, PRSP, to VRE such resistant strains, to exhibit strong antibacterial activity It is known (for example, Patent Document 1).
WO 2005/026147
Patent Document 1, as the physicochemical characteristics of the compound (1) only has been shown to be a light brown free crystals. Also, Patent Document 1, the solubility in water of Compound (1), stability, no disclosure whatsoever information including characteristics of the crystal.
The present invention aims to provide a technique capable of improving the solubility and storage stability in water of the compound (1).
(Reference Example 4)
Bis (acetato -O) – [6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylate -O 3, O 4] boron Under a nitrogen atmosphere, boric acid (catalyst preparation) 86.4 g (1.40mol) was added acetic anhydride 17.9 L (190mol), and was heated and stirred for 30 minutes at 70.0 ~ 77.7 ℃. It was then cooling the mixture to an internal temperature of 24.7 ℃ (hot water set temperature 23.0 ℃). Subsequently, it was added portionwise boric acid to 4 times to the mixture. Specifically, the addition of boric acid (1 time) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.7 ~ 27.4 ℃. The addition of boric acid (second) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.3 ~ 26.3 ℃. In addition boric acid (third time) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 24.3 ~ 26.8 ℃. In addition boric acid (4 th) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 25.1 ~ 28.3 ℃. The mixture was stirred for 30 minutes at 50.0 ~ 54.9 ℃, was with boric acid triacetate adjusted solution.
In the boric acid triacetate adjusted solution, 6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylic acid ethyl ester 4.60kg (14. In a reaction preparation solution are added 0mol), and stirred for 3 hours at 53.7 ~ 56.9 ℃. The reaction preparation was cooled to 30.0 ℃, and allowed to stand overnight at room temperature. The reaction preparation was allowed to dissolve with heating to precipitate up to 55.0 ℃, acetone 13.8L was added and the reaction solution (1).
Separately, under nitrogen atmosphere, it is mixed Tsunemizu 161L and aqueous ammonia (28%) 28.2L (464mol), and cooled the mixture to 1.6 ℃. To the mixture, it was added the reaction solution of the above (1), to obtain a crude crystal acquisition solution crowded washed with acetone 9.20L. After cooling the crude crystal acquisition solution to 15.0 ℃, it was stirred for 1 hour at 6.2 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with Tsunemizu 46.0L, to give 9.07kg of wet crude crystals. Set temperature 65.0 to about 16 hours and dried under reduced pressure at ℃, the crude crystals were obtained 5.89kg.
Under a nitrogen atmosphere, it is mixed acetone and 29.5L crude crystal, the resulting mixture was heated and dissolved (melting temperature 52.6 ℃). When heated, it was dropped until the crystallization of diisopropyl ether 58.9L in a mixture (dropping amount 10.0L; 52.8 → 48.7 ℃; crystallization temperature 49.0 ℃). After crystallization confirmation, stirred for 15 minutes the mixture at 49.0 ~ 50.1 ℃, it was dropped the rest of diisopropyl ether to the mixture (50.1 → 46.4 ℃), 46.7 ~ 51.7 It was stirred for 15 minutes mixture at ℃. After cooling the mixture to 15 ℃, it was stirred for 30 minutes at 8.1 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with acetone and diisopropyl ether 5.89L 11.8L, to obtain 6.19kg of wet crystals. For about 20 hours drying under reduced pressure at warm water set temperature 65.0 ℃, bis (acetato -O) – [6,7-difluoro-1- (2-fluoroethyl) -8-methoxy-4-oxo-1,4- dihydro-3-carboxylate -O 3, O 4] was obtained 5.42kg boron (90.4% yield).
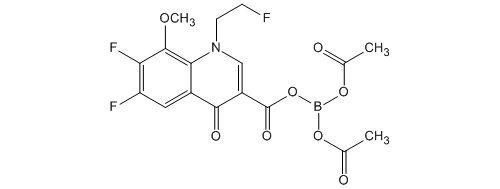
Melting point: 183 ~ 185 ℃ (dec).
Elemental analysis (%): calculated as C 17 H 15 BF 3 NO 8: C, 47.58; H, 3.52; N, 3.26.
Measured value: C, 47.91; H, 3.44; N, 3.04.
1 H-NMR (CDCl 3, 400 MHz) δ: 2.04 (6H, s), 4.22 (3H, d, J = 2.4Hz), 4.88 (2H, dt, J = 47.0 , 4.4Hz), 5.21 (2H, dt, J = 24.9,4.4Hz), 8.17 (1H, t, J = 8.8Hz), 9.11 (1H, s).
ESI MS (positive) m / z: 430 (M + H) +.
IR (KBr) cm -1: 3080,1703.
………………………………………….
WO 2005026147
http://www.google.com/patents/EP1666477A1?cl=en
KEY INTERMEDIATE

604798-54-1
3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-
| Chemical Name:3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-CAS: 604798-54-1Molecular Formula: C8H15FN2Molecular Weight: 158.2165032 |
………………………….
KEY INTERMEDIATE
CAS 848498-67-9
-8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ
キシ]ボラン
| 化学物質名 | ビス(アセチルオキシ)[6,7-ジフルオロ-1-(2-フルオロエチル) -8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ キシ]ボラン |
|---|---|
| 構造別分類コード番号 | F60622212422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
|
| 安衛法官報通し番号 | 21534 |
| 安衛法官報公示整理番号 | 8-(1)-3764 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 848498-67-9 |
| 出典 | 厚生労働省 |
……………………………….
KEY INTERMEDIATE
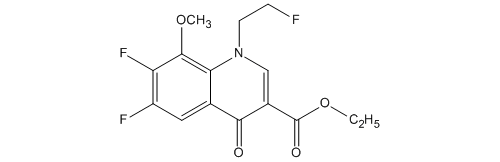
3-Quinolinecarboxylic acid, 6,7-difluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, ethyl ester
114214-60-7
C15H14F3NO4
ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル
| 化学物質名 | 6,7-ジフルオロ-1-(2-フルオロエチル)-8-メトキシ-4-オキ ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル |
|---|---|
| 構造別分類コード番号 | F60622322422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
 |
| 安衛法官報通し番号 | 21467 |
| 安衛法官報公示整理番号 | 8-(1)-3758 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 114214-60-7 |
| 出典 | 厚生労働省 |
| WO2003076428A1 * | 8 Mar 2002 | 18 Sep 2003 | Toshifumi Akiba | Quinolonecarboxylic acid derivative |
| WO2005026147A1 | 8 Sep 2004 | 24 Mar 2005 | Yoshikazu Asahina | 7-(4-substituted 3- cyclopropylaminomethyl-1 pyrrolidinyl) quinolonecarboxylic acid derivative |
| WO2007082471A1 * | 18 Jan 2007 | 26 Jul 2007 | Guangzhou Baiyunshan Pharmaceu | Anti-infective compound, preparation method thereof and use thereof |
| CN1158846A * | 9 May 1995 | 10 Sep 1997 | 昆山市康壮达兽药厂 | Synthesis technology of norfluxacini hydrochloride |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014174846A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174847A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174848A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Tablet |
- Kyorin. Kyorin—Main R&D Activities-1 (4 February 2013 Release). Available online: http://www.kyorin-pharm.co.jp/en/business/pdf/main_rd_activities_20130204_en.pdf (accessed on 4 February 2013).
- Kyorin. Drug discovery, development, and lcm with medical professionals and patients in mind. Available online: http://www.kyorin-gr.co.jp/en/business/gensen/r_and_d.shtml (accessed on 11 April 2013).
-
……….
![]()



Ochyanomizu Sola City 16F,
Kanda Surugadai 4-6, Chiyoda-ku,
Tokyo 101-8311 Japan
TEL: 03-3525-4711
Access
One-minute walk from the Hijiribashi exit of Ochanomizu station on JR Chuo and Sobu lines
One-minute walk from the B2 exit of Shin-Ochanomizu station on Tokyo Metro Chiyoda line
Four-minutes walk from the No.1 exit of Ochanomizu station on Tokyo Metro Marunouchi line
Six-minutes walk from the B3 exit of Ogawamachi station on Toei Subway Shinjuku line
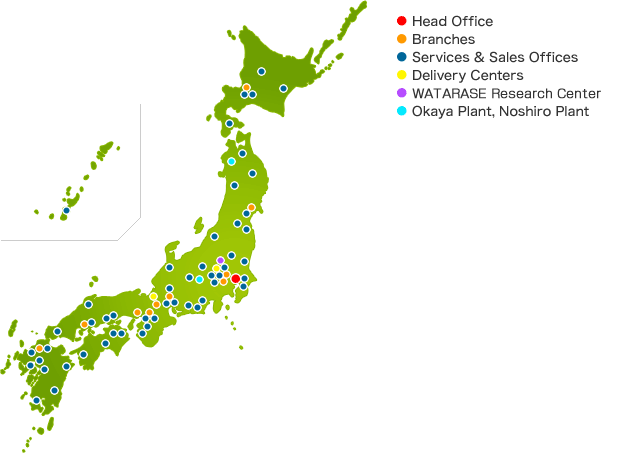
| Trade Name | KYORIN Pharmaceutical Co.,Ltd. |
|---|---|
| Business | Manufacture and sales of prescription medicines |
| Head Office | Ochyanomizu Sola City 16F, Kanda Surugadai 4-6, Chiyoda-ku, Tokyo 101-8311 Japan (Access Map) |
| Telephone | 03-3525-4711 |
| Foundation | 1923 |
| Establishment | 1940 |

 Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park –
Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park – .
.

Filed under: Phase2 drugs, Uncategorized Tagged: JAPAN, KRP-AM1977, KYORIN, lascufloxacin, Shimotsuga-gun, Tochigi







